

Abstract
Glioblastoma (GBM) is the most common and lethal primary malignant tumor of the central nervous system, and effective therapeutic options are lacking. The phosphatidylinositol 3-kinase (PI3K) pathway is frequently dysregulated in many human cancers, including GBM. Agents inhibiting PI3K and its effectors have demonstrated preliminary activity in various tumor types and have the potential to change the clinical treatment landscape of patients with solid tumors. In this review, we describe the activation of the PI3K pathway in GBM, explore why inhibition of this pathway may be a compelling therapeutic target for this disease, and provide an update of the data on PI3K inhibitors in clinical trials and from earlier investigation.
Keywords: AKT; glioblastoma; GBM; mTOR, PI3K
Glioblastoma (GBM; WHO grade IV astrocytoma) is the most common and aggressive primary malignant tumor of the CNS.1,2 Approximately 10 000 persons (3.19 cases per 100 000 population) receive a diagnosis of GBM each year in the United States.2 Because GBM tumors are characterized by invasive and diffuse growth, surgical control of the disease is difficult. Currently, the standard treatment for GBM entails maximal surgical resection, followed by radiation with concurrent and adjuvant chemotherapy to treat the residual infiltrative component of the tumor.3,4 Despite these aggressive treatment modalities, GBMs are notoriously resistant to radiation and chemotherapy, and, invariably, the disease returns. Thus, GBM presents a grave prognosis and patients have an expected 1-year survival rate of 34% and a 5-year survival rate of only 5%.2
In contrast to other tumor types, such as chronic myeloid leukemia5 or gastrointestinal stromal tumors,6 targeted therapy has made only a minor impact on the natural history and clinical management of GBM. Advances in the understanding of the pathogenesis of GBM and of several of the different molecular aberrations in GBM, including those occurring in phosphatase and tensin homolog (PTEN),7 epidermal growth factor receptor (EGFR), p16/p19, and p53, have led to a new era of exciting possibilities for the treatment of GBM.8 In 2009, for example, the humanized anti–vascular endothelial growth factor (anti-VEGF) monoclonal antibody bevacizumab (Avastin) received accelerated approval for GBM, representing the only new therapy to be approved in GBM in more than a decade and the first targeted agent to be approved to treat this disease.9 Although bevacizumab treatment has achieved significantly increased rates of tumor response and progression-free survival, compared with historical controls,10,11 the benefit in overall survival has been more modest. Nonetheless, these data provide an encourage further research of targeted agents to treat GBM. However, the development of efficacious targeted therapies for GBM is impeded by the complex, heterogeneous nature of the disease and the difficulty of passing through the blood-brain barrier. To successfully develop new therapeutic approaches for GBM, combination approaches targeting several molecular pathways will likely yield more effective treatments. Despite our advancing knowledge of the genetic alterations involved in the disease, the identification of multiple attractive molecular targets, and the availability of bevacizumab, GBM remains associated with significant morbidity and mortality, underscoring the need for improved therapeutic approaches.
The essential role of phosphatidylinositol 3-kinase (PI3K) signaling in proliferation, cellular metabolism, and apoptosis may represent a therapeutic target with the potential for improvement in clinical outcome for GBM. Inhibiting the PI3K pathway alone or in combination with other oncogenic pathways could induce a cytostatic effect and induction of cell death and may also have more far-ranging outcomes, including anti-angiogenic effects. Here, we describe the PI3K pathway in GBM, the characteristics that make it a compelling therapeutic target, and the data of PI3K inhibitors in clinical trials and those under investigation for the treatment of patients with this disease. We further discuss the evaluation of potential biomarkers for appropriate patient selection and the assessment of effects of PI3K inhibitors that have been incorporated into ongoing clinical trials.
The PI3K Pathway in GBM
The PI3K family of intracellular lipid kinases is involved in diverse signaling pathways that regulate proliferation, differentiation, migration, metabolism, and survival.12–14 PI3Ks are grouped into 3 classes (I–III) according to their substrate preference and sequence homology, and each class has distinct roles in cellular signaling. Class I PI3Ks are further divided into 2 subgroups (A and B). The pathway described in this section will focus on class IA PI3Ks.
Upstream receptor tyrosine kinases (RTKs), such as EGFR, activate class IA PI3Ks. The binding of the p85 regulatory subunit of PI3K to phosphotyrosine residues on activated RTKs leads to a conformational change in p85, releasing the inhibition of the catalytic subunit p110 (α, β, and δ isoforms) of PI3K. PI3K localizes to the plasma membrane and catalyzes the formation of phosphatidylinositol 3,4,5-trisphosphate (PIP3) through the phosphorylation of phosphatidylinositol 4,5-bisphosphate (PIP2). PIP3 is a critical activator of the serine/threonine kinase AKT (also known as protein kinase B). Binding of PIP3 to AKT leads to the membrane recruitment of AKT and subsequent phosphorylation by PDK1 (3-phosphoinositide-dependent kinase) and the mammalian target of rapamycin complex 2 (mTORC2). After activation, AKT is recruited to the cellular membrane and, in turn, phosphorylates, activates, or inhibits numerous target proteins involved in regulating cellular metabolism, survival and motility, protein synthesis, and vesicle sorting (Fig. 1).13–16
Fig. 1.
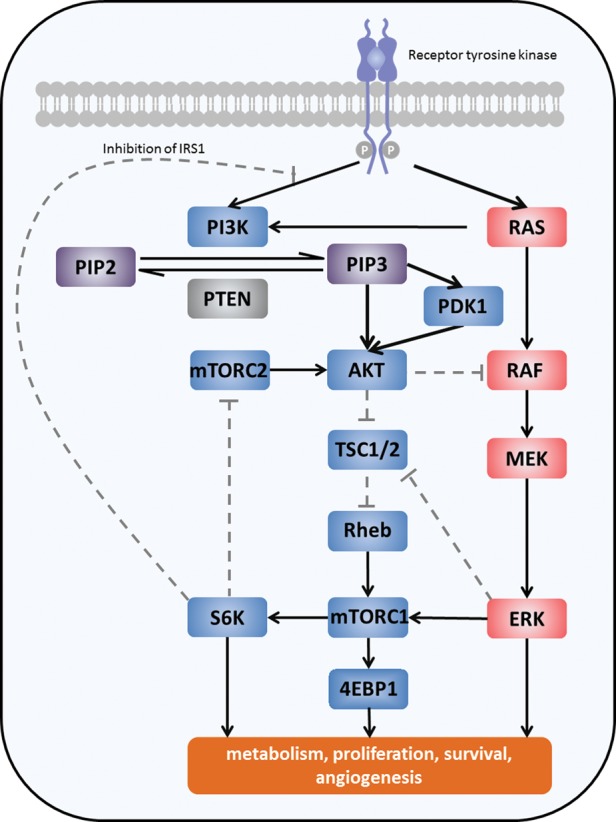
Schematic representation of the PI3K pathway and its main components. Solid lines represent activation; dashed lines represent inhibition. 4EBP1, eukaryotic initiation factor 4E binding protein 1; ERK, extracellular signal-related kinase; IRS1, insulin receptor substrate 1; MEK, mitogen-activated protein/ERK kinase; mTORC1/2, mammalian target of rapamycin complex 1/2; PI3K, phosphatidylinositol 3-kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PDK1, 3-phosphoinositide dependent protein kinase-1; PTEN, phosphatase and tensin homolog; RAS, rat sarcoma oncogene; Rheb, rat homolog enriched in the brain; S6K, S6 kinase; TSC, tuberous sclerosis protein.
The PI3K pathway is negatively regulated by various proteins. PTEN and the SH2 domain-containing inositol phosphatases antagonize PI3K activity through the dephosphorylation of PIP3 to the biologically inactive PIP2. In addition, activation of the mTOR complex1 (mTORC1) effector, p70 S6 kinase (S6K), modulates PI3K activation through negative feedback inhibition.17,18
Because the signaling components of the PI3K/AKT/mTOR pathway are central regulators of cell proliferation, growth, differentiation, survival, and angiogenesis, it is not surprising that dysregulation of this pathway through genetic alterations in several proteins in the PI3K signaling pathway, including p85, p110, PTEN, and AKT, has been demonstrated to play a pivotal role in the pathogenesis of cancer.19–24 Tyrosine kinase expression and subsequent signaling, specifically EGFR signaling, have long been implicated in the pathogenesis of GBM.25,26 Amplification of EGFR in GBM leads to activation of the PI3K pathway and has been noted in approximately 45% of GBM cases.27 Amplification or activating mutations of PIK3CA, the gene encoding the p110α subunit of PI3K, or PIK3R1, the gene encoding the p85 regulatory subunit of PI3K, have been demonstrated in approximately 15% of patients with GBM.21,24,28 Similarly, loss of function mutations, chromosomal deletions, or epigenetic gene silencing of PTEN have been found in approximately 40% of GBM cases29 and have been shown to lead to poor survival.30 In fact, alterations of at least one of the EGFR, PTEN, or PIK3CA genes have been detected in 63%–86% of primary and 31% of secondary GBM.28,29,31 Taken together, these data highlight the importance of this pathway in the pathophysiology of this disease.
PI3K Pathway Inhibitors and Their Preliminary Clinical Results
Because of the aberrant hyperactivation of the PI3K pathway, inhibition of its components presents an attractive target for cancer therapeutics. There has been a tremendous effort to develop PI3K pathway inhibitors for the treatment of cancer (Table 1). For example, the rapamycin analogs everolimus (Afinitor) and temsirolimus (Torisel), both of which inhibit mTORC1, have been approved for the treatment of advanced renal cell carcinoma.32,33 Everolimus is also indicated for the treatment of subependymal giant cell astrocytoma (SEGA) associated with tuberous sclerosis and progressive neuroendocrine tumors of pancreatic origin (PNET) in some patients.34,35 In patients with GBM, these rapalogs, as single agents or in combination with other agents and/or radiation, have yielded mostly infrequent and short-lived responses.36–39 However, the results of these studies have led to a more profound understanding of the PI3K pathway in GBM and the development of potentially more efficacious and better tolerated agents. Here, we will briefly summarize data of select PI3K pathway inhibitors currently in clinical development and highlight studies that investigate some of these inhibitors specifically in GBM.
Table 1.
PI3K/AKT/mTOR pathway inhibitors currently in clinical development
| Agent, manufacturer | Target | Tumor types investigated | Clinical development |
|---|---|---|---|
| Pan-PI3K inhibitors | |||
| BKM120, Novartis | pan-PI3K inhibitor | Solid tumors, prostate cancer, CRC, ER+ mBC, Her2+ mBC, GBM, hematologic malignancies, NSCLC, and endometrial cancer | Phase I/II |
| XL147, Exelixis | pan-PI3K inhibitor | GBM, endometrial cancer, ovarian cancer, NSCLC, Her2+ breast cancer, lymphoma, and solid tumors | Phase II |
| PX866, Oncothyreon | pan-PI3K inhibitor | GBM, NSCLC, CRC, HNSCC, prostate cancer, and solid tumors | Phase II |
| GDC-0941, Genentech | pan-PI3K inhibitor | ER+ mBC, Her2+ mBC, NSCLC, NHL, and solid tumors | Phase II |
| GSK2126458, GlaxoSmithKline | pan-PI3K inhibitor | Lymphoma and solid tumors | Phase I |
| BAY 80-6946, Bayer | pan-PI3K inhibitor | Solid tumors | Phase I |
| Isoform-specific inhibitors | |||
| BYL719, Novartis | PI3K p110α | Solid tumors | Phase I |
| GS-1101 (CAL-101), Gilead/Calistoga | PI3K p110δ | CLL, NHL, and hematologic malignancies | Phase II |
| Dual PI3K/mTOR inhibitors | |||
| BEZ235, Novartis | PI3K/mTOR | Solid tumors and HR+ mBC | Phase I/II |
| XL765, Exelixis | PI3K/mTOR | GBM, NSCLC, and solid tumors | Phase Ib/II |
| GDC-0980, Genentech | PI3K/mTOR | mBC, NHL, and solid tumors | Phase II |
| SF1126, Semafore Pharmaceuticals | PI3K/mTOR | Solid tumors | Phase I |
| PF-4691502, Pfizer | PI3K/mTOR | Solid tumors | Phase I |
| AKT inhibitors | |||
| Perifosine (KRX-0401), Keryx | AKT | CRC, MM, glioma, hematologic malignancies, lymphoma, ovarian cancer, NSCLC, RCC, sarcoma, and solid tumors | Phase III |
| GDC-0068, Genentech | AKT | Solid tumors | Phase I |
| MK2206 (Merck) | AKT | Solid tumors | Phase II |
| RX-0201, Rexahn Pharmaceuticals | AKT anti-sense | Pancreatic cancer | Phase II |
| GSK2110183, GlaxoSmithKline | AKT | MM, hematologic malignancies, and Langerhans cell histiocytosis | Phase I |
| GSK2141795, GlaxoSmithKline | AKT | Lymphoma and solid tumors | Phase I |
| PBI-05204, Phoenix Biotechnology | AKT | Solid tumors | Phase I |
| VD-0002, VioQuest Pharmaceutical | AKT | Solid tumors and hematologic malignancies | Phase I |
| Dual AKT/mTOR inhibitors | |||
| MKC-1, EntreMed | AKT/mTOR | Ovarian cancer, NSCLC, breast cancer, metastatic pancreatic cancer, and metastatic CRC | Phase II |
| mTORC inhibitors | |||
| Everolimus, Novartis | mTORC1 | RCC, HCC, lymphoma, mBC, and GBM | Approved for RCC, PNET and SEGA |
| Temsirolimus, Wyeth | mTORC1 | Various tumor types | Approved for RCC |
| Ridaforolimus, Merck/Ariad | mTORC1 | Sarcoma, endometrial cancer, mBC, prostate cancer, Her2+ mBC, NSCLC, sarcoma, and solid tumors | Phase III |
| OSI-027, Astellas/OSI Pharmaceutical | mTORC1/2 | Solid tumors and lymphoma | Phase I |
| AZD8055, AstraZeneca | mTORC1/2 | GBM, HCC, and solid tumors | Phase I/II |
| AZD2014, AstraZeneca | mTORC1/2 | Solid tumors | Phase I |
| INK128, Intellikine | mTORC1/2 | mBC, NHL, MM, and solid tumors | Phase I |
| CC-223, Celgene | mTORC1/2 | NHL and MM | Phase I |
CLL, chronic lymphocytic leukemia; CRC, colorectal carcinoma; ER, estrogen receptor; GBM, glioblastoma; HCC, hepatocellular carcinoma; Her2, human epidermal growth factor receptor-2; HNSCC, head and neck squamous cell carcinoma; HR, hormone receptor; mBC, metastatic breast cancer; MM, multiple myeloma; NHL, Non-Hodgkin's lymphoma; NSCLC, non-small cell lung cancer; PI3K, phosphatidylinositol 3-kinase; PNET, pancreatic neuroendocrine tumors; RCC, renal cell carcinoma; SEGA, subependymal giant cell astrocytoma.
PI3K Inhibitors
Two types of PI3K inhibitors target the p110 catalytic subunit: however, there are both pan-PI3K inhibitors and isoform-specific PI3K inhibitors. Pan-PI3K inhibitors are active against all family members of PI3K and include wortmannin derivatives, whereas isoform-specific inhibitors selectively inhibit p110α, β, or δ.
BKM120 (Novartis) is a pan-class I PI3K inhibitor without mTOR and Vps34 activity. It inhibits wild-type PI3Kα with an IC50 of 35 nM and also inhibits the other 3 PI3K paralogs (PI3Kβ, -γ, -δ) with IC50 values of 108–348 nM. Preclinical and early clinical data demonstrate that BKM120 has anti-proliferative and pro-apoptotic activity in a variety of tumor cell lines, xenografts, and patients with cancers associated with PI3K activating mutations.40–43 BKM120 demonstrated a dose-dependent growth inhibition in GBM cell lines (including U87, U251, LN229, LN18, and D54) regardless of their PTEN and/or EGFR status.40,41,43 Furthermore, oral BKM120 was well-tolerated by mice harboring intracerebral U87 xenografts and prolonged their survival.43 BKM120 can also penetrate the blood-brain barrier, making it an attractive option for the treatment of GBM. In a single-agent, multi-center, open-label, dose-escalation phase Ia study, with a maximum tolerated dose (MTD) expansion arm in patients with advanced solid tumors, BKM120 was generally well-tolerated and showed biologic and anti-tumor activity.44 The MTD was 100 mg/day. Dose-limiting toxicities (DLTs) were experienced in 9 patients: hyperglycemia (grade 4 in 2 patients), upper abdominal pain (grade 3 in 1 patient), skin rash (grade 3 in 1 patient), and mood alteration (grade 2 in 2 patients and grade 3 in 1 patient). Of the evaluable patients, 3 (5%) of 66 patients had partial responses (2 unconfirmed, 1 confirmed), 28 (42%) of 66 had stable disease as a best response, and 20 patients experienced durable response (more than 16 weeks). Of the partial responses, 2 patients had breast cancer; 1 patient had a triple-negative (estrogen receptor-negative [ER-], progesterone receptor-negative [PR-], and human epidermal growth factor receptor-2-negative [Her2-]) tumor, and the other had an ER+/PR+/Her2- tumor. The third partial response was in a patient with parotid gland ductal carcinoma. Frequent adverse events in this trial were those common to most PI3K inhibitors: fatigue, rash, and gastrointestinal toxicities (including nausea, diarrhea, and decreased appetite). The only grade 3/4 adverse event suspected to be related to the study drug was the other had an hepatotoxicity (transaminase increase, hyperbilirubinemia; 11%), which occurred in 5% or more of patients. A unique adverse event noted with BKM120 that has not been observed clinically with other PI3K inhibitors to date was mood changes (including mood alterations, emotional disorders, and affective disorders), although its relationship to BKM120 is still being analyzed. In addition, hyperglycemia occurred in approximately 30% of the patients, with only 4 (5%) experiencing grade 3 events. Hyperglycemia is not necessarily an unexpected toxicity for PI3K pathway inhibitors, because this pathway is involved in cellular metabolism and glucose/insulin regulation, and hyperglycemia has been noted in preclinical and clinical studies.45–51 Overall, there are approximately 20 ongoing and upcoming phase I and II trials with BKM120 administered as either a single agent or in combination therapy. In GBM, there is an ongoing phase II trial of BKM120 among patients in first or second recurrence with evidence of activation of the PI3K pathway (NCT01339052). Patients in this study are stratified according to their PTEN and PIK3CA mutational status to determine which of these subgroups may be more sensitive to PI3K inhibition. In addition, a phase I trial of BKM120 with radiation therapy and temozolomide in patients with newly diagnosed GBM (NCT01473901) and a phase I/II trial of BKM120 with bevacizumab in patients with recurrent GBM (NCT01349660) are ongoing. Phase II trials of BKM120 are also ongoing in endometrial cancer, castration-resistant metastatic prostate cancer, and non-small cell lung cancer (NSCLC).
PX-886 (Oncothyreon) is a semi-synthetic derivative of wortmannin and irreversibly inhibits PI3K through the formation of a covalent bond with PI3K. The primary metabolite of PX-866, 17-OH, is even more potent than the parent compound against the α and β isoforms of PI3K and has improved potency against forms of PI3Kα that contain activating mutations. In a panel of human tumor xenografts, the presence of PI3KCA mutations and the loss of PTEN activity were positive predictors of response to PX-866, whereas oncogenic RAS mutations were a predictor for resistance.52 In glioma cells, PX-866 dramatically inhibited proliferation in a variety of cell lines, with greater sensitivity seen in PTEN-negative cell lines, where IC50 values were 3-fold lower (low μM range) than in PTEN-positive cell lines. PX-866 also resulted in increased autophagy and decreased the invasive and angiogenic potential of cells. In human U87 mouse xenograft models, PX-866 inhibited subcutaneous tumor growth and increased the median survival time of animals with intracranial tumors.53 Results from a single-agent, phase I open-label, dose-escalation study of PX-866 in patients with advanced solid tumors who had failed or were intolerant to standard therapies demonstrated that PX-866 was well-tolerated using both intermittent (once daily on days 1–5 and 8–12 of a 28-day cycle) and continuous daily dosing.54 Overall, 13 (22%) of 60 patients treated with PX-866 had stable disease after a median 57 days (range, 4–235 days) on study. The most common adverse events were diarrhea, nausea, vomiting, fatigue, and alanine aminotransferase and aspartate aminotransferase level elevation (the latter with continuous dosing). The MTD was 12 and 8 mg with intermittent and continuous dosing, respectively. A phase II trial is evaluating the efficacy and safety of daily PX-866 in patients with relapsed GBM at first relapse as assessed by objective response and early progression rates (NCT01259869).
XL147 (SAR245408; sanofi), another pan-PI3K inhibitor, has shown single-agent preclinical activity in human breast cancer cell lines and xenograft models with an IC50 of approximately 6 μM and has demonstrated synergistic activity with other therapeutics.55–57 In an open-label, phase I dose-escalation study of XL147 in patients with advanced solid tumors and lymphomas, the MTD of XL147 was 600 mg/day with either intermittent (21 days on/7 days off) or continuous dosing schedules.58 The DLT for the intermittent dosing schedule was rash. Overall, all-grade rash occurred in 13 (21%) of 62 patients, and grade 3 rash occurred in 2 (3%) of 62 patients. Of the 75 evaluable patients, 13 (17%) patients had stable disease and 1 (1%) patient with NSCLC had a partial response. In another phase I trial of XL147 in combination with erlotinib (Tarceva), 7 (27%) of 26 patients with advanced solid tumors had stable disease and 1 (4%) patient with NSCLC had a confirmed partial response.56 Furthermore, in a phase I trial of XL147 in combination with paclitaxel (Taxol) and carboplatin (Paraplatin), anti-tumor activity was shown in 10 (46%) of 22 evaluable patients, including 5 patients with confirmed complete or partial responses.57 An exploratory pharmacodynamic trial is ongoing in patients with recurrent GBM who are candidates for surgical resection. These patients will receive XL147 prior to surgery, and tumor tissue will be analyzed to determine whether therapeutic drug levels can be achieved within the tumor and for evidence of inhibition of the PI3K pathway (NCT01240460).
In addition to pan-class I PI3K inhibitors, PI3K inhibitors selective for specific p110 isoforms are currently in development. It is hoped that isoform-specific p110 inhibition may lead to more targeted inhibition of pathogenic PI3K signaling and, therefore, have an improved safety profile. Isoform-selective inhibitors for p110α are currently in phase I/II clinical trials in patients with advanced solid tumors and include BYL719, GDC0032, and INK1117.
Dual PI3K/mTOR Inhibitors
The catalytic domains of p110 and mTOR are structurally similar, and because PI3K/AKT pathway-activating mutations also activate the mTOR pathway, it has been suggested that dual inhibition of these pathways may be a more effective therapeutic approach. In general, the dual PI3K/mTOR inhibitors act in an ATP-competitive and reversible manner. Some of the dual PI3K/mTOR inhibitors currently being investigated include BEZ235, XL765, GDC-0980, GDC0084, SF1126, and PF-4691502.
BEZ235 (Novartis) is a potent dual PI3K/mTORC1/2 inhibitor that inhibits PI3K and mTOR kinase activity by binding to the ATP binding cleft of these enzymes with IC50 values of 4, 75, 7, 5, and 21 nmol/L against p110α, p110β, p110γ, p110δ, and mTOR, respectively.59 It has been extensively studied in preclinical models, including refractory breast cancer, glioma, renal cell carcinoma, sarcoma, and NSCLC.60–66 In glioma cell lines, treatment with BEZ235 led to G1 cell-cycle arrest, induced autophagy, and reduced VEGF expression. In vivo, BEZ235 significantly prolonged the survival of tumor-bearing mice.67 In a phase I/IIb study, 28 patients with advanced solid tumors were treated with a special delivery system (SDS) capsule formulation of BEZ235 as a single agent or in combination with trastuzumab.68 A stable response was observed in 6 (40%) of the 15 evaluable patients, 4 of whom were treated for longer than 12 weeks. Overall, BEZ235 was well-tolerated, and the most common adverse events considered to be related to the study drug included nausea, diarrhea, vomiting, and fatigue. Twice-daily administration of the SDS sachet formulation is being further investigated in a phase I study in patients with advanced solid tumors. Several other trials of BEZ235, either as a single agent or in combination, in patients with advanced solid tumors are ongoing.
XL765 (SAR245408; sanofi) exhibits dose-dependent cytotoxicity in GBM cell lines coupled with specific PI3K signaling inhibition, with IC50 values against the α, β, δ, and γ isoforms of p110 of 39, 113, 43, and 9 nM, respectively.69,70 In addition, in luciferase-tagged GBM xenograft models, XL765 resulted in a 92% reduction in median tumor bioluminescence compared with the control. Furthermore, data from this study demonstrated synergistic activity when XL765 was combined with temozolomide and a trend toward improved survival, compared with temozolomide alone.69 In a phase I single-agent study in 79 patients, XL765 was well-tolerated and exhibited pharmacodynamic activity in a range of solid tumors.71 The MTDs were 50 mg twice daily and 90 mg once daily. The most common related adverse events with XL765 were nausea (31%), diarrhea (20%), anorexia (12%), elevated liver enzymes (12%), rash (10%), and vomiting (13%). Drug-related grade 3 or more transaminase level elevation was observed in 4 (5%) patients. XL765 has also been investigated in combination with erlotinib in patients with refractory advanced solid tumors.72 No MTD was established in this phase I study. Overall, the combination was well-tolerated, without major pharmacokinetic interaction between the drugs. Similarly, no MTD was established in an ongoing study in patients with malignant glioma (n = 22) for XL765 given in combination with adjuvant temozolomide. Nonetheless, because of grade 3 or greater study-drug-related adverse events including thrombocytopenia (15%), lymphopenia (10%), brain edema (5%), leukopenia (5%), transaminase increases (5%), and rash (5%), the optimal administered dose was determined to be 60 mg daily. Overall, 11 patients remained in the study for 16 weeks or more.73 This study has recently been expanded to evaluate the MTD of XL765 with radiation therapy and temozolomide in patients with newly diagnosed GBM. A phase I exploratory trial is investigating the effect of XL765 on tumor tissue in patients with recurrent GBM who are candidates for surgical resection (NCT01240460).
Another oral dual PI3K/mTOR inhibitor, GDC-0980 (Genentech), with IC50 values against p110α, β, δ, γ, and mTOR of 4.8, 26.8, 6.7, 13.8, and 17.3 nM, respectively, has demonstrated a favorable safety profile and preliminary anti-tumor activity in a phase I dose-escalation trial on a 21/28-day schedule in patients with solid tumors or non-Hodgkin's lymphoma.74,75 The MTD was 70 mg with a DLT of maculopapular rash. Of the 33 patients enrolled, 3 patients with mesothelioma demonstrated 23%–28% decreases in tumor activity by the response evaluation criteria in solid tumors (RECIST), and of patients evaluated by fluorodeoxyglucose-positron emission tomography (FDG-PET), 5 of 6 patients had 29%–64% decreases in maximal standardized uptake value (SUVmax).75 Phase Ib clinical trials evaluating GDC-0980 in combination with bevacizumab, trastuzumab, and paclitaxel and in combination with paclitaxel and carboplatin are ongoing and planned, respectively. Finally, a phase I clinical trial evaluating GDC-0980 in combination with a fluoropyrimidine, oxaliplatin (Eloxatin), and bevacizumab in patients with advanced solid tumors is ongoing.
GDC-0084 is a potent, selective inhibitor of Class I PI3K and mTOR with good penetration across the blood-brain barrier. Preclinical studies show inhibition in a panel of GBM cell lines and orthotopic U87 tumors. A first-in-man phase I trial in high-grade gliomas is ongoing NCT01547546.
AKT Inhibitors
Because the principal role of AKT is to facilitate cellular survival and suppress apoptotic cell death and because activation has been demonstrated to be a key event in tumorigenesis, the inhibition of AKT is a promising therapeutic target for GBM. AKT inhibitors generally either compete for the ATP-binding in the catalytic domain or act as allosteric inhibitors that bind to the plexstrin homology domain of AKT preventing its translocation to the cell membrane.
Several AKT inhibitors have entered clinical trials, but perifosine (Keryx Biopharmaceuticals), also known as KRX-0401, is the most advanced. Perifosine is an allosteric inhibitor of AKT and has been shown to block phosphorylation of AKT but does not have an effect on PI3K activation.76 Perifosine has been investigated in numerous phase I and II clinical trials, either as a single agent or in combination with agents, such as bortezomib, capecitabine, sorafenib, and rapamycin.77–87 In combination, perifosine has demonstrated modest anti-tumor activity, whereas, as a single agent, perifosine has had mixed results but does appear to be efficacious in patients with Waldenstrom's macroglobulinemia and sarcoma.79,87 The most common adverse events with perifosine are fatigue and gastrointestinal toxicities. In addition to ongoing phase I and II trials, perifosine is currently being explored in 2 phase III studies (colorectal cancer and multiple myeloma). On the basis of promising preclinical data,88 a phase II trial of perifosine in recurrent GBM was conducted but unfortunately showed minimal single-agent activity (NCT00590954). Data suggesting that the combined inhibition of AKT and mTOR may be more effective than inhibition of these targets alone has led to an ongoing phase I/II trial of perifosine with temsirolimus in recurrent high-grade gliomas (NCT01051557).
MK-2206 is an orally active, highly selective non-ATP competitive allosteric AKT inhibitor that is equally potent toward AKT1 and AKT2 and approximately 20% as potent against human AKT3 (IC50 = 8, 12, and 65 nM, respectively). Phase I studies have evaluated the drug using every-other-day and once-weekly schedules. The MTD for the every-other-day schedule was 60 mg, with skin rash as the DLT.89 The MTD for the once-weekly dosing was 200 mg, with rash again being the DLT. Limited single-agent activity has been seen with MK2206 in these studies. Currently multiple combination studies are being conducted. A study in recurrent high-grade gliomas is being considered, although there are some questions regarding the ability of the drug to pass through the blood-brain barrier.
mTOR Inhibitors
mTOR mediates signaling through the canonical PI3K pathway using 2 distinct functional complexes, mTORC1 and mTORC2. mTORC1 (rapamycin-sensitive) positively regulates cell growth and proliferation through phosphorylation of the translational regulators S6K1 and 4EBP1, whereas mTORC2 (rapamycin-insensitive) regulates AKT signaling.90 As described above, the rapamycin analogs everolimus and temsirolimus are mTORC1 inhibitors that have been approved for treatment of several neoplasms. Whereas rapamycin and its derivatives impede the activity of mTORC1, ATP-competitive mTOR inhibitors target the common kinase domain, and as a result, both mTORC1 and mTORC2 activities are repressed. Increased tumor-cell proliferation, observed in some malignancies following treatment with rapamycin, an inhibitor of mTORC1 but not mTORC2, was postulated to be related to the negative-feedback regulation of mTORC2 on PI3K signaling.39,91 For this reason, simultaneous inhibition of both mTORC1 and mTORC2 may provide a therapeutic advantage. Current mTORC1/2 inhibitors in clinical trials include AZD8055 and AZD2014 (Astra Zeneca), CC-223 (Celgene), INK128 (Intellikine), and OSI-027 (OSI Pharmaceuticals).
Preliminary data from a phase I study of AZD8055 was presented at the 2011 ASCO Annual Meeting.92 Overall, 49 patients (42 evaluable) with advanced solid tumors were enrolled in the study. The MTD was 90 mg twice daily, and the DLT was limited to transaminase level elevation. Unlike other rapalogs, rash and mucositis were not dose-limiting. There is an ongoing phase I/II study of this agent in Asian patients with advanced-stage hepatocellular carcinoma with mild or moderate hepatic impairment and an upcoming phase I study in patients with recurrent gliomas not receiving enzyme-inducing anti-epileptic drugs. A trial of CC-223 in recurrent GBM is also planned.
Potential Biomarkers of Response
Prediction of response to targeted agents through the use of histopathology and molecular markers has become integral in the treatment of patients with various types of cancer. In GBM, no biomarker assays linked to targeted therapeutics have been validated or standardized; however, recent clinical trials have made an extensive effort to identify markers that could potentially predict and evaluate response, resistance, and/or recurrence.
An important step in clinical trial design is the selection and screening of patients for activation of pathways relevant to the targeted therapy of interest. For PI3K inhibitors, preclinical data have suggested that activating mutations of PIK3CA and inactivation or loss of PTEN represent the most relevant molecular alterations to predict response to PI3K inhibition in multiple cancers.52 An elegant study by Di Nicolantonio et al. demonstrated that patients with tumors that contained PIK3CA mutations and loss of PTEN activity displayed clinical benefit with everolimus (mTOR inhibitor) treatment, except when KRAS mutations were present. This finding was consistent with data from preclinical cell and tumor xenograft models (generated through the subcutaneous injection of HKe-3 or ME-180 cells into immunocompromised mice).93 In another study, a retrospective analysis of 7 trials was performed to evaluate the effect of BRAF mutations in independent patient populations with colorectal cancer. This study found that 8.3% of patients with BRAF mutations, compared with 38% of patients without BRAF mutations (P = .0012), responded to treatment with the EGFR inhibitor cetuximab.94 In addition, in a retrospective study of 86 patients with multiple cancer types being treated in clinical trials with agents targeting the PI3K pathway, higher response rates were observed in patients with PIK3CA mutations and/or PTEN loss (mutation or complete loss of staining by immunohistochemistry), along with wild-type KRAS, compared with unselected patients or patients with PIK3CA mutations and/or PTEN loss and simultaneous KRAS mutations.95 Additional alterations, which occur fairly commonly in GBM and remain to be evaluated with respect to response predictions, are mutations in PIK3R1 and AKT and gene amplification of EGFR and PDGFRA, which may strongly activate the PI3K pathway. Taken together, these studies demonstrate that prospective screening for mutational status is warranted to design and implement trials such that informative patient mutation profiles are addressed. Multiplex somatic mutation genotyping and copy number assays for efficient screening of patients have recently entered into clinical pathology practice and should aid in more effective trial design,96–98 but similar prospective studies are needed in patients with GBM to validate this approach.
Pharmacodynamic biomarkers offer the ability to determine whether agents have indeed directly inhibited the intended target within the tumor and whether there has been modulation of the key pathway components predicted to effect response. Increasingly, these measurements are made on tumor or non-tumor tissue samples obtained just prior to and during treatment with targeted agents. For PI3K inhibitors, studies often use minimally invasive skin biopsies to examine hair follicles because of their high basal PI3K activity level. For example, investigators of the phase I trials with XL147 and XL765 have been able to demonstrate modulation of PI3K pathway components and a reduction in ERK/MAPK pathway signaling in skin biopsies, hair follicles, and tumor biopsies in a diverse spectrum of solid tumor types.58,70,71,99 In particular, reductions in the phosphorylation of AKT and its downstream targets proline-rich AKT substrate of 40 kilodaltons (PRAS40), eIF4E-binding protein 1(4EBP1), and S6, as well as MEK and ERK have been noted. Reduced PI3K signaling and phosphorylation of MEK and ERK were also shown to be associated with reductions in the proliferation marker Ki67 and increases in apoptosis in patients receiving XL147.58 Another surrogate marker of response to PI3K pathway inhibitors, which is currently being evaluated in clinical trials, is the impact of PI3K inhibitors on glucose homeostasis through the measurement of fasting glucose, insulin, or plasma C-peptides. Results from phase I studies with XL147 and XL765 have demonstrated that treatment with these drugs led to increased insulin levels in patients in an exposure-dependent manner but had minimal impact on glucose levels.70,100 In addition, in trials using BEZ235 and BKM120, dose-dependent increases in plasma C-peptide levels were noted.42,101 FDG uptake by PET has been widely used as a pharmacodynamic marker in BKM120 and BEZ235 trials and is being implemented in trials with GDC-0980 and CAL-101.42,44,75,101 For example, in the BKM120 phase I trial presented by Grana et al. at the 2011 ASCO Annual Meeting, inhibition of tumor metabolic activity was detected as early as cycle 1, and the percent change from baseline suggested a trend toward indicating response.44 However, for CNS cancers, neither skin biopsies nor peripheral glucose homeostasis are likely to serve as reliable surrogates for the measurement of drug levels and pathway inhibition in the brain and in brain tumors due to the blood-brain barrier. Furthermore, sampling of brain tumors before and after targeted therapy is challenging and not commonly performed. For CNS cancers, therefore, the effects of targeted therapy in the brain are measured using the most recent biopsy prior to therapy and compared with a post-therapy biopsy (if available). This practice of using a recent biopsy, rather than one taken directly prior to targeted therapy, assumes that most current therapies would have little effect on the tumor with respect to specific pathways from initial tumor evaluation to recurrent tumor samples.
Conclusions
Although we still face hurdles in fully understanding the complexity of signal transduction in the PI3K pathway and in implementating a standardized, validated routine diagnostic measurement, the future of GBM treatment with PI3K-taregeted agents is promising. Overall, recent research has made tremendous steps toward understanding the pathology of this disease and identifying therapeutic options. Furthermore, growing preclinical and clinical evidence suggests that PI3K pathway inhibitors will provide viable options for the treatment of cancer patients, including those with GBM. Future challenges may include determining which patient populations will benefit from these inhibitors (PTEN loss vs PIK3CA mutations) and optimizing the combination of PI3K pathway inhibitors with inhibitors of other pathways, such as MAP kinase, with inhibitors of tumor stem cells, and with radiation therapy, chemotherapy, and anti-angiogenic therapies. Continued exploration of potential biomarkers will also hopefully lead to more successful treatments for GBM patients.
Acknowledgments
We thank Candice L. Willmon, PhD, and Amanda Quinn, PhD of Articulate Science, for medical writing assistance.
Financial support: This work was supported by Novartis Pharmaceuticals.
Conflict of interest statement: P.Y.W. has received research support from Amgen, Astra Zeneca, Boehringer, Esai, Novartis, Genentech/Roche, Genzyme, Medimmune, Merck, Vascular Biogenics, Exelixis, and Sanofi-Aventis; has served on advisory boards for Merck, Novartis, and EMD Serono; and has received fees for lectures from Merck. E.Q.L. has served on advisory boards for Novartis and Genentech/Roche. D.A.R. has carried out consultant work for Novartis and Genentech/Roche. K.L.L. receives research funding from Plexxikon and Novartis. W.K.A.Y. receives research funding from Novartis and has performed consultant work for Eden.
References
- 1.Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol. 2006;1:97–117. doi: 10.1146/annurev.pathol.1.110304.100043. doi:10.1146/annurev.pathol.1.110304.100043. [DOI] [PubMed] [Google Scholar]
- 2.Central Brain Tumor Registry of the United States. Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2004–2007. 2011. Available at http://cbtrus.org/2007-2008/2007-20081.html. Accessed August 30, 2011.
- 3.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. doi:10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. doi:10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 5.Kantarjian HM, Baccarani M, Jabbour E, Saglio G, Cortes JE. Second-generation tyrosine kinase inhibitors: the future of frontline CML therapy. Clin Cancer Res. 2011;17:1674–1683. doi: 10.1158/1078-0432.CCR-10-2922. doi:10.1158/1078-0432.CCR-10-2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blay JY. A decade of tyrosine kinase inhibitor therapy: historical and current perspectives on targeted therapy for GIST. Cancer Treat Rev. 2011;37:373–384. doi: 10.1016/j.ctrv.2010.11.003. doi:10.1016/j.ctrv.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Knobbe CB, Merlo A, Reifenberger G. PTEN signaling in gliomas. Neuro Oncol. 2002;4:196–211. [PMC free article] [PubMed] [Google Scholar]
- 8.Kanu OO, Hughes B, Di C, et al. Glioblastoma Multiforme Oncogenomics and Signaling Pathways. Clin Med Oncol. 2009;3:39–52. doi: 10.4137/cmo.s1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen MH, Shen YL, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist. 2009;14:1131–1138. doi: 10.1634/theoncologist.2009-0121. doi:10.1634/theoncologist.2009-0121. [DOI] [PubMed] [Google Scholar]
- 10.Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. doi: 10.1200/JCO.2008.19.8721. doi:10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 11.Kreisl TN, Kim L, Moore K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27:740–745. doi: 10.1200/JCO.2008.16.3055. doi:10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. doi:10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 13.Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28:1075–1083. doi: 10.1200/JCO.2009.25.3641. doi:10.1200/JCO.2009.25.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. doi:10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 15.Dugani CB, Klip A. Glucose transporter 4: cycling, compartments and controversies. EMBO Rep. 2005;6:1137–1142. doi: 10.1038/sj.embor.7400584. doi:10.1038/sj.embor.7400584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. doi:10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 17.Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol. 2009;29:5657–5670. doi: 10.1128/MCB.00735-09. doi:10.1128/MCB.00735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harrington LS, Findlay GM, Gray A, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. doi:10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25:55–57. doi: 10.1038/75596. doi:10.1038/75596. [DOI] [PubMed] [Google Scholar]
- 20.Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998;12:3675–3685. doi: 10.1101/gad.12.23.3675. doi:10.1101/gad.12.23.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gallia GL, Rand V, Siu IM, et al. PIK3CA gene mutations in pediatric and adult glioblastoma multiforme. Mol Cancer Res. 2006;4:709–714. doi: 10.1158/1541-7786.MCR-06-0172. doi:10.1158/1541-7786.MCR-06-0172. [DOI] [PubMed] [Google Scholar]
- 22.Cheng CK, Fan QW, Weiss WA. PI3K signaling in glioma–animal models and therapeutic challenges. Brain Pathol. 2009;19:112–120. doi: 10.1111/j.1750-3639.2008.00233.x. doi:10.1111/j.1750-3639.2008.00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004;3:1221–1224. doi: 10.4161/cc.3.10.1164. doi:10.4161/cc.3.10.1164. [DOI] [PubMed] [Google Scholar]
- 24.Broderick DK, Di C, Parrett TJ, et al. Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res. 2004;64:5048–5050. doi: 10.1158/0008-5472.CAN-04-1170. doi:10.1158/0008-5472.CAN-04-1170. [DOI] [PubMed] [Google Scholar]
- 25.Hurtt MR, Moossy J, Donovan-Peluso M, Locker J. Amplification of epidermal growth factor receptor gene in gliomas: histopathology and prognosis. J Neuropathol Exp Neurol. 1992;51:84–90. doi: 10.1097/00005072-199201000-00010. doi:10.1097/00005072-199201000-00010. [DOI] [PubMed] [Google Scholar]
- 26.Wong AJ, Ruppert JM, Bigner SH, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci USA. 1992;89:2965–2969. doi: 10.1073/pnas.89.7.2965. doi:10.1073/pnas.89.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mellinghoff IK, Wang MY, Vivanco I, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–2024. doi: 10.1056/NEJMoa051918. doi:10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 28.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. doi:10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masica DL, Karchin R. Correlation of somatic mutation and expression identifies genes important in human glioblastoma progression and survival. Cancer Res. 2011;71:4550–4561. doi: 10.1158/0008-5472.CAN-11-0180. doi:10.1158/0008-5472.CAN-11-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koul D. PTEN signaling pathways in glioblastoma. Cancer Biol Ther. 2008;7:1321–1325. doi: 10.4161/cbt.7.9.6954. doi:10.4161/cbt.7.9.6954. [DOI] [PubMed] [Google Scholar]
- 31.Kita D, Yonekawa Y, Weller M, Ohgaki H. PIK3CA alterations in primary (de novo) and secondary glioblastomas. Acta Neuropathol. 2007;113:295–302. doi: 10.1007/s00401-006-0186-1. doi:10.1007/s00401-006-0186-1. [DOI] [PubMed] [Google Scholar]
- 32.Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–456. doi: 10.1016/S0140-6736(08)61039-9. doi:10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 33.Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alpha, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–2281. doi: 10.1056/NEJMoa066838. doi:10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 34.Krueger DA, Care MM, Holland K, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–1811. doi: 10.1056/NEJMoa1001671. doi:10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 35.Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514–523. doi: 10.1056/NEJMoa1009290. doi:10.1056/NEJMoa1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galanis E, Buckner JC, Maurer MJ, et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol. 2005;23:5294–5304. doi: 10.1200/JCO.2005.23.622. doi:10.1200/JCO.2005.23.622. [DOI] [PubMed] [Google Scholar]
- 37.Chang SM, Wen P, Cloughesy T, et al. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest New Drugs. 2005;23:357–361. doi: 10.1007/s10637-005-1444-0. doi:10.1007/s10637-005-1444-0. [DOI] [PubMed] [Google Scholar]
- 38.Sarkaria JN, Galanis E, Wu W, et al. North Central Cancer Treatment Group Phase I trial N057K of everolimus (RAD001) and temozolomide in combination with radiation therapy in patients with newly diagnosed glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 2011;81:468–475. doi: 10.1016/j.ijrobp.2010.05.064. doi:10.1016/j.ijrobp.2010.05.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cloughesy TF, Yoshimoto K, Nghiemphu P, et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5:e8. doi: 10.1371/journal.pmed.0050008. doi:10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Voliva CF, Pecchi S, Burger M, et al. Biological characterization of NVP-BKM120, a novel inhibitor of phosphoinosotide 3-kinase in Phase I/II clinical trials [abstract]. Proceedings of the 101st Annual Meeting of the American Association for Cancer Research; Apr 17–21;2010; Washington, DC. Philadelphia (PA):. AACR; 2010. Abstract nr 4498. [Google Scholar]
- 41.Maira M, Menezes D, Pecchi S, et al. NVP-BKM120, a novel inhibitor of phosphoinosotide 3-kinase in Phase I/II clinical trials, shows significant antitumor activity in xenograft and primary tumor models [abstract]. Proceedings of the 101st Annual Meeting of the American Association for Cancer Research; Apr 17–21; 2010; Washington, DC Philadelphia (PA). AACR; 2010. Abstract nr 4497. [Google Scholar]
- 42.Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, Demanse D, De Buck SS, Ru QC, Peters M, Goldbrunner M, Baselga J. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30:282–290. doi: 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 43.Koul D, Fu J, Shen R, LaFortune TA, Wang S, Tiao N, Kim YW, Liu JL, Ramnarian D, Yuan Y, Garcia-Echevrria C, Maira SM, Yung WK. Antitumor activity of NVP-BKM120--a selective pan class I PI3 kinase inhibitor showed differential forms of cell death based on p53 status of glioma cells. Clin Cancer Res. 2012;18:184–195. doi: 10.1158/1078-0432.CCR-11-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grana B, Burris HA, Rodon Ahnert J, et al. Oral PI3 kinase inhibitor BKM120 monotherapy in patients (pts) with advanced solid tumors: an update on safety and efficacy [abstract] J Clin Oncol. 2011;29:3043. [Google Scholar]
- 45.Ihle NT, Lemos R, Schwartz D, et al. Peroxisome proliferator-activated receptor gamma agonist pioglitazone prevents the hyperglycemia caused by phosphatidylinositol 3-kinase pathway inhibition by PX-866 without affecting antitumor activity. Mol Cancer Ther. 2009;8:94–100. doi: 10.1158/1535-7163.MCT-08-0714. doi:10.1158/1535-7163.MCT-08-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.She QB, Chandarlapaty S, Ye Q, et al. Breast tumor cells with PI3K mutation or HER2 amplification are selectively addicted to Akt signaling. PLoS One. 2008;3:e3065. doi: 10.1371/journal.pone.0003065. doi:10.1371/journal.pone.0003065. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Tabernero J, Rojo F, Calvo E, et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol. 2008;26:1603–1610. doi: 10.1200/JCO.2007.14.5482. doi:10.1200/JCO.2007.14.5482. [DOI] [PubMed] [Google Scholar]
- 48.Cherrin C, Haskell K, Howell B, et al. An allosteric Akt inhibitor effectively blocks Akt signaling and tumor growth with only transient effects on glucose and insulin levels in vivo. Cancer Biol Ther. 2010;9:493–503. doi: 10.4161/cbt.9.7.11100. doi:10.4161/cbt.9.7.11100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Elizalde VS, Russillo M, Scaltriti M, et al. Dual-targeting of AMPK and PI3K/mTOR in a panel of breast cancer cell lines [abstract]. Proceedings of the 101st Annual Meeting of the American Association for Cancer Research; Apr 17–21 2010; Washington, DC. Philadelphia (PA). AACR; 2010. Abstract nr 4477. [Google Scholar]
- 50.O'Donnell A, Faivre S, Burris HA, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol. 2008;26:1588–1595. doi: 10.1200/JCO.2007.14.0988. doi:10.1200/JCO.2007.14.0988. [DOI] [PubMed] [Google Scholar]
- 51.Wolpin BM, Hezel AF, Abrams T, et al. Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J Clin Oncol. 2009;27:193–198. doi: 10.1200/JCO.2008.18.9514. doi:10.1200/JCO.2008.18.9514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ihle NT, Lemos R, Wipf P, et al. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009;69:143–150. doi: 10.1158/0008-5472.CAN-07-6656. doi:10.1158/0008-5472.CAN-07-6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koul D, Shen R, Kim YW, et al. Cellular and in vivo activity of a novel PI3K inhibitor, PX-866, against human glioblastoma. Neuro Oncol. 2010;12:559–569. doi: 10.1093/neuonc/nop058. doi:10.1093/neuonc/nop058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jimeno A, Herbst RS, Falchook GS, et al. Final results from a phase I, dose-escalation study of PX-866, an irreversible, pan-isoform inhibitor of PI3 kinase [abstract] J Clin Oncol. 2010;28:3089. [Google Scholar]
- 55.Chakrabarty A, Sanchez V, Kuba MG, Rinehart C, Arteaga CL. Breast Cancer Special Feature: feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc Natl Acad Sci USA. 2012;109:2718–2723. doi: 10.1073/pnas.1018001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moldovan C, Soria J, LoRusso P, et al. A phase I safety and pharmacokinetic (PK) study of the PI3K inhibitor XL147 (SAR245408) in combination with erlotinib in patients (pts) with advanced solid tumors [abstract] J Clin Oncol. 2010;28:3070. [Google Scholar]
- 57.Traynor AM, Kurzrock R, Bailey HH, et al. A phase I safety and pharmacokinetic (PK) study of the PI3K inhibitor XL147 (SAR245408) in combination with paclitaxel (P) and carboplatin (C) in patients (pts) with advanced solid tumors [abstract] J Clin Oncol. 2010;28:3078. [Google Scholar]
- 58.Edelman G, Bedell C, Shapiro G, et al. A phase I dose-escalation study of XL147 (SAR245408), a PI3K inhibitor administered orally to patients (pts) with advanced malignancies [abstract] J Clin Oncol. 2010;28:3004. [Google Scholar]
- 59.Maira SM, Stauffer F, Brueggen J, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. doi:10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 60.Serra V, Markman B, Scaltriti M, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68:8022–8030. doi: 10.1158/0008-5472.CAN-08-1385. doi:10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 61.Konstantinidou G, Bey EA, Rabellino A, et al. Dual phosphoinositide 3-kinase/mammalian target of rapamycin blockade is an effective radiosensitizing strategy for the treatment of non-small cell lung cancer harboring K-RAS mutations. Cancer Res. 2009;69:7644–7652. doi: 10.1158/0008-5472.CAN-09-0823. doi:10.1158/0008-5472.CAN-09-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cao P, Maira SM, Garcia-Echeverria C, Hedley DW. Activity of a novel, dual PI3-kinase/mTor inhibitor NVP-BEZ235 against primary human pancreatic cancers grown as orthotopic xenografts. Br J Cancer. 2009;100:1267–1276. doi: 10.1038/sj.bjc.6604995. doi:10.1038/sj.bjc.6604995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.De PKR, De N, Leyland-Jones B. In vitro evaluation of dual pan-PI3-kinase/mTOR inhibitor in HER2 overexpressing breast cancer cells [abstract]. Proceedings of the 101st Annual Meeting of the American Association for Cancer Research; Apr 17–21 2010; Washington, DC. Philadelphia (PA). AACR; 2010. Abstract nr 337. [Google Scholar]
- 64.Bhende PM, Park SI, Lim MS, Dittmer DP, Damania B. The dual PI3K/mTOR inhibitor, NVP-BEZ235, is efficacious against follicular lymphoma. Leukemia. 2010;24:1781–1784. doi: 10.1038/leu.2010.154. doi:10.1038/leu.2010.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leung E, Kim JE, Rewcastle GW, Finlay GJ, Baguley BC. Comparison of the effects of the PI3K/mTOR inhibitors NVP-BEZ235 and GSK2126458 on tamoxifen-resistant breast cancer cells. Cancer Biol Ther. 2011;11:938–946. doi: 10.4161/cbt.11.11.15527. doi:10.4161/cbt.11.11.15527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fan QW, Cheng C, Hackett C, et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal. 2010;3:ra81. doi: 10.1126/scisignal.2001017. doi:10.1126/scisignal.2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu TJ, Koul D, LaFortune T, et al. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol Cancer Ther. 2009;8:2204–2210. doi: 10.1158/1535-7163.MCT-09-0160. doi:10.1158/1535-7163.MCT-09-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peyton JD, Rodon Ahnert J, Burris H, et al. A dose-escalation study with the novel formulation of the oral pan-class I PI3K inhibitor BEZ235, solid dispersion system (SDS) sachet, in patients with advanced solid tumors [abstract] J Clin Oncol. 2011;29:3066. [Google Scholar]
- 69.Prasad G, Sottero T, Yang X, et al. Inhibition of PI3K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro Oncol. 2011;13:384–392. doi: 10.1093/neuonc/noq193. doi:10.1093/neuonc/noq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.LoRusso P, Markman B, Tabernero J, et al. A phase I dose-escalation study of the safety, pharmacokinetics (PK), and pharmacodynamics of XL765, a PI3K/TORC1/TORC2 inhibitor administered orally to patients (pts) with advanced solid tumors [abstract] J Clin Oncol. 2009;27:3502. [Google Scholar]
- 71.Brana I, LoRusso P, Baselga J, et al. A phase I dose-escalation study of the safety, pharmacokinetics (PK), and pharmacodynamics of XL765 (SAR245409), a PI3K/TORC1/TORC2 inhibitor administered orally to patients (pts) with advanced malignancies [abstract] J Clin Oncol. 2010;28:3030. [Google Scholar]
- 72.Cohen RB, Janne PA, Engelman JA, et al. A phase I safety and pharmacokinetic (PK) study of PI3K/TORC1/TORC2 inhibitor XL765 (SAR245409) in combination with erlotinib (E) in patients (pts) with advanced solid tumors[abstract] J Clin Oncol. 2010;28:3015. doi: 10.1200/JCO.2009.26.1347. doi:10.1200/JCO.2009.26.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nghiemphu PL, Omuro AM, Cloughesy T, et al. A phase I safety and pharmacokinetic study of XL765 (SAR245409), a novel PI3K/TORC1/TORC2 inhibitor, in combination with temozolomide (TMZ) in patients (pts) with newly diagnosed malignant glioma [abstract] J Clin Oncol. 2010;28:3085. [Google Scholar]
- 74.Dolly S, Wagner AJ, Bendell JC, et al. A first-in-human, phase l study to evaluate the dual PI3K/mTOR inhibitor GDC-0980 administered QD in patients with advanced solid tumors or non-Hodgkin's lymphoma [abstract] J Clin Oncol. 2010;28:3079. [Google Scholar]
- 75.Wagner AJ, Bendell JC, Dolly S, et al. A first-in-human phase I study to evaluate GDC-0980, an oral PI3K/mTOR inhibitor, administered QD in patients with advanced solid tumors [abstract] J Clin Oncol. 2011;29:3020. [Google Scholar]
- 76.Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA, Roy KK. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol Cancer Ther. 2003;2:1093–1103. [PubMed] [Google Scholar]
- 77.Friedman DR, Davis PH, Lanasa MC, et al. Pre-clinical and interim results of a Phase II trial of perifosine in patients with relapsed or refractory chronic lymphocytic leukemia (CLL) [abstract] Blood. 2010;116:1842. [Google Scholar]
- 78.Carlo-Stella C, Guidetti A, Viviani S, et al. Clinical activity and safety of the combined therapy with the AKT inhibitor perifosine and the multikinase inhibitor sorafenib in heavily pretreated patients with relapsed/refractory lymphomas: preliminary results of a Phase II trial [abstract] Blood. 2010;116:2861. doi: 10.1182/blood-2010-07-294595. doi:10.1182/blood-2010-07-294595. [DOI] [PubMed] [Google Scholar]
- 79.Ghobrial IM, Roccaro A, Hong F, et al. Clinical and translational studies of a phase II trial of the novel oral Akt inhibitor perifosine in relapsed or relapsed/refractory Waldenstrom's macroglobulinemia. Clin Cancer Res. 2010;16:1033–1041. doi: 10.1158/1078-0432.CCR-09-1837. doi:10.1158/1078-0432.CCR-09-1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chee KG, Longmate J, Quinn DI, et al. The AKT inhibitor perifosine in biochemically recurrent prostate cancer: a phase II California/Pittsburgh cancer consortium trial. Clin Genitourin Cancer. 2007;5:433–437. doi: 10.3816/CGC.2007.n.031. doi:10.3816/CGC.2007.n.031. [DOI] [PubMed] [Google Scholar]
- 81.Unger C, Berdel W, Hanauske AR, Sindermann H, Engel J, Mross K. First-time-in-man and pharmacokinetic study of weekly oral perifosine in patients with solid tumours. Eur J Cancer. 2010;46:920–925. doi: 10.1016/j.ejca.2009.12.028. doi:10.1016/j.ejca.2009.12.028. [DOI] [PubMed] [Google Scholar]
- 82.Becher OJ, Trippett TM, Kolesar J, et al. Phase I study of single-agent perifosine for recurrent pediatric solid tumors [abstract] J Clin Oncol. 2010;28:9540. [Google Scholar]
- 83.Greco FA, Infante JR, Burris HA, et al. Safety and pharmacokinetic (PK) study of perifosine plus capecitabine (P-CAP) in patients (pts) with refractory metastatic colorectal cancer (mCRC) [abstract] J Clin Oncol. 2010;28:e14086. [Google Scholar]
- 84.Richards DA, Nemunaitis JJ, Vukelja SJ, et al. Final results of a randomized phase II study of perifosine in combination with capecitabine (P-CAP) versus placebo plus capecitabine (CAP) in patients (pts) with second- or third-line metastatic colorectal cancer (mCRC) [abstract] J Clin Oncol. 2010;28:3531. doi: 10.1200/JCO.2009.27.4787. doi:10.1200/JCO.2009.27.4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Campos LT, Nemunaitis J, Stephenson J, et al. Phase II study of single agent perifosine in patients with hepatocellular carcinoma (HCC) [abstract] J Clin Oncol. 2009;27:e15505. [Google Scholar]
- 86.Vogelzang NJ, Hutson TE, Samlowski W, et al. Phase II study of perifosine in metastatic renal cell carcinoma (RCC) progressing after prior therapy (Rx) with a VEGF receptor inhibitor [abstract] J Clin Oncol. 2009;27:5034. [Google Scholar]
- 87.Bailey HH, Mahoney MR, Ettinger DS, et al. Phase II study of daily oral perifosine in patients with advanced soft tissue sarcoma. Cancer. 2006;107:2462–2467. doi: 10.1002/cncr.22308. doi:10.1002/cncr.22308. [DOI] [PubMed] [Google Scholar]
- 88.Momota H, Nerio E, Holland EC. Perifosine inhibits multiple signaling pathways in glial progenitors and cooperates with temozolomide to arrest cell proliferation in gliomas in vivo. Cancer Res. 2005;65:7429–7435. doi: 10.1158/0008-5472.CAN-05-1042. doi:10.1158/0008-5472.CAN-05-1042. [DOI] [PubMed] [Google Scholar]
- 89.Yap TA, Yan L, Patnaik A, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol. 2011;29:4688–4695. doi: 10.1200/JCO.2011.35.5263. doi:10.1200/JCO.2011.35.5263. [DOI] [PubMed] [Google Scholar]
- 90.Carracedo A, Baselga J, Pandolfi PP. Deconstructing feedback-signaling networks to improve anticancer therapy with mTORC1 inhibitors. Cell Cycle. 2008;7:3805–3809. doi: 10.4161/cc.7.24.7244. doi:10.4161/cc.7.24.7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.O'Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. doi:10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Banerji U, Aghajanian C, Raymond E, et al. First results from a phase I trial of AZD8055, a dual mTORC1 and mTORC2 inhibitor [abstract] J Clin Oncol. 2011;29:3096. [Google Scholar]
- 93.Di Nicolantonio F, Arena S, Tabernero J, et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J Clin Invest. 2010;120:2858–2866. doi: 10.1172/JCI37539. doi:10.1172/JCI37539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lin JS, Webber EM, Senger CA, Holmes RS, Whitlock EP. Systematic review of pharmacogenetic testing for predicting clinical benefit to anti-EGFR therapy in metastatic colorectal cancer. Am J Cancer Res. 2011;1:650–662. [PMC free article] [PubMed] [Google Scholar]
- 95.Janku F, Garrido-Laguna I, Wheler JJ, et al. Screening for PIK3CA mutations, PTEN loss, and RAS/RAF mutations in early-phase protocols with PI3K/mTOR pathway inhibitors [abstract] J Clin Oncol. 2011;29:10507. [Google Scholar]
- 96.MacConaill LE, Campbell CD, Kehoe SM, et al. Profiling critical cancer gene mutations in clinical tumor samples. PLoS One. 2009;4:e7887. doi: 10.1371/journal.pone.0007887. doi:10.1371/journal.pone.0007887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dias-Santagata D, Akhavanfard S, David SS, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010;2:146–158. doi: 10.1002/emmm.201000070. doi:10.1002/emmm.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ducray F, De Reynies A, Chinot O, et al. An ANOCEF genomic and transcriptomic microarray study of the response to radiotherapy or to alkylating first-line chemotherapy in glioblastoma patients. Mol Cancer. 2010;9:234. doi: 10.1186/1476-4598-9-234. doi:10.1186/1476-4598-9-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Papadopoulos KP, Markman B, Tabernero J, et al. A phase I dose-escalation study of the safety, pharmacokinetics (PK), and pharmacodynamics (PD) of a novel PI3K inhibitor, XL765, administered orally to patients (pts) with advanced solid tumors [abstract] J Clin Oncol. 2008;26:3510. [Google Scholar]
- 100.Shapiro G, Kwak E, Baselga J, et al. Phase I dose-escalation study of XL147, a PI3K inhibitor administered orally to patients with solid tumors [abstract] J Clin Oncol. 2009;27:3500. [Google Scholar]
- 101.Burris H, Rodon J, Sharma S, et al. First-in-human phase I study of the oral PI3K inhibitor BEZ235 in patients (pts) with advanced solid tumors [abstract] J Clin Oncol. 2010;28:3005. [Google Scholar]