

Abstract
Objective
This study is to determine if functional suppression of the catalytic domain of activation-induced cytidine deaminase (AID) can suppress the hyper-reactive germinal center responses in BXD2 mice.
Methods
A transgenic (Tg) BXD2 mouse expressing a dominant negative (DN) form of AID (Aicda-DN) at the somatic hypermutation (SHM) site was generated. Real-time RT-PCR was used to determine the expression of Aicda and DNA damage/repair genes. ELISA was used to measure sera levels of autoantibodies and immune complexes. Development of GCs and antibody containing immune complexes (ICs) as well as proliferative and apoptotic cells were determined using flow cytometry and/or immunohistochemistry analyses. Development of arthritis and kidney disease was evaluated histologically in 6-8 month-old mice.
Results
Suppression of the SHM function of AID resulted in a significant decrease in autoantibody production without affecting the expression of DNA damage-related genes in GC B cells of BXD2-Aicda-DN Tg mice. There was decreased proliferation, increased apoptosis, increased expression of caspase 9 mRNA in GC B cells, and lower numbers of GCs in the spleen of BXD2-Aicda-DN Tg mice. Decreased GC response was associated with lower IgG containing ICs. Anti-IgM and anti-Ig plus anti-CD40-induced B cell proliferative responses were decreased in BXD2-Aicda-DN Tg mice.
Conclusion
Inhibition of AID SHM function in BXD2 mice suppressed development of spontaneous GCs, generation of autoantibody producing B cells, and autoimmunity in BXD2 mice. Suppression of AID catalytic function that limits selection-based survival of GC B cells could become a novel therapy for the treatment of autoimmune disease.
Introduction
In both systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA), autoantibodies can be present for years before the onset of disease (1, 2). Importantly, autoantibodies have been shown to be directly pathogenic and can target different tissues including the kidney (3) or joint (4). High affinity self-reactive antibody production requires B cells to undergo somatic hypermutation (SHM) and class switch recombination (CSR), both of which require activation-induced cytidine deaminase (AID) in the germinal center (GC) (5, 6). High titers of anti-CCP autoantibodies have been shown to be correlated with increased AID in the synovium and peripheral blood of patients with RA (7).
Rituximab is an anti-CD20 antibody that depletes CD20+ B cells, including B-cell precursors and mature B cells but not plasma cells (8). Rituximab has been shown to be effective in RA (9, 10) but not SLE clinical trials, although it may be effective in treatment in SLE patients (11, 12). Sera titers of autoantibodies increase in the majority of patients after cessation of therapy (13) can result in the regeneration of a diverse and highly mutated polyclonal heavy-chain repertoire in certain RA patients (14). Patients who experienced a relapse of RA on return of B cells also tended to show repopulation with higher numbers of memory B cells (15). A therapy capable of intervening at the B-cell antibody development stage is important since this approach could prevent the generation of plasma/memory B cells or prolong the effects of Rituximab. Development of such therapy for autoimmune disease, however, remains a challenge. Blockade of CD40/CD154 interaction in SLE using a humanized anti-CD154 antibody has been associated with vascular complications including arterial occlusions (16). Another approach is blockade of follicular dendritic cell networks by the use of a decoy receptor, LT-β receptor-IgG1 lymphotoxin (baminercept, BG9924) (17) to disruption GC responses, however, this can exacerbate autoimmune arthritis by enhancing the Th1 response (18).
We have previously shown that BXD2 mice develop lupus, erosive arthritis, and large spontaneous GCs in the spleen that produce pathogenic autoantibodies as a result of increased expression of AID in B cells in the spleens of BXD2 mice (19, 20). This mouse model therefore can be used to evaluate the effects of AID inhibition in autoimmune conditions. Nonetheless, a complete deficiency of Aicda in normal mice has been shown to lead to enlarged GCs following exogenous antigen stimulation (21-23). This is thought to be due to a lack of AID-induced double stranded DNA strand breaks (DSBs) which results in abnormally low apoptosis of B cells in AID−/− mice (21-23). These results further suggest that complete elimination of AID in mice is not an ideal model to understand the potential therapeutic effects of AID suppression in humans.
To circumvent the difficulties associated with complete elimination of AID, we have generated BXD2 mice expressing an Aicda-S38A-H56R/E58Q dominant-negative (DN) transgene (BXD2-Aicda-DN Tg). The Aicda-H56R/E58Q DN transgene has been previously shown to specifically inhibit the post-cleavage step of the somatic hypermutation process (24). Therefore, functional suppression of AID using an Aicda-DN approach should overcome the perplexing problem of low apoptosis and enlarged GCs as seen in the AID−/− mice. To prevent the excessive CSR function of the Aicda-DN Tg, an additional Serine 38 (AGT) to Alanine 38 (GCT) mutation was introduced into the PKA phosphorylation site (25), leading to the generation of a S38A and H56R/E58Q-triple mutant of the Aicda-DN Tg. In the present study, we show that targeted inhibition of the catalytic domain of AID in BXD2 mice using the Aicda-DN Tg approach resulted in decreased autoantibody production and smaller GCs. Smaller GCs were the result of both decreased proliferation and increased apoptosis that were associated with dramatically lower levels of IgG containing immune complexes in the spleen follicles. Our results show that normalization of AID catalytic function can be used as a novel target to suppress autoimmune GC responses.
Materials and Methods
Aicda-DN construct
The wild-type (WT) Aicda gene was obtained by TA cloning and sequencing of the murine Aicda gene. The full length gene was obtained by primers (forward primer 5′- AGAAAGTCACGCTGGAGACC-3′ reversed primer 5′-ATGTTGCACAGCAAGCTCAG-3′). Single base pair mutations were introduced into WT Aicda using the GeneTailor Site-Directed Mutagenesis kit (Invitrogen). Mutations were introduced into the catalytic domain Histidine 56 (CAC) to Arginine 56 (CGC)/Glutamic acid 58 (GAA) to Glutamine 58 (CAA) (H56R/E58Q) using the following primer pair: Forward 5′-ACAAGTCTGGCTGCC(G)CGTG(C)AATTGTTGTT-3′; Reverse 5′-GGCAGCCAGACTTGTTGCGAAGGTGGCC-3′. The Serine 38 (AGT) to Alanine 38 (GCT) mutation was introduced into the PKA phosphorylation site using the following primer pair: Forward 5′-GGTGAAGAGGAGAGAT(GC)TGCCACCTCCTG-3′ and Reverse 5′-ATCTCTCCTCTTCACCACGTAGCAGAGG-3′. All mutations have been confirmed by sequencing. To induce constitutive expression of the Aicda-DN transgene, the chicken β actin (CAG) promoter was used to drive the expression of the Tg.
Mice
The BXD2-Aicda-DN Tg mouse was generated at the UAB Gene Targeting Core Facility (GTCF) using the standard protocol. All other mice were either bred at UAB or purchased from the Jackson Laboratory (Bar Harbor, Maine). All animal procedures were approved by The University of Alabama at Birmingham Institutional Animal Care and Use Committee.
Quantitative real-time PCR analysis (qRT-PCR)
qRT-PCR was carried out using the method we previously described (20, 26). Wild-type (WT) Aicda specific primers were designed at the 3′ untranslated region of WT Aicda. This sequence is not present in the Aicda-DN Tg: TTGGGTCGTGAATGATGC (F); CCTGAAAGTGAGCCTTAGAG (R); For the Aicda-DN specific transcript, Aicda specific primers were designed to recognize the 3′ non-coding segment of the transgene: TCTGTAGCGACCCTTTGC (F); TTCCACAACTATCCAACTCAC (R). Primer sequence used for other gene expression determination by real-time PCR is shown in Supplementary Table 1.
High affinity anti-NP response analysis
Mice were immunized i.p. with 50 μg of chicken γ globulin haptenated with 4-hydroxy-3-nitrophenylacetyl chicken gamma globulin (NP-CGG) (BioSearch Technologies) adsorbed to 1.3 mg alum (Sigma-Aldrich) in a total volume of 100 μl NP-CGG alum/PBS. High affinity anti-NP antibodies in the serum were measured by ELISA using NP2-bovine serum albumin (NP2-BSA, a low hapten density to detect high affinity anti-NP antibodies) (Biosearch Technologies) as the target antigens (27).
ELISA and ELISPOT
Measurement of serum levels of autoantibodies and determination of autoantibody producing B cells in the spleens was quantitated using an ELISPOT assay as we previous described (19, 20, 28). For both ELISA and ELISPOT, each well was coated with 5 μg/ml of the tested autoantigen. BiP was purchased from Assay Designs, Inc (Ann Arbor, Michigan). All other autoantigens were purchased from Sigma-Aldrich.
Circulating Immune Complexes (CIC) and urinary albumin measurement
A mouse CIC ELISA kit (Alpha Diagnostic International) was used to determine serum titers of IgG-containing CICs (28). The amount CIC was determined by incubation with isotype-specific secondary antibody (IgG2b and IgG2c). Albumin in urine was measured by a competitive ELISA (Albuwell M; Exocell, Philadelphia, PA) according to the manufacturer's instructions.
Immunohistochemical analysis of tissue sections
The tissues were fixed in 10% formaldehyde/PBS and paraffin-embedded. Sections (5 μm) were incubated with a rabbit monoclonal anti-mouse Ki67 antibody (clone SP6, Lab Visions/ Neomarkers, Fremont, CA) or HRP-conjugated goat anti-mouse IgG (Southern Biotech, Birmingham, AL). For the Ki67 staining, this was followed by a secondary goat anti-rabbit (111-065-144 from Jackson ImmunoResearch Laboratories, West Prove, PA, in 1 to 1000 dilution) with an AP- or HRP-streptavidin leveling (Covance Research Product, Inc., Denver, PA). The sections were then subjected to a standard streptavidin-peroxidase technique with the reaction being developed using a 3,3′-diaminobenzidine (DAB) substrate kit, ScyTek Laboratories, Logan, UT) or a FAST RED Chromogen System (SIGNET, Covance). Negative controls included omission of the primary antibodies. Quantitation of IgG+ glomeruli was carried out using the ImageJ program (developed at the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/). Background intensity was subtracted for each image.
TUNEL staining for the detection of apoptotic cells on tissue sections was carried out using the ApopTag Peroxidase in situ Apoptosis Detection Kit (S7100) according to the manufacture's instructions (Chemicon/Millipore, Billerica, MA).
Histologic evaluation of inflammatory cell infiltration and bone erosions
Joints (knee, elbow, ankle, and wrist) tissue and sections using the method we previously described (29). Joints were scored by a blinded observer for chronic arthritis damage based on the criteria we described previously (29). Five high power fields (hpf) were randomly selected for each joint for the evaluation of the average number of infiltrating inflammatory cells, synovial hyperplasia (SH), and Marginal erosions (ME). Each parameter was evaluated to obtain the average score of each parameter/hpf/joint/mouse.
Flow cytometry analysis and sorting
B cells were purified from single-cell spleen preparations using a positive selection column (Miltenyi Biotech). Flow cytometry was performed on fluorescently-labeled single cell suspensions derived from spleens using the method we described previously (19, 20, 30). For the detection or isolation of GC B cells, cells were labeled with biotin-conjugated PNA (Vector Laboratories, Burlingame, CA), PE-conjugated anti-Fas (clone Jo2), Alexa700 conjugated anti-CD19 (clone 6D5), and PE-Cy5- or Alexa450-conjugated streptavidin (SA) (Biolegend, San Diego, CA). For the detection of various cell types, including pDCs and B cells, fluorescent –conjugated anti-PDCA1 (eBioscience, San Diego, CA), anti-I-A/I-E (eBioscience), and anti-CD11c, anti-CD19, anti-IgD (all Biolegend Inc, San Diego CA), anti-IgM (Southern Biotech, Birmingham, AL), anti-CD21/35, anti-CD23, and anti-CD93/AA4 antibodies, anti-CD86 (clone GL1), anti-CD40 (clone 3/23), and anti-mouse B220 (clone RA3-6B2), were employed. Acquisition and gating of marginal zone (MZ) and marginal zone precursor (MZ-P) B cells was carried out based on the method we previously described (26). MZ B cells were defined as CD23null+lo sIgMhi sIgDlo CD21hi CD1dhi CD19+ cells; MZ-P B cells were defined as CD23hi sIgMhi sIgDhi CD21hi CD1dhi CD19+ cells (31).
Intracellular analysis of cytokines produced by the CD4+ T cells was carried out using the protocol we previously described (20). Cells were stained extracellularly with Alexa700–conjugated anti-CD4 (RM4-5), then fixed and permeabilized with Cytofix/Cytoperm solution (BD Biosciences) prior to intracellular staining with APC-conjugated anti-IFN-γ (XMG1.2) and PE-conjugated anti-IL-17 (TC11-18H10).
Intracellular BrdU staining was carried out using an Alexa 647-anti-BrdU antibody following the methods provided BD-Biosciences. Cells (≥100,000/sample) were analyzed by flow cytometry on a BD-LSR-II Flow Cytometer ® (Becton Dickenson). BrdU was administered 7 hrs and 1 hr prior to sacrifice of the mouse. The analysis was performed using FlowJo Software (Tree Star, Inc, Ashland, OR).
In vitro proliferative responses
Single-cell suspensions prepared from the spleens of mice were cultured in triplicate wells (2×105 cells/well) and stimulated with anti-IgM (2 μg/ml), anti-CD40 (HM40-3; Biolegend, 2 μg/ml) plus rabbit-anti-mouse Ig (clone 187.1, BD-Biosciences, 4 μg/ml), anti-CD3 (clone 145-2C11, 2 μg/ml) + anti-CD28 (clone 37.51, 5 μg/ml) or LPS (Escherichia coli 055:B5; Sigma-Aldrich, 10 μg/ml). The proliferative response was measured by a standard [3H]-thymidine incorporation assay at the 72 hr time point.
Statistical analysis
All results were shown as mean ± standard error of the mean (SEM). A two-tail t test was used when two groups were compared for statistical differences. ANOVA test was used when more than 2 groups were compared for statistical differences. A chi-square test was used to determine the differences in the incidence of arthritis development between WT BXD2 and BXD2-Aicda-DN Tg mice. P values less than 0.05 were considered significant.
Results
Aicda-DN Tg suppresses generation of high-affinity antibodies in BXD2 mice
AID has been shown to be mainly expressed by GC B cells (32, 33). We have determined the expression of Aicda in PNA+Fas+CD19+ GC B cells, PNA-Fas-CD19+ non-GC B cells, and CD19- non-B cells and found that there was 10-fold increased expression of Aicda in GC versus non-GC B and non-B cells in the spleen of BXD2 mice (Figure 1A), suggesting that the major actions of AID take place in GC B cells in BXD2 mice.
Figure 1.
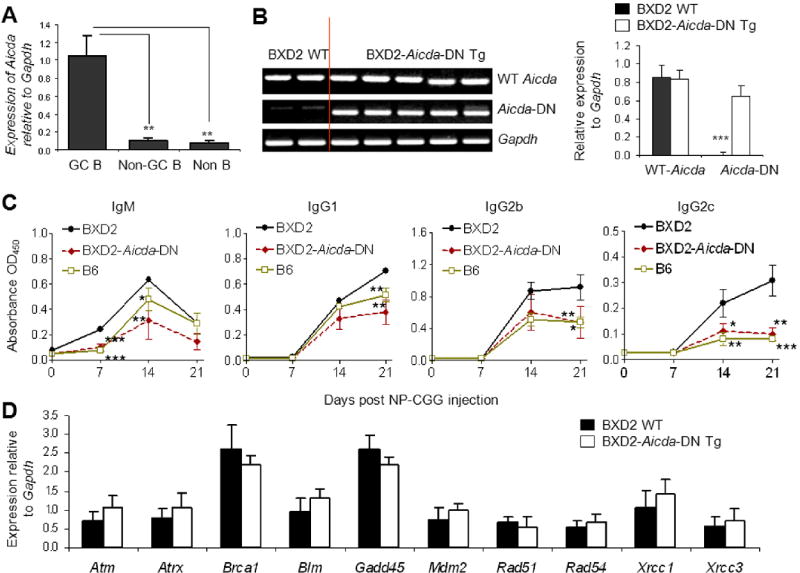
Aicda-DN Tg normalized antibody responses without affecting DNA damage response genes in GCs. A, qRT-PCR analysis of the expression of Aicda in FACS sorted GC B cells (PNA+Fas+CD19+), non-GC B cells (PNA-Fas-CD19+) and non-B cells (CD19-) in the spleens of 5-6-mo-old BXD2 mice. The results are shown as the expression of Aicda after normalization with the expression of Gapdh (** P < 0.01; N = 5 per group). B, C, D, An Aicda-DN Tg construct was injected directly into BXD2 single-cell embryos to generate BXD2-Aicda-DN Tg mice. B, Left: Gel electrophoresis of splenocyte RNA analysis of the expression of Aicda-DN Tg. Right: qRT-PCR analysis of the expression of WT-Aicda and Aicda-DN Tg (*** P <0.0001; N ≥ 6 per group). C, ELISA analysis of the anti-NP2 antibody response in NP-CGG-injected mice. Sera were collected at the indicated days after the immunization (* P < 0.05; ** P < 0.01; *** P < 0.005 between the indicated strains versus WT BXD2 mice; N ≥ 4 per group). D, qRT-PCR analysis of the expression of the indicated genes in FACS isolated PNA+Fas+ CD19+ GC B cells from the spleens of 5-6-mo-old mice (N ≥ 5 per group).
To specifically demonstrate that suppression of the SHM function of AID can suppress autoimmune disease in BXD2 mice, BXD2 Aicda-DN Tg mice were produced by introducing three mutations into mouse Aicda. The Aicda-DN Tg specific primers for a non-coding Tg sequence were used to specifically detect the levels of Aicda-DN Tg. There were nearly equivalent levels of wild-type (WT) Aicda expressed in the spleen of WT BXD2 compared to BXD2-Aicda-DN Tg mice (Figures 1B). Expression of the Aicda-DN Tg was not detected in WT BXD2 mice (Figures 1B).
We next evaluated if the Aicda-DN Tg suppressed antibody affinity maturation function in B cells of BXD2 mice. Antibody affinity maturation was determined by analysis of the production of high affinity antibody production after immunization of B6, BXD2, and BXD2-Aicda-DN Tg mice with NP-CGG. Affinity maturation of the anti-NP response after immunization with NP-CGG, including the IgM isotype, has been shown to require SHM of the V186.2/V3 genes of the J558 family to generate high-affinity anti-NP antibodies (34). There was more rapid development of the high affinity IgM anti-NP2 response at day 7 in WT BXD2 compared to that in normal B6 mice (Figure 1C). This transient increase in the IgM anti-NP2 antibodies in BXD2 mice was followed by increased levels of the high-affinity IgG1, IgG2b and IgG2c anti-NP2 antibodies from days 7 to 21 in BXD2 mice, compared to B6 mice, suggesting strong SHM and CSR responses in BXD2 mice. Interestingly, there was a significant decrease in all IgG isotypes (IgG1, IgG2b and IgG2c) high-affinity anti-NP2 antibody in the BXD2-Aicda-DN Tg mice compared to WT-BXD2 mice at day 21 (Figure 1C). Furthermore, the levels of the high-affinity IgG1, IgG2b and IgG2c anti-NP2 antibodies in BXD2-Aicda-DN Tg mice were equivalent to those in normal control B6 mice, suggesting that the presence of Aicda-DN Tg did not completely eliminate the anti-NP2 antibody response but suppressed it to normal levels.
Previous results by Papavasiliou and Schatz (24) indicated that catalytic functional suppression of AID by the H56R/E58Q DN form of AID did not perturb the AID mediated DSBs over the Ig variable regions. We determined the expression 10 genes that are important for regulation of DNA damage/repair responses, including Atm, Atrx, Brca1, Blm, Gadd45, Mdm2, Rad51, Rad54, Xrcc1 and Xrcc3 in PNA+Fas+ GC B cells isolated from the spleens of WT BXD2 and BXD2-Aicda-DN Tg mice. The results indicated that there was no significance difference in the expression of these genes comparing GC B cells from the two groups of mice (Figure 1D), suggesting that the presence of Aicda-DN Tg in BXD2 mice did not alter the expression of genes that regulates DNA damage and repair responses in GC B cells.
Decreased autoantibodies and ICs in BXD2 Aicda-DN Tg mice
The development of autoimmunity in the BXD2 mice is spontaneous and follows the emergence of pathogenic highly mutated autoantibodies that can induce the development of glomerulonephritis and arthritis (19, 28, 35). As compared with age-matched WT BXD2 mice, BXD2-Aicda-DN-Tg mice exhibited lower serum titers of IgG autoantibodies against BiP, histone, and DNA (Figure 2A). There were also lower numbers of IgG anti-BiP, anti-histone, and anti-DNA autoantibody-producing B cells in the spleen as determined by an ELISPOT assay (Figure 2B). There was also significantly lower circulating levels IgG2b, and IgG2c isotypes of C1q-containing ICs (Figure 2C) and decreased trapping of high-affinity IgG autoantibody-containing ICs in the spleen follicles of BXD2-Aicda-DN-Tg mice comparted to that in WT BXD2 mice (Figure 2D).
Figure 2.

Decreased ICs and autoantibody responses in 6-8-mo-old BXD2-Aicda-DN Tg mice. A, ELISA analysis of circulating autoantibodies in the sera (* P < 0.05; ** P < 0.01; and *** P < 0.005 between the indicated strains versus WT BXD2 mice; N ≥ 6 per group). B, ELISPOT analysis of the autoantibody producing B cell against the indicated autoantigens using B cells purified from the spleen of mice (* P < 0.05; ** P < 0.01; and *** P < 0.005 between the indicated strains versus WT BXD2 mice; N ≥ 6 per group). C, ELISA analysis of C1q containing ICs in the sera in 6-8-mo-old mice (* P < 0.05; ** P < 0.01; *** P < 0.005 comparing BXD2 vs. B6 and BXD2-Aicda-DN Tg mice; N ≥ 6 per group). D, Immunohistochemical staining of IgG+ cells in the spleen of 6-8-mo-old BXD2 and BXD2-Aicda-DN Tg mice. The objective lens used for acquiring each image is indicated. The photomicrographs shown are representative of the results obtained on analysis of tissue sections from 4 mice in each group.
Aicda-DN Tg suppresses autoimmunity in BXD2 mice
There were significantly decreased IgG glomerular deposits in the kidney of BXD2-Aicda-DN-Tg mice (Figure 3A). The proteinuria that developed in WT BXD2 mice was reduced significantly in the BXD2-Aicda-DN-Tg mice (Figure 3B). The arthritis in WT BXD2 mice is characterized by inflammatory infiltration (II), extensive synovial hyperplasia (SH) and marginal erosions (ME) of bone (Figure 3C). In the BXD2-Aicda-DN-Tg mice, there was minimal inflammatory infiltration, synovial hyperplasia and marginal erosions (Figure 3C). Less than 25% of BXD2-Aicda-DN-Tg mice had developed arthritis although the incidence of arthritis in WT BXD2 mice at this age (6-8-mo-old) is greater than 70% (Figure 3D). These results demonstrate that suppression of AID catalytic function not only suppresses autoantibody production but also inhibits development of arthritis and autoimmune renal disease in BXD2 mice.
Figure 3.

Decreased autoimmune disease in 6-8-mo-old BXD2-Aicda-DN Tg mice. A, Left: Immunohistochemistry staining of IgG antibody deposits on the glomerulus. The objective lens used for acquiring each image is indicated. Right: Quantitation of the intensity of IgG deposits in glomeruli (Results are shown as mean ± SEM, *** P < 0.005; at least 10 glomeruli were quantitated for each mouse, N ≥ 6 per group). B, Urinary albumin in WT BXD2 mice or BXD2-Aicda-DN Tg mice (mean ± SEM; * P < 0.05; N=10 mice/group). C, Left: H&E staining of joint sections in WT BXD2 and BXD2-Aicda-DN Tg mice. ME, marginal erosion; SH, synovial hyperplasia; II, Inflammatory Infiltrate (N ≥ 6 per group). Right: Inflammatory infiltration, synovial hyperplasia, and marginal erosions of the joints were scored using a 0 – 4 scoring system (* represents P < 0.05 and ** represents P < 0.005; N ≥ 6 per group). D, The incidence of arthritis determined by bilateral symmetric erythema and swelling of either the ankle or forelimb joint was determined in BXD2 or BXD2-Aicda-DN Tg mice at 6-8 month of age (*** represents P < 0.001; N ≥ 10 per group; Chi-square test).
Aicda-DN Tg suppresses GC responses in BXD2 mice
We found that the spleen of BXD2-Aicda-DN-Tg mice, at 5-6-mo of age, were considerably smaller than those of the age-matched WT BXD2 mice and there were significantly lower numbers of total cell count and CD19+ B cells in the spleen (Figure 4A). Functional inhibition of the abnormally upregulated AID resulted in a lower percentage of GC B cells (Figure 4B) in the spleen of BXD2 Aicda-DN Tg mice. The reduced percentage of GC B cells was associated with a significantly lower percentage of both BrdU+ cycling B cells and cycling PNA+Fas+ GC B cells in the spleen of the BXD2-Aicda-DN Tg mice (Figure 4B). There was a dramatic decrease in the number of proliferating GCs in BXD2-Aicda-DN Tg mice indicated by significantly fewer Ki67 positive follicles in the spleens (Figure 4C).
Figure 4.
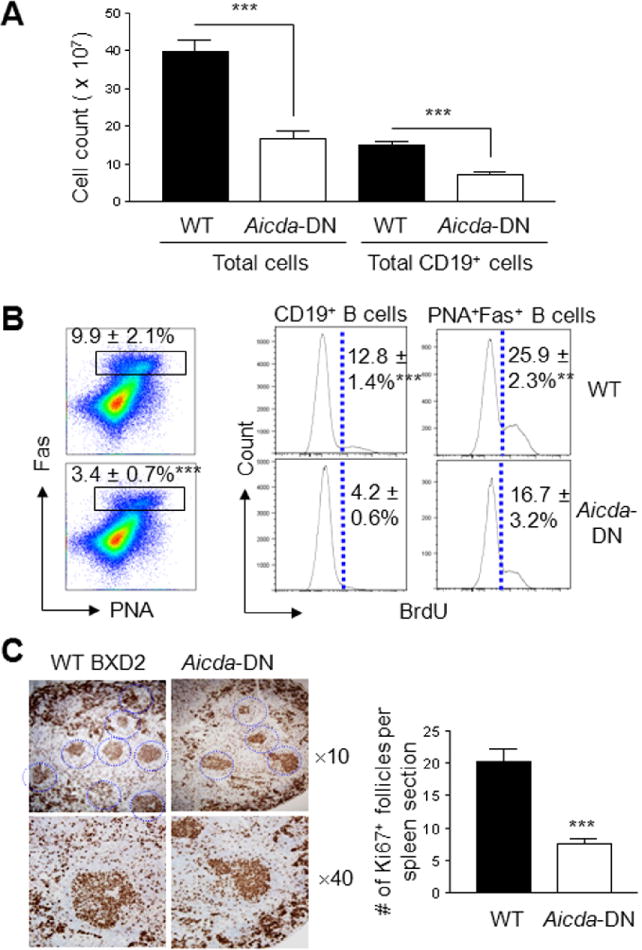
The Effects of Aicda-DN on in vivo GCs and B-cell proliferative responses. Spleen cellular composition and GCs were characterized in WT BXD2 and BXD2-Aicda-DN Tg mice at 5-6-mo of age. A, Hemocytometry quantitation of total spleen cell count and FACS quantitation of total spleen CD19+ B-cell count (*** P < 0.005; N ≥ 6 per group). B, Left: FACS analysis of percent of PNA+Fas+ B220+ GC B cells in WT BXD2 and BXD2-Aicda-DN Tg mice (*** P < 0.005; N ≥ 6 per group). Right: In vivo proliferation as determined by BrdU incorporation for CD19+ B cells and PNA+Fas+ CD19+GC B cells in WT BXD2 and BXD2-Aicda-DN Tg mice (*** P < 0.005; N ≥ 6 per group). C, Immunohistological analysis (left) and quantitation of the number of Ki67+ follicles per microscopic view (right, objective lens ×10 and ×40) in the spleen of WT BXD2 and BXD2-Aicda-DN Tg mice (*** P < 0.005; at least 4 randomly chosen views per section were counted; N ≥ 6 per group).
Aicda-DN Tg did not alter the autoimmune phenotypes at the pre-GC stage
To further demonstrate that the Aicda-DN-Tg acted specifically on GC B cells, we examined several other major autoimmune cellular pathogenic features that have been described to lead to increased autoreactive GC development in BXD2 mice (19, 20, 26). Interestingly, none of these pathways were altered in BXD2 mice by the Aicda-DN-Tg, including the percent of marginal zone precursor (MZ-P) B cells (Figure 5A), the percent of plasmacytoid dendritic cells (Figure 5B), the percent of IL-17 producing CD4 T cells (Figure 5C), and the percent of CD86 in both GC and non-GC B cells in the spleen of Aicda-DN Tg versus WT BXD2 mice (Figure 5D).
Figure 5.

Aicda-DN Tg did not alter the major immune abnormalities that predispose BXD2 mice to spontaneous GCs. A, FACS quantitation of the percent of CD23lo MZ and CD23hi MZ-P B cells within the IgMhiCD21hi compartment of B cells in the spleens (P > 0.05; N ≥ 6 per group). B, FACS quantitation of the percent of pDCs defined as PDCA1+ (left) or as MHC-II+CD11clow (right) cells in the indicated strains of mice (P > 0.05; N ≥ 6 per group). C, FACS analysis of IL-17 producing TH-17 or IFN-γ producing TH-1 CD4 T cells in the spleens (P > 0.05; N ≥ 6 per group). The represented plots were gated within the CD4+ T cells. D, FACS analysis of the percent of CD86+ GC (PNA+Fas+ B220+, solid curve) and non-GC (PNA-Fas- B220+, dashed curve) in the spleens of the indicated strains of mice. Representative histograms are shown in the left and the average of the percentage of CD86+ cells is shown in the right (P > 0.05; N ≥ 6 per group). All experiments were carried out using 4-mo-old mice.
Aicda-DN Tg enhances GC apoptosis in BXD2 mice
Ligation of the B cell antigen receptor (BCR) and CD40 plays a key role in promoting the GC reaction (36). Stimulation of spleen cells with anti-IgM or anti-Ig plus anti-CD40 revealed that the proliferative responses from BXD2-Aicda-DN Tg mice was dramatically lower compared to that from WT BXD2 cells (Figure 6A) although B cells from the spleen of WT BXD2 mice and BXD2-Aicda-DN Tg mice expressed equivalent levels of CD40 (Figure 6B). In contrast, stimulation of spleen cells from both strains with anti-CD3+anti-CD28 or LPS resulted in equivalent levels of the proliferative responses (Figure 6A), suggesting that the presence of Aicda-DN Tg in BXD2 mice mainly affected B cells and it diminished the responses of B cells to antigen and T-cell stimulation.
Figure 6.

Increased in vivo GC apoptosis in the spleens of BXD2-Aicda-DN Tg mice. A, Thymidine incorporation analysis of the splenocyte proliferative response by the indicated stimulation in 4-mo-old WT BXD2 and BXD2-Aicda-DN Tg mice (* P < 0.05; ** P < 0.01; N=6 per group). B, FACS analysis of the percent of CD40+ B cells in 4-mo-old mice. Representative histograms and the average of the percentage of CD40+ (gated within CD19+ cells) are shown (P > 0.05; N ≥ 6 per group). C, In situ TUNEL staining (left) and quantitation of TUNEL+ cells per follicle of the spleen (right) of 5-6-mo-old WT BXD2 and BXD2-Aicda-DN Tg mice (* P < 0.05; N ≥ 6 per group). D, qRT-PCR analysis of the expression of Casp9 in FACS isolated PNA+Fas+CD19+ GC B cells from 5-6-mo-old mice (*** P < 0.005; N ≥ 6 per group).
Following CD40L stimulation, apoptosis operates to eliminate GC-B-cells, unless activated GC-B-cells encounter a second signal via B-cell Ig receptors (37). The dramatically lower proliferative response to anti-IgM or anti-CD40 plus anti-Ig stimulation and the dramatic lower levels of IgG containing ICs in spleen follicles in BXD2 Aicda-DN Tg mice suggest the likelihood that B cells from BXD2 Aicda-DN Tg mice may exhibit increased apoptosis, compared to B cells from WT BXD2 mice. Indeed, there was a significantly higher number of TUNEL+ apoptotic cells per follicle in the spleen of the BXD2-Aicda-DN-Tg mice, compared to that in the spleen follicles of WT BXD2 mice (Figure 6C). Consistent with the increased number of apoptotic cells, the expression of caspase 9 (Casp9), which is involved in antigen-stimulated cell death of GC B cells (38, 39), was approximately 4-fold elevated in GC B cells isolated from BXD2-Aicda-DN-Tg spleen, compared to that in the spleen of WT BXD2 mice (Figure 6D).
Discussion
AID was identified by Dr. Honjo and colleagues as a critical enzyme for antibody maturation (32). To enable SHM or CSR processes, AID induces point mutations or S region DSBs, respectively, of the Ig genes (5, 6). Regulation of Aicda RNA expression is complex (40), and abnormal expression of AID is associated with the development of T-dependent GC responses, including the development of the ectopic GCs that occur in autoimmune patients (7, 41). In this regard, AID has emerged as a potential target for preventing the development of high affinity autoantibodies based on mouse models of autoimmune disease (19, 42, 43). Complete or partial deficiency of AID inhibits the development of autoimmunity in Aicda-/- or Aicda+/- MRL-Faslpr/lpr mice (42, 43). Complete deficiency of AID resulted in deficiency of SHM and CSR, although IgM antibodies that were produced were somewhat protected against certain organisms (22, 44, 45). Paradoxically, complete deficiency of AID in normal mice lead to enlarged GCs following exogenous antigen stimulation (21, 23), suggesting that undesirable side effects may arise with complete elimination of AID expression.
To evaluate the therapeutic effects of AID suppression for autoimmune disease, we have generated BXD2 mice expressing an Aicda-S38A-H56R/E58Q-DN Tg to avoid deficiency of DSBs and enlargement of GCs that are frequently associated with complete lack of AID (21-23). The expression of DNA damage and repair genes was equivalent between GC B cells of WT BXD2 and BXD2-Aicda-DN Tg mice, suggesting that the presence of Aicda-DN Tg in BXD2 mice did not affect DSB damage responses in GC B cells. The present study further showed that hyperactivity of AID is important for the induction of autoimmune disease in BXD2 mice. Suppression of the abnormally increased SHM function but not complete elimination of the expression levels of AID prevented the production of pathogenic autoantibodies and ameliorated the development of autoimmune joint and kidney disease. BXD2-Aicda-DN Tg mice exhibited >90% 1 year survival compared to approximately 50% 1 year survival for BXD2 mice. The most interesting finding in the present study, however, was the dramatically suppressed GC response which was characterized by suppressed B-cell proliferation and enhanced GC apoptosis in the spleen of BXD2-Aicda-DN Tg mice.
It may appear paradoxical that there are large GCs and decreased apoptosis in both Aicda-/- mice and BXD2 mice that have high Aicda expression. Survival of GC B cells is a unique process in that it depends on two mechanisms. One mechanism is DNA damage survival based on the balance between DNA damage and repair. The second mechanism is selection survival from apoptosis based on BCR affinity selection. Expression of Aicda has an opposite effect on these two survival mechanisms. Therefore, large GCs can result from conditions of low or no AID expression due to low DNA damage-related apoptosis (21-23), whereas large GCs can also result from conditions of high AID expression because of higher BCR selection survival.
Our data suggest that large GCs in BXD2 mice result from high AID expression together with higher BCR selection survival, which is normalized in BXD2-Aicda-DN Tg mice. BXD2-Aicda-DN Tg mouse B cells exhibited a lower proliferation in vivo, and after in vitro stimulation with anti-IgM or anti-CD40 plus anti-Ig. During the primary immune response, low-affinity antibodies of the IgM subclass are produced. Following antigen stimulation of B cells through the B-cell antigen receptor and CD40, B cells enter the GC microenvironment and begin to proliferate and undergo clonally expansion (46, 47). The presence of anti-Ig is essential as deficiency of this signal is associated with Fas-mediated apoptosis of GC B cells (37). Our results suggest that inhibition of the catalytic function of AID in BXD2-Aicda-DN Tg mouse is associated with impaired proliferation and survival signaling through BCR and CD40. An increased rate of cell death is a major characteristic of lower affinity clones of B cells in the GCs (48), and activation of caspase 9 has been associated with antigen receptor-mediated apoptosis of GC B cells (38, 39). These results are consistent with the finding of high Casp9 and higher number of apoptotic cells in the GCs of BXD2-Aicda-DN Tg mouse spleens. In contrast, BXD2 GC B cells have high survival based on affinity maturation and selection survival. SHM is diminished in BXD2-Aicda-DN Tg mice compared to WT BXD2 mice. In addition, there was no change in DNA damage/repair genes comparing GC B cells from BXD2 and BXD2-Aicda-DN Tg mice. These results suggest that decreased GC size in BXD2-Aicda-DN Tg mice are based on decreased GC B cells selection survival, whereas DNA damage-related apoptosis is unchanged compared to WT BXD2 mice.
Almost all features of autoimmune disease found in BXD2 mice, including increased marginal zone precursor B cells, increased plasmacytoid dendritic cells, increased numbers of IL-17 producing CD4 T cells, and increased expression of CD86 (19, 20, 26), were also seen in the BXD2 Aicda-DN-Tg mouse. These results suggest that the most likely target of Aicda-DN Tg to suppress autoimmune response in BXD2 mice is at the GC stage of B-cell development. Excessive levels of AID overdrives SHM and production of pathogenic autoantibodies as a central and necessary component of autoantibody production. Our results therefore suggest that AID overexpression during GC B cell development may act as one important control point for development of autoimmunity.
In summary, while our previous findings suggested that formation of autoimmune GCs promotes the expression of Aicda in the B cells of the WT BXD2 mice (20), the current studies support the concept that AID catalytic function plays an important role to sustain the spontaneous GCs in the BXD2 mice. Our studies further indicate that inhibitors that can specifically block the abnormally increased AID SHM function, or inhibitors of the B cell selection survival pathway, can be developed into a GC-targeted therapy to prevent autoantibody mediated autoimmune diseases. Such therapeutic strategy not only will be effective in preventing generation of hypermutated autoantibodies but also be effective in eliminating the GC response that foster the development of these autoantibody producing B cells. A specific block of over-reactive AID catalytic function may therefore be complementary to other B-cell therapy to eliminate the generation of plasma B cells (49, 50).
Supplementary Material
Acknowledgments
We thank Dr. Tim Townes at UAB for providing the CAG promoter. We thank Ms. Enid Keyser of the Analytic and Preparative Cytometry Facility - UAB Rheumatic Diseases Core Facility (P30 AR48311) and Mr. Marion L. Spell of the UAB Center for AIDS FACS Core (P30 AI027767) for operating the FACS instrument. We thank Mr. Larry Johnson and Mr. Jinju Zhang of the UAB Gene Targeting Core Facility (P30 AR48311) of generation of the BXD2-Aicda-DN Tg mice. We also thank Dr. Fiona Hunter for expert review of the manuscript and Ms. Carol Humber for excellent secretarial assistance.
This work was supported by the American College of Rheumatology Research and Education Foundation for the Within Our Reach: Finding a Cure for Rheumatoid Arthritis campaign, the Alliance for Lupus Research – Target Identification in Lupus program, the Department of Veterans Affairs Merit Review Grant 1I01BX000600-01, Daiichi-Sankyo, the National Institutes of Health Grants 1AI 071110-01A1 and ARRA 3RO1AI71110-02S1 (all to J.D.M.), the Lupus Research Institute Novel Research project and the Arthritis Investigator Award supported by the Arthritis Foundation (both to H.-C.H).
Abbreviations
- AID
activation-induced cytidine deaminase
- CSR
class-switch recombination
- DN
dominant negative
- DSBs
DNA double-strand breaks
- GC
germinal center
- IC
immune complexes
- NP-CGG
4-hydroxy-3-nitrophenylacetyl chicken gamma globulin
- RA
rheumatoid arthritis
- SHM
somatic hypermutation
- SLE
systemic lupus erythematosus
Footnotes
All authors claim to have no financial interests which could create a potential conflict of interest or the appearance of a conflict of interest with regard to the work.
Author Contributions: H.-C.H., and J.D.M. contributed to experimental design, data analysis and manuscript preparation; P.Y., Q.W. G.J. T.G., and J.L. contributed to all sample preparation, FACS analysis, and generation of BXD2-Aicda-DN Tg mice. J.W., C.R.S., and W.E.G. contributed to all histology sample preparation, imaging capture, and histology data analysis. T.L. and D.D.C. contributed to the anti-NP response analysis.
- Supplementary Table 1. qRT-PCR primers, amplicon size, and annealing temperature used.
References
- 1.Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349(16):1526–33. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- 2.Rantapaa-Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48(10):2741–9. doi: 10.1002/art.11223. [DOI] [PubMed] [Google Scholar]
- 3.Li QZ, Xie C, Wu T, Mackay M, Aranow C, Putterman C, et al. Identification of autoantibody clusters that best predict lupus disease activity using glomerular proteome arrays. J Clin Invest. 2005;115(12):3428–39. doi: 10.1172/JCI23587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferucci ED, Majka DS, Parrish LA, Moroldo MB, Ryan M, Passo M, et al. Antibodies against cyclic citrullinated peptide are associated with HLA-DR4 in simplex and multiplex polyarticular-onset juvenile rheumatoid arthritis. Arthritis & Rheumatism. 2005;52(1):239–246. doi: 10.1002/art.20773. [DOI] [PubMed] [Google Scholar]
- 5.Longerich S, Basu U, Alt F, Storb U. AID in somatic hypermutation and class switch recombination. Current Opinion in Immunology. 2006;18(2):164–174. doi: 10.1016/j.coi.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 6.Honjo T, Muramatsu M, Fagarasan S. Aid: How does it aid antibody diversity? Immunity. 2004;20(6):659–668. doi: 10.1016/j.immuni.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 7.Xu X, Hsu HC, Chen J, Grizzle WE, Chatham WW, Stockard CR, et al. Increased expression of activation-induced cytidine deaminase is associated with anti-CCP and rheumatoid factor in rheumatoid arthritis. Scand J Immunol. 2009;70(3):309–16. doi: 10.1111/j.1365-3083.2009.02302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keystone E. B cell targeted therapies. Arthritis Res Ther. 2005;7 3:S13–8. doi: 10.1186/ar1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350(25):2572–81. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 10.Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54(9):2793–806. doi: 10.1002/art.22025. [DOI] [PubMed] [Google Scholar]
- 11.Terrier B, Amoura Z, Ravaud P, Hachulla E, Jouenne R, Combe B, et al. Safety and efficacy of rituximab in systemic lupus erythematosus: results from 136 patients from the French AutoImmunity and Rituximab registry. Arthritis Rheum. 2010;62(8):2458–66. doi: 10.1002/art.27541. [DOI] [PubMed] [Google Scholar]
- 12.Merrill JT, Neuwelt CM, Wallace DJ, Shanahan JC, Latinis KM, Oates JC, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62(1):222–33. doi: 10.1002/art.27233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cambridge G, Leandro MJ, Edwards JC, Ehrenstein MR, Salden M, Bodman-Smith M, et al. Serologic changes following B lymphocyte depletion therapy for rheumatoid arthritis. Arthritis Rheum. 2003;48(8):2146–54. doi: 10.1002/art.11181. [DOI] [PubMed] [Google Scholar]
- 14.Rouziere AS, Kneitz C, Palanichamy A, Dorner T, Tony HP. Regeneration of the immunoglobulin heavy-chain repertoire after transient B-cell depletion with an anti-CD20 antibody. Arthritis Res Ther. 2005;7(4):R714–24. doi: 10.1186/ar1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leandro MJ, Cambridge G, Ehrenstein MR, Edwards JCW. Reconstitution of peripheral blood B cells after depletion with rituximab in patients with rheumatoid arthritis. Arthritis & Rheumatism. 2006;54(2):613–620. doi: 10.1002/art.21617. [DOI] [PubMed] [Google Scholar]
- 16.Looney RJ, Anolik J, Sanz I. B cells as therapeutic targets for rheumatic diseases. Curr Opin Rheumatol. 2004;16(3):180–5. doi: 10.1097/00002281-200405000-00003. [DOI] [PubMed] [Google Scholar]
- 17.Daridon C, Burmester GR, Dorner T. Anticytokine therapy impacting on B cells in autoimmune diseases. Curr Opin Rheumatol. 2009;21(3):205–10. doi: 10.1097/BOR.0b013e32832a0760. [DOI] [PubMed] [Google Scholar]
- 18.Han S, Zhang X, Marinova E, Ozen Z, Bheekha-Escura R, Guo L, et al. Blockade of lymphotoxin pathway exacerbates autoimmune arthritis by enhancing the Th1 response. Arthritis Rheum. 2005;52(10):3202–9. doi: 10.1002/art.21341. [DOI] [PubMed] [Google Scholar]
- 19.Hsu HC, Wu Y, Yang P, Wu Q, Job G, Chen J, et al. Overexpression of activation-induced cytidine deaminase in B cells is associated with production of highly pathogenic autoantibodies. J Immunol. 2007;178(8):5357–65. doi: 10.4049/jimmunol.178.8.5357. [DOI] [PubMed] [Google Scholar]
- 20.Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9(2):166–75. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 21.Fagarasan S, Muramatsu M, Suzuki K, Nagaoka H, Hiai H, Honjo T. Critical Roles of Activation-Induced Cytidine Deaminase in the Homeostasis of Gut Flora. Science. 2002;298(5597):1424–1427. doi: 10.1126/science.1077336. [DOI] [PubMed] [Google Scholar]
- 22.Hase K, Takahashi D, Ebisawa M, Kawano S, Itoh K, Ohno H. Activation-induced cytidine deaminase deficiency causes organ-specific autoimmune disease. PLoS One. 2008;3(8):e3033. doi: 10.1371/journal.pone.0003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaheen A, Boulianne B, Parsa JY, Ramachandran S, Gommerman JL, Martin A. AID constrains germinal center size by rendering B cells susceptible to apoptosis. Blood. 2009;114(3):547–54. doi: 10.1182/blood-2009-03-211763. [DOI] [PubMed] [Google Scholar]
- 24.Papavasiliou FN, Schatz DG. The activation-induced deaminase functions in a postcleavage step of the somatic hypermutation process. J Exp Med. 2002;195(9):1193–8. doi: 10.1084/jem.20011858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basu U, Chaudhuri J, Alpert C, Dutt S, Ranganath S, Li G, et al. The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature. 2005;438(7067):508–11. doi: 10.1038/nature04255. [DOI] [PubMed] [Google Scholar]
- 26.Wang JH, Li J, Wu Q, Yang P, Pawar RD, Xie S, et al. Marginal Zone Precursor B Cells as Cellular Agents for Type I IFN-Promoted Antigen Transport in Autoimmunity. J Immunol. 2009 doi: 10.4049/jimmunol.0900870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelsoe G, Reth M, Rajewsky K. Control of idiotope expression by monoclonal anti-idiotope and idiotope-bearing antibody. Eur J Immunol. 1981;11(5):418–23. doi: 10.1002/eji.1830110513. [DOI] [PubMed] [Google Scholar]
- 28.Hsu HC, Zhou T, Kim H, Barnes S, Yang P, Wu Q, et al. Production of a novel class of polyreactive pathogenic autoantibodies in BXD2 mice causes glomerulonephritis and arthritis. Arthritis Rheum. 2006;54(1):343–55. doi: 10.1002/art.21550. [DOI] [PubMed] [Google Scholar]
- 29.Hsu HC, Zhang HG, Song GG, Xie J, Liu D, Yang PA, et al. Defective Fas ligand-mediated apoptosis predisposes to development of a chronic erosive arthritis subsequent to Mycoplasma pulmonis infection. Arthritis Rheum. 2001;44(9):2146–59. doi: 10.1002/1529-0131(200109)44:9<2146::AID-ART368>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 30.Xie S, Li J, Wang JH, Wu Q, Yang P, Hsu HC, et al. IL-17 activates the canonical NF-kB signaling pathway in autoimmune B cells of BXD2 mice to upregulate the expression of RGS16. J Immunol. 2010 doi: 10.4049/jimmunol.0903133. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allman D, Pillai S. Peripheral B cell subsets. Curr Opin Immunol. 2008;20(2):149–57. doi: 10.1016/j.coi.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, et al. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem. 1999;274(26):18470–6. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 33.Tran TH, Nakata M, Suzuki K, Begum NA, Shinkura R, Fagarasan S, et al. B cell-specific and stimulation-responsive enhancers derepress Aicda by overcoming the effects of silencers. Nat Immunol. 2010;11(2):148–54. doi: 10.1038/ni.1829. [DOI] [PubMed] [Google Scholar]
- 34.Takahashi Y, Dutta PR, Cerasoli DM, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. V Affinity maturation develops in two stages of clonal selection. J Exp Med. 1998;187(6):885–95. doi: 10.1084/jem.187.6.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mountz JD, Yang P, Wu Q, Zhou J, Tousson A, Fitzgerald A, et al. Genetic segregation of spontaneous erosive arthritis and generalized autoimmune disease in the BXD2 recombinant inbred strain of mice. Scand J Immunol. 2005;61(2):128–38. doi: 10.1111/j.0300-9475.2005.01548.x. [DOI] [PubMed] [Google Scholar]
- 36.Voort Rvd, Taher TEI, Keehnen RMJ, Smit L, Groenink M, Pals ST. Paracrine Regulation of Germinal Center B Cell Adhesion through the c-Met-Hepatocyte Growth Factor/Scatter Factor Pathway. J Exp Med. 1997;185(12):2121–2131. doi: 10.1084/jem.185.12.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi Y. Differentiation and apoptosis of human germinal center B-lymphocytes. Immunologic Research. 1997;16(2):161–174. doi: 10.1007/BF02786360. [DOI] [PubMed] [Google Scholar]
- 38.Bouchon A, Krammer PH, Walczak H. Critical role for mitochondria in B cell receptor-mediated apoptosis. Eur J Immunol. 2000;30(1):69–77. doi: 10.1002/1521-4141(200001)30:1<69::AID-IMMU69>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 39.Herold MJ, Kuss AW, Kraus C, Berberich I. Mitochondria-dependent caspase-9 activation is necessary for antigen receptor-mediated effector caspase activation and apoptosis in WEHI 231 lymphoma cells. J Immunol. 2002;168(8):3902–9. doi: 10.4049/jimmunol.168.8.3902. [DOI] [PubMed] [Google Scholar]
- 40.Lee-Theilen M, Chaudhuri J. Walking the AID tightrope. Nat Immunol. 2010;11(2):107–9. doi: 10.1038/ni0210-107. [DOI] [PubMed] [Google Scholar]
- 41.Humby F, Bombardieri M, Manzo A, Kelly S, Blades MC, Kirkham B, et al. Ectopic lymphoid structures support ongoing production of class-switched autoantibodies in rheumatoid synovium. PLoS Med. 2009;6(1):e1. doi: 10.1371/journal.pmed.0060001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang C, Foley J, Clayton N, Kissling G, Jokinen M, Herbert R, et al. Abrogation of lupus nephritis in activation-induced deaminase-deficient MRL/lpr mice. J Immunol. 2007;178(11):7422–31. doi: 10.4049/jimmunol.178.11.7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang C, Zhao ML, Diaz M. Activation-induced deaminase heterozygous MRL/lpr mice are delayed in the production of high-affinity pathogenic antibodies and in the development of lupus nephritis. Immunology. 2009;126(1):102–13. doi: 10.1111/j.1365-2567.2008.02882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kasahara Y, Kaneko H, Fukao T, Terada T, Asano T, Kasahara K, et al. Hyper-IgM syndrome with putative dominant negative mutation in activation-induced cytidine deaminase. J Allergy Clin Immunol. 2003;112(4):755–60. doi: 10.1016/s0091-6749(03)01860-8. [DOI] [PubMed] [Google Scholar]
- 45.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102(5):553–63. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 46.Bergthorsdottir S, Gallagher A, Jainandunsing S, Cockayne D, Sutton J, Leanderson T, et al. Signals that initiate somatic hypermutation of B cells in vitro. J Immunol. 2001;166(4):2228–34. doi: 10.4049/jimmunol.166.4.2228. [DOI] [PubMed] [Google Scholar]
- 47.Kallberg E, Jainandunsing S, Gray D, Leanderson T. Somatic mutation of immunoglobulin V genes in vitro. Science. 1996;271(5253):1285–9. doi: 10.1126/science.271.5253.1285. [DOI] [PubMed] [Google Scholar]
- 48.Anderson SM, Khalil A, Uduman M, Hershberg U, Louzoun Y, Haberman AM, et al. Taking Advantage: High-Affinity B Cells in the Germinal Center Have Lower Death Rates, but Similar Rates of Division, Compared to Low-Affinity Cells. J Immunol. 2009;183(11):7314–7325. doi: 10.4049/jimmunol.0902452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Isenberg DA. B cell targeted therapies in autoimmune diseases. J Rheumatol Suppl. 2006;77:24–8. [PubMed] [Google Scholar]
- 50.Silverman GJ. Targeting of B cells in SLE: rationale and therapeutic opportunities. Bull NYU Hosp Jt Dis. 2006;64(1-2):51–6. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.