

Abstract
The National Center for Biotechnology Information (NCBI) is well known for the nucleotide sequence archive, GenBank and sequence analysis tool BLAST. However, NCBI integrates many types of biomolecular data from variety of sources and makes it available to the scientific community as interactive web resources as well as organized releases of bulk data. These tools are available to explore and compare fungal genomes. Searching all databases with Fungi [organism] at http://www.ncbi.nlm.nih.gov/ is the quickest way to find resources of interest with fungal entries. Some tools though are resources specific and can be indirectly accessed from a particular database in the Entrez system. These include graphical viewers and comparative analysis tools such as TaxPlot, TaxMap and UniGene DDD (found via UniGene Homepage). Gene and BioProject pages also serve as portals to external data such as community annotation websites, BioGrid and UniProt. There are many different ways of accessing genomic data at NCBI. Depending on the focus and goal of research projects or the level of interest, a user would select a particular route for accessing genomic databases and resources. This review article describes methods of accessing fungal genome data and provides examples that illustrate the use of analysis tools.
Keywords: Bioinformatics, fungal genomics, comparative genomics, protein clusters
Introduction
In 1988 the National Center for Biotechnology (NCBI) was created to facilitate a better understanding of diseases in humans by managing data in a more efficient manner. The mission then and now is to conduct basic research in computation biology, build databases and provide world-wide access to biomedical information. Over the years these databases increasingly accommodated a great number of non-human centred information and as part of the International Nucleotide Sequence Database Collaboration (INSDC) contain one of the largest collections of sequence data.
Motivation to sequence fungal genomes stem from the fact that Fungi significantly impacts human wellbeing as it relates to medicine, health care, biotechnology, food security, alternative energy and maintaining the ecological integrity. With their relatively compact genomes Fungi have the largest and broadest set of sequenced genomes amongst eukaryotes. However, a wealth of data is only valuable if it is accompanied with user-friendly bioinformatic tools designed to address questions of biological importance. NCBI has data mining tools that assist fungal biologists in various disciplines (systematics, genetics, plant pathology, evolutionary biology etc) towards their goal of searching genomic and other ‘omic’ data for biological significance. These databases and tools are listed in Tables 1, 2 and 3 with URL links, the utility of each link and Entrez query examples. There are multiple ways to access sequence data and precomputed results using NCBI web sites. With the number of information resources and new databases growing very fast, navigation through the web pages could be quite challenging and a recent review address this issue by describing educational resources available at NCBI via the NCBI Education page (http://www.ncbi.nlm.nih.gov/Education/) (Cooper et al., 2010).
Table 1.
A list of selected Entrez Databases at NCBI as it apply to fungi, website URLs and their utility.
| Resource | Data type | URL | URL Utility | Entrez query example |
|---|---|---|---|---|
| All Databases | http://www.ncbi.nlm.nih.gov/gquery/?term=Fungi+%5BORGN%5D | Displays record counts from all databases relevant to Fungi. |
Fungi [ORGN] | |
| EST | primary sequence |
http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=helpsequences&part=sequencesquickstart | Tips on accessing EST data. | Magnaporthe oryzae [ORGN] |
| GSS | primary sequence |
http://www.ncbi.nlm.nih.gov/dbGSS/dbGSS_access.html | Tips on accessing GSS data. | Glomerella graminicola [ORGN] AND mitochondrion [Filter] |
| Nucleotide | primary & derived sequence |
http://www.ncbi.nlm.nih.gov/nuccore | Retrieve archived nucleotide sequences using a text search with Entrez. |
srcdb tpa ddbj/embl/genbank[prop] AND Fungi [organism] |
| Protein | Primary & derived sequence |
http://www.ncbi.nlm.nih.gov/protein | Retrieve protein sequences using a text search with Entrez |
GenBank[Filter] AND Fungi [ORGN] AND 10:600 [slen] AND pheromone [TITLE] NOT receptor [TITLE] NOT “protein nuccore wgs”[Filter] |
| SRA | primary sequence |
http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?cmd=show&f=search_blast&m=search&s=blast&m=search&s=blast | Search 454 data using a sequence based approach with Blast. |
Fungi [orgn] AND “Illumina Genome Analyzer II” |
| Probe | primary sequence |
http://www.ncbi.nlm.nih.gov/probe | Search Trace Assembly by species name. |
FUNGI [ORGN] AND mutants |
| GEO | primary functional |
http://www.ncbi.nlm.nih.gov/geo/ | A portal to browse DataSets or query DataSets and Gene profiles. |
gds pubmed[Filter] AND Fungi [ORGN] |
| Peptidome | primary functional |
http://www.ncbi.nlm.nih.gov/peptidome/ | Retrieve tandem mass spectrometry peptide and protein identification data. |
Fungi [ORGN] AND peroxisome |
| PubMed | primary non- sequence |
http://www.ncbi.nlm.nih.gov/pubmed | Retrieve citations using a text search with Entrez |
(fungi AND genomics [Title/Abstract]) AND “review”[Publication Type] NOT virus NOT bacteria |
| Taxonomy | primary non- sequence |
http://www.ncbi.nlm.nih.gov/taxonomy | Search the Taxonomy Database. |
Fungi [ORGN] AND Sordariomycetes [Lineage] |
| Genome Project |
primary & derived non- sequence |
http://www.ncbi.nlm.nih.gov/genomes/leuks.cgi?p1=10%3A0&p3=11:Fungi%7C12%3A&taxgroup=11:Fungi%7C12%3A& | A list of fungal genomes submitted or registered for submission at NCBI. |
Fungi [ORGN] AND “has chr”[Properties] NOT “has wgs” [Properties] |
| Gene | derived | http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=helpgene&part=EntrezGene | Description of data display in Gene and how to search. |
Pezizomycotina [ORGN] AND GeneRif [prop] NOT NEWENTRY |
| UniGene | derived | http://www.ncbi.nlm.nih.gov/unigene | Search UniGene clusters with Entrez system. |
Fungi [ORGN] 500:1200[Sequence Count] |
| HomoloGene | derived | http://www.ncbi.nlm.nih.gov/homologene/ | Search homologs among the eukaryotes listed. |
MCM7[Gene Name]) AND Saccharomyces cerevisiae [ORGN] |
| Protein Clusters |
derived | http://www.ncbi.nlm.nih.gov/proteinclusters/ | Search related proteins with shared function in Protein Clusters. |
“het c”[Domain Name] |
| CDD | derived | http://www.ncbi.nlm.nih.gov/Structure/cdd/docs/cdd_search.html | Summary and access to all possible search options in CDD. |
“pfam”[Database] AND pf07217[Alternative Accession] |
| BioSystems | derived | http://www.ncbi.nlm.nih.gov/biosystems | Hints on finding a BioSystem and search with Entrez system. |
“organism specific biosystem”[Filter] AND Fungi [ORGN] AND siderophore |
Table 2.
Additional resources at NCBI as it apply to fungi, website URLs and their utility.
| Resource | Data type | URL | URL Utility |
|---|---|---|---|
|
| |||
| Other resources | |||
| Trace Archive | primary sequence |
http://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Nucleotides&PROGRAM=blastn&BLAST_SPEC=TraceArchive&BLAST_PROGRAMS=megaBlast&PAGE_TYPE=BlastSearch | Search the Trace Archive using a sequence based approach with Blast. |
| Trace Assembly Archive | primary sequence |
http://www.ncbi.nlm.nih.gov/Traces/assembly/assmbrowser.cgi?cmd=show&f=tree&m=main&s=tree | Search Trace Assembly by species name. |
| RefSeq | derived | http://www.ncbi.nlm.nih.gov/sites/entrez?Db=genomeprj&Cmd=DetailsSearch&Term=%22Fungi%22[Organism]+AND+type_refseq[prop] | A list of Fungi RefSeq projects. |
| TPA | derived | http://www.ncbi.nlm.nih.gov/genbank/TPA.html | Detailed information about what can and cannot be submitted as Third Party Annotation. |
| Gnomon gene prediction | derived | http://www.ncbi.nlm.nih.gov/genome/guide/gnomon.shtml | Description of the single genome Gnomon gene prediction pipeline. |
| GeneRIF | http://www.ncbi.nlm.nih.gov/projects/GeneRIF/GeneRIFhelp.html | Information on how the research community can add functional annotation to genes in Entrez Gene. |
|
| Organelle Genome Resources |
derived | http://www.ncbi.nlm.nih.gov/genomes/GenomesGroup.cgi?taxid=4751&opt=organelle | A list of fungi with complete mitochondrial genomes and links to data. |
Table 3.
A list of selected tools at NCBI as it apply to fungi, website URLs and their utility.
| Resource | URL | URL Utility |
|---|---|---|
| Entrez data retrieval system |
http://www.ncbi.nlm.nih.gov/gquery/ | Search all Entrez databases simultaneously. |
| BLAST | http://www.ncbi.nlm.nih.gov/sutils/genom_table.cgi?organism=fungi | A dedicated BLAST page to search only fungal genomes. |
| BLink | http://www.ncbi.nlm.nih.gov/sutils/static/blinkhelp.html | Detailed help on the BLink output format. |
| TaxMap | http://www.ncbi.nlm.nih.gov/sutils/taxik.cgi?gi=18521&mtax=36629&cut=95&cutp=10&x=449&y=42 | TaxMap example of Aspergillus fumigatus chr8 and region selected. |
| TaxPlot | http://www.ncbi.nlm.nih.gov/sutils/taxik2.cgi | The default comparison page of TaxPlot. |
| UniGene Digital Differential Display |
http://www.ncbi.nlm.nih.gov/UniGene/ddd.cgi?TAXID=5141&ACT=new | EST profiles available for comparison from Neurospora crassa as an example. |
| FLink | http://www.ncbi.nlm.nih.gov/Structure/flink/docs/flink_about.html | Description of the FLink tool and a step-by- step example. |
This review focus on highlighting and demonstrating resources and tools useful to new and regular NCBI users in the Fungi community around the world. There are three major approaches to access data directly: (i) find an object of interest using a text query (for example, gene symbol, organism name, author name etc); (ii) use a sequence query to find related data and (iii) use tools to find an answer to your question. Examples of all three approaches are provided.
Data submission, storage and public representation
NCBI serves as a major public repository of nucleic acid sequences which receives data through international collaboration (INSDC: http://www.insdc.org/) with the DNA databank of Japan (DDBJ) (Kaminuma et al., 2010) and the European Nucleotide Archive (ENA) (Leinonen et al., 2010a) as well as from large sequencing centers and individual researchers worldwide. According to its mission, to collect and disseminate bio-medical information, NCBI accepts primary experimental information for various types of biological data. It provides data retrieval systems and computational power for the analysis of submitted data and supports a data integration system by calculating relationships and providing cross-references between different data types.
The term ‘database’ is being used in various contexts and deserves some clarification. In a general way a database can be defined as storage for organized information, it can be implemented as a collection of simple ‘flat’ files or as a more sophisticated relational database. In the latter definition ‘database’ refers to the storage of data objects at NCBI that serve as a ‘back-end’ to public representation of the data and thus is a database accessed by users indirectly through an application (eg. web browser). However, a database can also be defined as an application that manages data and allows fast storage and retrieval of that data for example Entrez. Entrez is the text-based search and retrieval system used at NCBI for all of the major databases, including, GenBank, PubMed, Taxonomy, OMIM and many others. These are also referred to as “source databases”. An Entrez “node” on the other hand is a collection of data that is grouped together and indexed together. It is usually referred to as an Entrez database. Entrez integrates data from a large number of sources, formats, and databases into a uniform information model and retrieval system. The actual databases from which records are retrieved and on which the Entrez indexes are based have different designs, based on the type of data, and reside on different machines. For example the Entrez nucleotide database retrieves data from various sources such as GenBank, RefSeq, TPA etc. At the time of writing the number of Entrez databases reached 40 and the list keeps growing (Sayers et al., 2010).
By reviewing tools and resources at NCBI we also hope to encourage fungal researchers to submit new submissions and updates to NCBI. The type of data submission will determine the submission procedure. This webpage: (http://www.ncbi.nlm.nih.gov/guide/howto/submit-sequence-data/) provides a summary of types of data accepted for submission, tools to use and documents to consult. Genomic assembly submissions are divided in four categories, small complete genomes (eg. mitochondria), large complete genomes (eg. chromosomes), incomplete genomes (eg. WGSs), high throughput genome sequences (eg. BACs). Both large complete and incomplete genomes require project registration as a first step (http://www.ncbi.nlm.nih.gov/genomes/mpfsubmission.cgi). It is best to consult the NCBI website for the latest information on WGS http://www.ncbi.nlm.nih.gov/genbank/wgs.html and complete eukaryotic genome submissions http://www.ncbi.nlm.nih.gov/genbank/eukaryotic_genome_submission.html, since the submission process is continually tweaked to optimize processing.
Primary data archives
The data collected by NCBI come from the experimental laboratories, individual researchers and large sequencing centers all over the world. Next-generation sequencing technologies have already changed dataflow dramatically; the amount of primary sequence data is growing so rapidly that the storage and maintenance of data become a challenge to central data archives. Recent technological and computational advancements have improved our understanding of biology to a far greater level than was deemed possible even 10 years ago. New research strategies create new data types and integrated data models. Metagenome sequencing of whole ecological communities, model organism re-sequencing, RNA-seq and other strategies are changing the way data is managed and analyzed. Some examples of traditional and novel databases are discussed below.
Sequence data
Traditionally nucleotide sequence data submitted to GenBank have been archived in different databases represented as different GenBank divisions and displayed as different Entrez databases (Benson et al., 2010). That includes organism specific GenBank divisions, for example, MAM for mammals, VRT for vertebrates, PLN for plants (historically, fungi belong to PLN division and even though the classification has been changed a long time ago, GenBank rules stay the same). Other divisions of GenBank are organized by sequence type or technology method, some examples relevant to this publication includes EST (Expressed Sequence Tags), GSS (Genome Survey Sequences) and WGS (Whole Genome Shotgun). For a complete list of divisions see the GenBank Release Notes: ftp://ftp.ncbi.nih.gov/genbank/gbrel.txt.
The EST database
The EST database (Table 1), a special division of GenBank, consists of expressed sequence tag records which contain relatively short “single-pass” cDNA sequences and other information. Large collections of ESTs enable the assembly of nucleotide sequence contigs and, if the genomic sequence is available, a means of mapping the species genome and ultimately assist in gene and pathway discovery. Due to the large volume and specific nature of EST sequences they are stored in a special ‘source’ database and represented as a separate searchable Entrez database. More than 200 fungal species (~2.7 million records) are represented in the EST database with 10 most abundant species associated with more than 50,000 records each.
The GSS database
The GSS database (Table 1) is similar to the EST division but contains only sequences that are genomic in origin. It holds the following types of data, random “single pass read” genome survey sequences, cosmid/BAC/YAC end sequences, exon trapped genomic sequences and transposon-tagged sequences. The GSS database contains data from more than 90 fungal species including three fungi (Glomerella graminicola, Schizosaccharomyces pombe and Ustilago maydis) that have more than 100,000 records each.
WGS data
Whole genome shotgun data are accessible via the Entrez nucleotide database but a list of WGS sequencing projects is also available here: http://www.ncbi.nlm.nih.gov/projects/WGS/WGSprojectlist.cgi. The link to this page can be found on the “Whole Genome Shotgun Sequence Submissions” page which is the first link when searched for by these words in the “NCBI Web Site” database using Entrez. Genome projects are listed in alphabetical order according to a stable 4-letter WGS accession prefix which does not change as the project is updated. All sequences belonging to the same project have accessions that share the same four letter prefix. The summary of WGS sequencing projects include project ID, annotation status and can be sorted by organism name, WGS prefix, number of contigs or the number of CON records (scaffolds or chromosomes). The summary include links to master records, Genome Projects, the Taxonomy Browser and complete genomes in the Entrez core nucleotide database. More than 125 fungal genomes are listed here of which about 60 are annotated.
Core nucleotide database
The Entrez nucleotide database (Table 1) contains sequences of various types representing individual genes and transcripts, genomic regions and whole genome assemblies from various source databases like GenBank, RefSeq and TPA. At the time of writing (end of 2010), the main nucleotide collection contains nuclear genome assemblies of more than 100 fungal species and some species such as Saccharomyces cerevisiae, Coccidioides posadasii and Ajellomyces capsulatus have genome assemblies from several different strains. The default display setting is the well-known GenBank flat file. However, other display options are available including a graphical display which may be more practical since many data points are given a frame of reference with the visualization of genes and its gene structure on the genome. In addition chromosomes in complete genomes are displayed with the major NCBI genome browser, Map Viewer (http://www.ncbi.nlm.nih.gov/projects/mapview/map_search.cgi?taxid=4751). Currently Map Viewer displays the genomes of seventeen Fungi.
Protein sequence database
Protein sequences are the fundamental determinants of biological structure and function and thus one of the major goals in a sequencing project is to predict these sequences from the genome. The protein database (Table 1) is a collection of sequences from several sources, including translations from annotated coding regions in source databases such as GenBank, RefSeq and TPA, as well as records from UniProt (which has subsumed Swiss Prot and PIR records) (The UniProt Consortium, 2010), PRF (http://www.prf.or.jp/index-e.html), and PDB (Berman et al., 2007). Therefore, a search in the protein database for a specific gene product may produce more than one record with identical sequence as a result of its presence in several source databases.
Trace Archive and Short Read Archive (SRA)
More recently, NCBI has created repositories for primary raw data coming directly from various sequencing machines: Trace Archive for traditional Sanger sequencing technology and SRA or short reads generated by new-generation sequencing technologies.
The Trace Archive serves as a repository of raw sequence reads from gel/capillary platforms such as Applied Biosystems ABI 3730. SRA stores sequencing data from next generation sequencing platforms such as Roche 454 GS System, Illumina Genome Analyzer, Applied Biosystems SOLiD System, Helicos Heliscope and others (Leinonen et al., 2010b) . The number of fungal taxa with data in the SRA database (132) is rapidly increasing compared with the number in the Trace Archive (101). Both the Trace Archive and SRA (454 data) can be queried using BLAST (Tables 1 & 2).
The link between raw sequence reads in the Trace Archive and the genome assembly as found in GenBank are captured in the Trace Assembly Archive (Salzberg et al., 2004) and alignments are visualized with a sequence viewer (Table 2). This repository provides users with the ability to access and evaluate assemblies. For example, observations such as frame shifts or SNPs can be validated by evaluating a genome region of interest for adequate coverage. At the time of writing, assemblies from four fungal taxa, Neosartorya fischeri NRRL 181, Talaromyces stipitatus ATCC 10500, Aspergillus clavatus NRRL 1 and Penicillium marneffei ATCC 18224 have been submitted to the Trace Assembly Archive and are available for investigation.
The Transcriptome Shotgun Assembly Sequence (TSA) database (http://www.ncbi.nlm.nih.gov/genbank/TSA.html) is an archive that contains computationally assembled transcript sequences from RNA-seq data submitted to SRA or the Trace Archive. The overlapping sequence reads from a complete transcriptome are assembled into transcripts by computational methods instead of by traditional cloning and sequencing of cloned cDNAs. The TSA database has records for, Claviceps purpurea - 4887), Puccinia triticina - 7662), and Alternaria alternata - 17).
NCBI Probe database
The NCBI Probe database (Table 1) is a public registry of nucleic acid reagents designed for use in a wide variety of applications such as gene expression, gene silencing, variation analysis and genome mapping using technologies such as PCR, Real Time qRT-PCR, microarrays and RNAi etc. The Probe database contain probe and primer records from more than 70 fungal species used in a variety of studies including transcript analysis, profiling microbial communities, differential gene expression experiments, genome mapping and more.
Functional genomic data
Gene Expression Omibus (GEO)
The GEO database (Table 1) serves as a public repository for data generated from high-throughput microarray experiments supporting MIAME-compliant (Minimum Information About a Microarray Experiment) data (Sayers et al., 2010). Data sets and profiles are searchable with text based and sequence based (BLAST) searches. Gene expression profiles from curated “DataSets” of seven fungal species are available in GEO and searchable by using free text, gene symbol, Gene Ontology (GO) terms, GenBank accessions or platform accession.
Peptidome
Peptidome (Table 1) is a new public repository at NCBI (Ji et al., 2010) that archives and freely distribute tandem mass spectrometry peptide and protein identification data generated by the scientific community. Data in this database can be searched by free text, peptide sequence, organism, accession, gene symbols, search engine and platform. Currently this repository contains seven studies from Saccharomyces cerevisiae. A Peptidome study record provides a description of the whole experiment and is composed of one or more sample records. A sample record contains a description of the biological material being investigated, protocols used to generate the data, the peptides and proteins that were identified, and the original spectra used to make the identifications. Search example and more information about this database are available on the home page (see Table 1).
Non-sequence data
PubMed
The PubMed database (Table 1) comprise approximately 20 million citation records from various sources including MEDLINE, online books and publishers of journals using LinkOut. PubMed Central (PMC) is a free digital archive of biomedical and life science journal literature. Journal submissions need to meet PMC’s standards for editorial quality and technical production. The PMC Journal List (http://www.ncbi.nlm.nih.gov/pmc/journals/) offers a view of all journal titles that have deposited or are currently depositing final, published articles into the PMC archive. A journal title will appear on the PMC journal list only when a journal’s article is publicly available in PMC. Some journals deposit all their articles to this archive. Examples of such journals related to fungi that fully participate in PMC include Persoonia and Studies in Mycology. Other publishers with a hybrid publishing model will deposit only selected articles.
Having all citations and full-text articles at one location and in one format facilitate quick searches and makes it possible to integrate the literature with a variety of other information resources at NCBI. Many citations and abstract relating to fungal research are available in PubMed with a subset directly linked to other databases (eg. DNA and protein sequences) which is made possible by the Entrez data retrieval system.
Taxonomy
The Entrez Taxonomy database should not be confused with the Taxonomy Browser. An Entrez search query in Table 1 will work in the Entrez Taxonomy search window but not in the Taxonomy Browser search pages (http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?mode=Root). However, clicking on links produced by a search in the Taxonomy database will automatically transfer you to the Taxonomy Browser. The Taxonomy Browser is very useful to provide a bird’s eye view regarding the data associated with registered species. Not only is this information accessible on each taxon page, but also on any taxonomic node by selecting checkboxes at the top of the page for databases of interest. The depth of the nodes can be adjusted and filtered to show for example only taxa with genome sequences (nuclear and mitochondrial) in a taxonomic framework.
Genome Project and BioProject
Genome Project (soon to be renamed to BioProject) is a collection of complete and incomplete (in-progress) large-scale sequencing, assembly, annotation, and mapping projects for cellular organisms. The database is organized into organism-specific overviews that function as portals from which all projects in the database pertaining to that organism can be browsed and retrieved.
BioProject database, a logical extension to Genome Project, will include more project types such as proteome, exome, genotype and phenotype studies, gene expression and many more. It will allow structural hierarchy for large initiatives such as Human Microbiome Project, 1000 Human Genome and GEBA (Genome Encyclopaedia of Bacteria and Archaea).
A summary of all the eukaryote genome projects are accessible via the Genome Project home page (http://www.ncbi.nlm.nih.gov/genomeprj) on the right hand side as the link Eukaryotic Projects and is directly accessible via Table 1. The data displayed can be filtered to show only fungal entries with the sequencing status and method of interest. This page provide organism specific (taxid specific) links to sequencing projects (GPID link) and an overview page (Organism link), which are part of the Genome Project database. In addition there are links to genome related information for each organism which include, GenBank accessions (GB), PubMed entries (PM), RefSeq accessions (genomic and mRNA), Entrez Gene entries (G), Trace Archive entries (T), BLAST database ; M - Map Viewer; F - FTP Sites (Table 1). An overview page gives a short description about the biology of the organism and is useful to consult if a data mining effort produced a record from an organism that is not well known to the user.
Derived (secondary) data
NCBI is adding value to primary data by providing computational analysis results and/or evaluating and improving data by manual curation by NCBI staff or external experts.
Refseq and TPA
Curated data can be found in the NCBI Reference Sequence collection (RefSeq) (Pruitt et al., 2009) and the Third Party Annotation (TPA) database (Cochrane et al., 2006). However, DDBJ/EMBL/GenBank contains primary sequence data and corresponding annotations submitted by laboratories that did the sequencing. The TPA database contains third-party assemblies of primary data with annotation which had been experimentally supported and published in a peer-reviewed scientific journal. Details about how to submit data, as well as examples of what can and cannot be submitted to TPA, are provided on the TPA home page (see Table 2).
RefSeq is a NCBI collection of reference sequences that is a curated, non-redundant set representing basic molecular types genomic DNA, mRNAs and proteins from whole genomes, high-quality genomic regions and targeted loci used for classification purposes. Primarily the annotated genome of a single fungal strain is selected to serve as the reference sequence (Table 2).
Gnomon gene prediction and annotation
Identifying putative genes is a basic essential step in the goal of converting genomic sequence data into biologically relevant information. Gnomon, the NCBI eukaryotic gene prediction tool is a combination of comparative genomics and ab initio modelling. Before a genome is annotated four data sets are collected. A cDNA collection is made from the organism to be annotated and in some cases cDNA from closely related organisms are also included. Then a Target protein set and a Search protein set is generated. The Target protein set includes known proteins from the organism to be annotated and other well studied genomes. The Search set is a much wider collection of eukaryotic proteins. In short the cDNA and Target protein sequences are aligned to the genome and together with ab initio modelling are consolidated into gene models. The first round of predicted models is compared with the Search protein set and the proteins found with good matches are aligned back on the genome to help refining the predicted gene models. With each round of gene prediction a trusted set of organism specific known genes serves as a training set to evaluate the significance of ab initio scores from predicted gene structures.
Recently, our gene prediction method Gnomon (Table 2) has been expanded to process several related genomes simultaneously. Multi-genome Gnomon is an iterative process that starts with a single genome annotation and uses protein similarity between predicted genes to gradually improve the annotation of each genome in the analysis. On each iteration step the best set of models is selected and used as a training set and evidence for the next step. The process usually converges to an optimized set of models at the third iteration. Finally the predicted gene products are annotated with appropriate protein names by using the UniProt protein naming document as a guideline.
Gnomon gene prediction and annotation is freely available to anyone who submits their eukaryotic genome to the public repository at NCBI. The Gnomon or multi-genome Gnomon annotation pipeline is computationally intensive, but genomes can be annotated through the pipeline at NCBI by request (genomes@ncbi.nlm.nih.gov).
RefSeq Targeted Loci Project
Targeted loci are specific molecular markers such as protein coding or ribosomal RNA genes that are used for phylogenetic analyses. Initially, this project contained curated sets of 16S sequences from type strains of prokaryotes but more recently it has been expanded to include other taxonomic groups. The 16S Ribosomal RNA Reference Sequence Similarity Search tool exemplify the functionality of this resources by allowing a visualization of BLAST hits from a single query sequence in the context of a pre-calculated phylogenetic tree using a maximum likelihood analysis of the 16S alignment.
NCBI is currently working to extend the RefSeq Targeted Loci Project (http://www.ncbi.nlm.nih.gov/genomes/static/refseqtarget.html), first to fungi but ultimately to include all groups of eukaryotes. An initial phylogenetically diverse set of 18S (GPID:39195) and 28S sequences (GPID:51803) have been selected, from sequences produced by Assembling the Fungal Tree of Life (AFTOL) project (http://aftol.org/). This collection will eventually be expanded to ex-type and well validated fungal sequences. The goal is to engage the fungal community to provide feedback on reliable sequences in order to finally produce a gold standard set of reference sequences for various sequence analysis applications.
Gene
The goal of Entrez Gene (Maglott et al., 2010) (Table 1) is to visualize and link gene-centred information, which include genome location (visualized by sequence viewer), sequence, expression, protein structure, function, interaction, pathways and homology. The source of information originates internally or externally (e.g. BioGrid). The service LinkOut (http://www.ncbi.nlm.nih.gov/projects/linkout/index.html) at NCBI also facilitate access to external online resources that is relevant to a gene and these links are maintained by external providers in order to extend, clarify, and supplement information found in Entrez databases.
Biologists in the fungal community also have the opportunity to add to the functional annotation of genes described in Entrez Gene. This functional annotation is called GeneRIF (Table 2) and requires only three types of information, a concise phrase describing a function or functions, a PubMed ID and a valid e-mail address from the annotator. Not all taxa are represented in Entrez Gene, and the current scope matches that of NCBI’s Reference Sequence group. To submit a GeneRIF follow the “Submit GeneRIF” link (under the Feedback heading) in the right hand column on the Entrez Gene web page for a gene of interest. At the time of writing the Entrez Gene database included 586 257 loci from Fungi of which 3 406 had a GeneRIF entry associated.
UniGene
Despite the fragmentary and inaccurate nature of EST sequences it is still a valuable source of gene discovery. The aim of the UniGene database (Table 1) is to organize transcript sequences into clusters that appear to come from the same transcription locus (Sayers et al., 2010). These clusters may represent coding and non-coding loci. A UniGene cluster from a coding locus contains sequences that represent a unique gene. The UniGene Cluster page summarizes the sequences in the cluster and a variety of derived information, such as protein similarity and gene expression in various morphological features, developmental stages and under different conditions. Organisms for which there are 70 000 or more EST sequences available are listed in UniGene and this includes five Fungi namely Coccidioides posadasii, Gibberella moniliformis, Magnaporthe oryzae, Neurospora crassa, and Filobasidiella neoformans.
HomoloGene
HomoloGene is a database of homologs (Table 1), generated by automated detection, among the annotated genes of several completely sequenced eukaryotic genomes (Sayers et al., 2010). Predictions are made across a taxonomically diverse group of eukaryotes from the protozoan Plasmodium falciparum to plants, fungi and animals. Fungi included in this analysis are Schizosaccharomyces pombe, Saccharomyces cerevisiae, Kluyveromyces lactis, Eremothecium gossypii, Magnaporthe oryzae and Neurospora crassa. A wider selection of fungi is represented by proteins in the Protein Cluster database.
Protein Clusters
This collection of related protein sequences (clusters) consist of reference sequence proteins and contain both curated and non-curated clusters (Klimke et al., 2009). The protein clusters database started out consisting only of proteins organized in four sets or groups: prokaryotes, phages, chloroplasts and mitochondria. Clusters in each group were created separately and later other groups of clusters were added which included protists and plants. The most recent group being added is the Fungi (Figure 1). Protein Clusters (Table 1) of related protein sequences are independently curated in each group.
Figure 1.

Screenshots showing a section of an alignment created by MUSCLE with matched conserved domains and a neighbor-joining tree of the fungal RPB1 protein cluster as displayed in the Genome Workbench application.
In short, proteins are compared by sequence similarity using an all against all BLAST search (E-value cutoff 10E-05; effective length of the search space set to 5 × 10E8). Each BLAST score is then modified by protein length × alignment length of the BLAST hit and the modified scores are sorted. Cluster members are reciprocal best hits of each other and have greater modified scores to cluster members than to proteins outside the cluster. Curated clusters are enriched with functional annotation, Enzyme Commission numbers identifying enzymatic function and publications describing function.
Conserved Protein Domains (CDD)
CDD (Conserved Domain Database) is a collection (Table 1) of sequence alignments and profiles representing protein domains conserved during molecular evolution (Marchler-Bauer et al., 2009). Alignments in the CDD are imported from two databases outside of NCBI, namely Pfam (Finn et al., 2010) and SMART (Letunic et al., 2009); from NCBI the collection of Protein Clusters (Klimke et al., 2009) and Clusters of Orthologous Groups (COG) (Tatusov et al., 2003).
BioSystems
The BioSystems database (Table 1) is a recent addition to NCBI and was developed as a complementary project to serve as a centralized repository of data, connecting biosystem records with associated literature, molecular, and chemical data (Geer et al., 2010). The BioSystems database contains biological pathways from four sources: KEGG ( Kanehisa et al., 2010), BioCyc (including its Tier 1 EcoCyc and MetaCyc databases, and its Tier 2 databases) (Caspi et al., 2010) and Reactome (Croft et al., 2010). Through these collaborations, the BioSystems database facilitates access to, and provides the ability to compute on, a wide range of biosystems data. Detailed diagrams and annotations for individual biosystems are available on web sites of source databases. Currently there are 5 245 BioSystems entries that apply to fungal species.
Access
Text search Entrez data retrieval system
Entrez (Sayers et al., 2010) is NCBI’s primary text search and retrieval system and comprises 40 molecular and literature databases (Table 1 and 3). The NCBI home page (http://www.ncbi.nlm.nih.gov/) displays the Entrez search system and has “All Databases” selected as default database, making it possible to search all databases simultaneously. A search term such as “Fungi [ORGN]” produces a page that displays record counts from each database that is relevant to Fungi (Table 1). Alternatively, by keeping the search box empty and clicking Go, the home page of each database can be easily reached where additional database-specific links are found.
The richest source of biological function information is in the literature and Entrez integrates the PubMed database of biomedical literature with 39 other literature and molecular databases including DNA and protein sequence, structure, gene, genome, genetic variation and gene expression, etc. The Entrez search interface features powerful options for constructing precise searches and managing results. Options include popular configurable Limits and pre-set filters to help focus on specific kinds of results and “Advanced Search” interface that facilitates constructing more sophisticated queries. Specialized search fields are available for each database and can be browsed and selected in the “Search Builder” section of the “Advanced Search” interface. Most importantly Entrez integrates data with links within and between databases. Not only does this interconnectivity enhance navigation and allow search results to be quickly focused or expanded, but also, more importantly, these relationships often expose unexpected connections that can lead to scientific discoveries. Alternatively, the user can search, link, and download sequences programmatically by using NCBI e-utilities (http://www.ncbi.nlm.nih.gov/entrez/query/static/eutils_help.html).
BLAST: regular and customized databases
The well-known BLAST sequence analysis tool (Altschul et al., 1990) finds regions of local similarity between sequences and is frequently used to search for sequences in a known set (database) that are similar to a query sequence of interest. A database in the context of this program is a specially formatted collection of sequences. For fast searching the sequences are divided in words of various length and word positions are indexed. A more detailed description can be found in The NCBI Handbook chapter: http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=handbook&part=ch16. Customized BLAST databases are available for sequences in the RefSeq collection and a link (Genomic BLAST: Eukaryotic) on the Genomes home page (http://www.ncbi.nlm.nih.gov/genome) provides access. A dedicated BLAST page with only fungal genomes is listed in Table 3.
In addition to basic BLAST there are also specialized BLAST searches available at NCBI, for example Primer-BLAST, COBALT (Papadopoulos and Agarwala, 2007) and more (http://blast.ncbi.nlm.nih.gov/Blast.cgi). COBALT is a constraint based protein multiple alignment tool and the alignment is aided by a collection of pairwise constraints derived from conserved domain database, protein motif database, and local sequence similarity using RPS-BLAST, BLASTP, and PHI-BLAST, respectively.
BLAST results with Links (BLink)
BLink (Table 3) displays results of BLAST searches that have been done for every protein sequence in the Entrez Proteins data domain. To access it, follow the BLink link displayed beside any hit in the results of an Entrez Proteins search. BLink offers links to various display options, including a distribution of hits by taxonomic grouping, best hits to each organism, protein domains in the query sequence, similar sequences that have known 3D structures, and more. Additional options allow you to filter out some taxa from a result list, increase or decrease the BLAST cut-off score, or filter BLAST hits to show only those from a specific source database, such as RefSeq or Swiss-Prot.
TaxMap
TaxMap reports the taxonomic distribution of best hits using BLAST to the “nr” database (excluding best hits within the same genus) for proteins annotated on each of the chromosomes of a genome. TaxMap displays a chromosome with protein encoding genes in the same order as they occur in the genome as a row of dots (maximum 100 proteins per row) that is colour-coded according to 4 taxonomic groups (Eukaryota, Eubacteria, Archae and Virus) to which a protein had the highest similarity (even by a very small margin). Absence of a coloured dot represents genes whose product does not show similarity to other proteins above the cut-off score (Cut-Off) and set margin (Cut-Off+). TaxMap is useful for identifying candidates that may have originated by a horizontal gene transfer event from other taxonomic lineages at some point in the evolutionary past of an organism. Fungal RefSeq genomes assembled to chromosome level are available in this tool which amount to 15 genomes mainly from the Saccharomycotina and a few other ascomycetes, one basidiomycete and two microsporidia. This tool is accessible via RefSeq chromosome accession links on RefSeq Project pages. An example of TaxMap’s functionality displaying chromosome 8 from Aspergillus fumigatus (Table 3) is discussed later within this review.
TaxPlot
TaxPlot (Table 3) is a tool that visualizes a 3-way comparison of taxa based on protein sequence similarity. To use TaxPlot, a reference genome is selected to which two other genomes are compared. Pre-computed BLAST results are then used to plot a point for each predicted protein in a reference genome, based on the best alignment with proteins in each of the two genomes being compared. The default comparison has Drosophila melanogaster as reference genome compared to Caenorhabditis elegans and Saccharomyces cerevisiae (all annotated strains at NCBI) with a cut-off score set to 10. However all parameters are user defined variables. This tool provides a genome-wide snapshot of protein similarity from a reference genome to other taxa of biological significance and could provide insight to shared functionality. Twenty two fungal genomes, which are complete or assembled in chromosomal scaffolds, are currently available for comparison in this tool. The use of TaxPlot and TaxMap has been demonstrated in a comparative genomics study involving food-borne pathogens (Bhagwat and Bhagwat, 2008).
UniGene Digital Differential Display (DDD)
The DDD tool (Table 3) list the title and tissue source for those libraries that have sequences in UniGene and allow comparisons between EST libraries within a specific organism. Two single libraries or two pools of libraries can be compared, for example transcripts obtained from a fungus grown under various conditions or life stages. UniGene entries that are statistically significant in EST counts between two conditions are displayed. Output includes for each gene, the frequency of its transcript in each library and the title of the gene’s corresponding UniGene cluster. Results are sorted by significance, with genes having the largest differences in frequencies displayed at the top.
FLink
The BioSystem database has a useful companion tool called FLink (Frequency weighted links) which is a tool that enables a user to traverse from a group of records in a source database (e.g., Proteins) to a ranked list of associated records in a destination database (e.g., BioSystems) (Table 3). Although FLink was initially developed as a companion tool for the BioSystems database, it can also be used in a similar way for other types of input and output data with the following supported databases, CDD, Gene, Protein, PubChem, Pubmed and Structure.
Examples
There are many ways of accessing fungal data at NCBI and depending on the focus and goal of a research project or the level of interest a user would select a particular route for accessing data. These may involve analysis using sequence similarity searches from BLAST (TaxMap, BLink, TaxPlot), direct browsing in the graphical sequence interface or text based searches in Entrez.
The usage scenarios outlined below illustrate some of the possible data mining avenues available at NCBI. The first example shows how a user can seamlessly traverse between several databases using pre-computed links, while making several discoveries along the way. The second scenario reveals the possibility of navigating directly from a PubMed record to the protein described in that publication and find similar proteins in his/her favourite fungus. The power of PHI-BLAST and PSI-BLAST are demonstrated while trying to find proteins with putative inter-zinc finger interactions in the third example.
Examples using sequence based searches and text based searches
Starting a search from a bioinformatic tool
A user may be interested to identify gene candidates that may have originated via a horizontal gene transfer event from other taxonomic lineages at some point in the evolutionary past of a fungus. TaxMap is a useful tool to find such candidates. For example, to access TaxMap for chromosome 8 of Aspergillus fumigatus, navigate to the RefSeq project page by selecting the Genome Project database in the Entrez search system and search for “Aspergillus fumigatus [organism]”. Under the RefSeq heading in the resulting overview page select Aspergillus fumigatus. Click on the RefSeq accession (NC_007201) and then on the TaxMap link. Figure 2A show the TaxMap tool and the taxonomic distribution of best hits using BLAST to the “nr” database with proteins on chromosome 8 of Aspergillus fumigatus strain Af293 as query. A. fumigatus proteins are color coded according taxonomic origin of the most similar hit. In this example most are pink indicating Eukaryota and 5 blue indicating Eubacteria. Twenty four proteins are not color coded which indicated there were no similar hits to proteins from fungi outside of the Genus Neosartorya (teleomorph Genus of A. fumigatus). Thus no score were higher than the BLAST threshold (Cut-Off) set at 95 and similar hits to a protein from a distant taxonomic group such as Eubacteria, Archae or Virus were not higher than the Cut-Off+ value which in this case was 105 (95+10). Hits that were in the grey zone e.g. between 95 and 105 are indicated with a dark filled circle and the rest by a plus sign. Clicking on the 5 most similar hits from the taxonomic group Eubacteria, show the resulting table with blast scores well above 105 (Figure 2B). These five proteins are candidates for possible horizontal gene transfer events. However, take note that the Cut-Off and Cut-Off+ parameters of the TaxMap tool are user defined variables which need to be adjusted to avoid false positives and this example only used default variables.
Figure 2.

(A) A TaxMap of chromosome 8 of Aspergillus fumigatus showing the taxonomic distribution of best hits (Eukaryotes = pink; Bacteria = blue) with (B) a list of best hits to bacteria produced by the link (number 5) in the Best column.
As an example the gene with the highest similarity hit in this group namely steroid monooxygenase may be of interest to a user and following the link under the Gene column (Figure 2B) will produce pre-computed BLAST results (a BLink page). Figure 3A displays a screenshot with the top 20 hits visible. A BLink page shows the distribution of all hits over major taxonomic groups in color coded boxes, a histogram of the % hits over a query sequence and distribution of CDD and BLASTP hits coverage over a query sequence. In this example the CDD link will produce a list of three domain hits of which the TrkA multi-domain hit, a predicted flavoprotein involved in K+ transport was most significant which mapped to the same region in the protein where all the best hits aligned.
Figure 3.
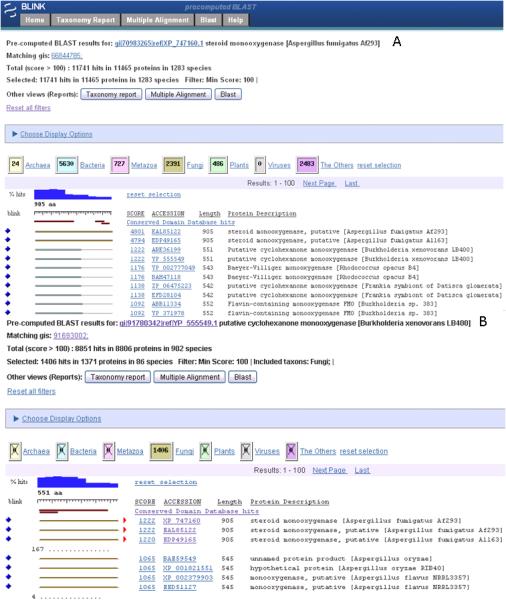
(A) Pre-computed BLAST results of the Aspergillus fumigatus gene with the highest similarity score in the list of 5 best hits to Bacteria (see Fig. 1). (B) A BLink report of the top bacterial hit as query (RefSeq accession YP_555549) and the results filtered to show only Fungi hits.
The best bacterial hit is that of a putative cyclohexanone monooxygenase [Burkholderia xenovorans LB400]. The biology of this bacterium may not be known to a mycologist but there is a quick way to read a short description about it. Click on the RefSeq accession (accession with the underscore) of this bacterial hit and on the resulting GenBank flat file follow the link to the Genome Project overview page for a short description.
Another BLink page with the “putative cyclohexanone monooxygenase“ hit as query can be obtained by following the blue diamond on the left (Figure 3A) and filtered to show other fungal hits buried in the BLAST results although with slightly lower scores (Figure 3B). The direction of a more than 10% overhang as compared to the hit range with the query is indicated by red arrows. Note, an overhang is on the C terminal end of the A. fumigatus protein but absent in other fungal proteins. The number and position of protein hits from non-fungal taxa that have been excluded by the filtering step (show Fungi Only) are indicated by a dotted line.
At this point the user may be interested to know if this gene is expressed. Using the RefSeq protein XP_747160 link on the BLINK page in Figure 3B produces the GenBank flat file. On this flat file page follow the BLAST link to analyze this sequence using tblastn and choose the database “Non-human, non-mouse ESTs (est_others)” to see if there are any matches to expressed gene sequences. There were no matches to A. fumigatus sequences but curiously the results show a 100% match to a sequence from a distant fungus Glomus intraradices on the C-terminal end (overlapping the region with a CDD hit, Methyltransferase in polyketide synthase (PKS)). Lower scoring hits from fungal ESTs were on the N-terminal end where all the previous best bacterial hits were. Upon further investigation this unexpected discovery may be of biological significance or merely a case of contamination. To view all the gene centred information for this accession follow the database cross reference (/db_xref) link to the Entrez Gene resource on the XP_747160 GenBank flat file (Figure 4). All headings are by default expanded to show all relevant information, but to fit all into a figure the last 6 headings were contracted. On the right hand side there are internal resources which specifically apply to this gene e.g. Conserved Domains and Map Viewer as well as external resources such as the KEGG database. In this web page (Figure 4), under the heading “Genomic regions, transcripts and products” is a link (“Open Full View”) to the full genome sequence viewer of the chromosome on which this gene of interest is located. A default view is seen in Figure 5A, however by increasing the range in the lower window so as to include more genes and pinning the info box of some genes on the viewer (when the mouse hovers over it), the view can be adjusted (Figure 5B). The steroid monooxygenase A. fumigatus gene is surrounded by genes involved in secondary metabolism e.g. polyketide synthases.
Figure 4.
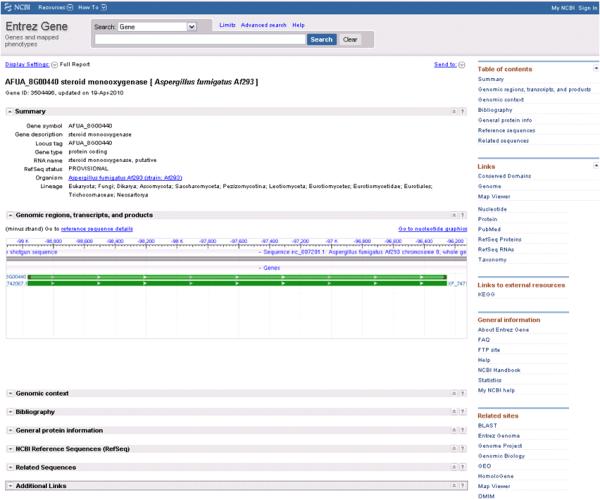
The Entrez Gene page of the steroid monooxygenase gene from Aspergillus fumigatus (locus tag AFUA_8G00440), showing gene related information including the sequence viewer and a link (Open Full View) to see the full view.
Figure 5.
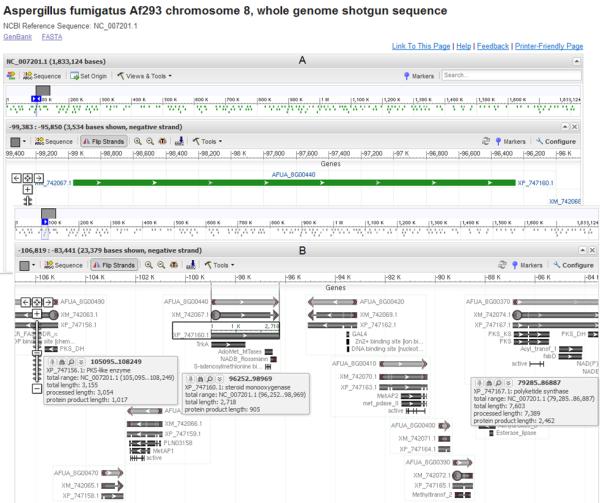
(A) The default view after following the sequence viewer link from the Entrez Gene page of locus AFUA_8G00440. (B) A graphical view of the genome neighbourhood around locus AFUA_8G00440 after modifying settings in the default view displayed above.
For more information about these secondary metabolites as it relates to bacteria and fungi do a global search using all databases in the Entrez Search Engine with the following query: “polyketide synthase” AND Bacteria AND Fungi. This search produces hits in several databases including for example PubMed, PMC, BioSystems etc (Figure 6).
Figure 6.
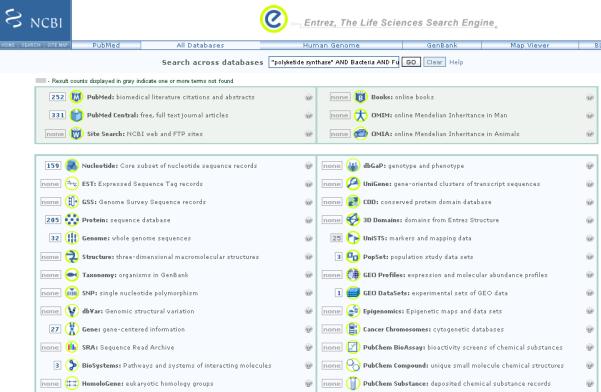
Results of a global Entrez search using the following as query, “polyketide synthase” AND Bacteria AND Fungi.
Alternatively, a user may wish to investigate which pathways these best hits (Figure 3A) are functioning in. By selecting the Blast button as “Other view (report)”, the format of the results page can be changed to a traditional BLAST report. In this format the user have the opportunity to select all hits and get these selected sequences. This action transfer a user to the protein database where a GI list of the selected protein sequences can be downloaded by clicking Send to, selecting File as destination and saving the GI list as format. After clearing the search window, the next step is to navigate to the BioSystem database by selecting the BioSystem database and clicking Go. The resulting default page provides a link to its companion tool FLink (http://www.ncbi.nlm.nih.gov/Structure/flink/flink.cgi) which takes the protein GI list as input and retrieves a ranked list of BioSystem records that contain proteins from the input list. Details about steps to follow in producing these results are available on the website of BioSystems: http://www.ncbi.nlm.nih.gov/Structure/flink/docs/flink_help.html#QuickStart. BioSystem entries for these hits involved caprolactam degradation (Figure 7A). The FLink companion tool also provides an option to link to other databases such as PubMed which if selected will list articles cited by protein sequence records in the search (Figure 7B).
Figure 7.
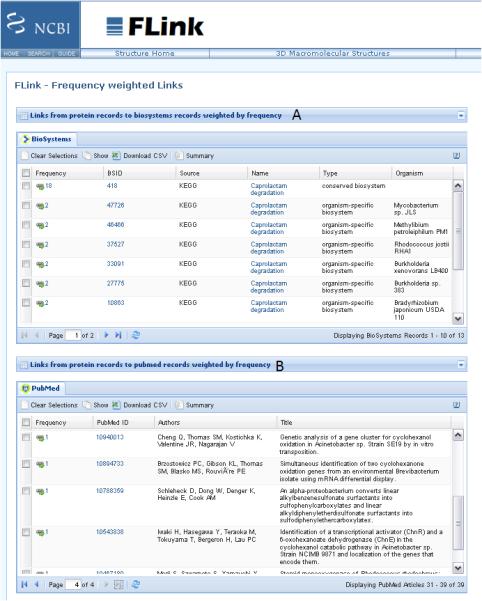
(A) A ranked list of biosystems weighted by the frequency of proteins present in the GI input list obtained from the BLAST results in Fig 2A. (B) A ranked list of PubMed records associated with the best BLAST hits in Fig 2A.
Starting a search from the PubMed database
In this scenario a user may be interested to find articles about fungal virulence factors and their interaction with reactive oxygen species during the infection of a plant. A search in the PubMed database using for example this Boolean query “ROS AND virulence AND fungus AND plant” delivered a list of 25+ papers with the most recent articles at the top. By selecting an article a user not only sees the abstract but also all available links from this record in the right hand column. Links such as related citations, Nucleotide, Protein, References for this PMC article and more are listed. Related information can be found one article at a time or several could be selected from a list and the resources of your choice selected in the right hand column. For this example the article with PubMed ID 19893627 was picked and the link followed to the described protein from Alternaria brassicicola. The protein TmpL is a FAD/NAD(P)-binding flavoprotein and involved in plant and animal fungal virulence (Kim et al., 2009). This action transferred a user to the Protein database as indicated in the search window at the top. In this page there is a “Run BLAST” link that will use this protein as a query in a BLAST search. On the BLAST page there are the usual parameters to be set but in this case we use default values and search similar proteins in the non-redundant protein sequence database. The resulting BLASTP search find several proteins with the top hits having an e-value = 0, >96% query coverage and >55% identity in the matched region.
Several of the top hits are annotated as non-ribosomal peptide synthetases (NPS) since the AMP-binding domain of this protein have similarity to adenylation domains of NPS proteins. However, as the research paper indicated this query protein and several homologs contain no thiolation and condensation domains which are typical components of a NPS module and not true NPS proteins. In addition the query protein contained FAD/NAD(P)-binding domains. To find FAD/NAD(P)-binding domain motifs in this query protein and sequences from your favorite fungus scroll down the BLAST page to the alignments and select protein sequences by clicking on the appropriate boxes. After getting the selected sequences change the display to fasta format by clicking on ‘Display Settings”, specifying FASTA (not FASTA text) and hit “Apply”. On the right hand side under the heading “Analyze these sequences” select “Find in these sequences”. This action opens a search box in the bottom navigation bar, wherein one can type any motif using protein single letter and ambiguity codes or Prosite patterns. In this case for example the pattern, “GSGIGP”, matching a putative NAD(P) binding domain motif in the TmpL protein were also found in protein hits from a close relative, Pyrenophora tritici-repentis. Matched pattern positions are indicated in the navigation bar.
Starting with a BLAST search
In this example a user may be interested to find a family of putative zinc finger (ZF) domain (ZFD) proteins (specifically PACC-like) that contain tandem CWCH2 sequence motifs that may be involved in inter-zinc finger interactions as suggested by Hatayama and Aruga, 2010. The Aspergillus niger gene PACC is listed as an example of a protein that contain two N-terminal ZFs which forms a structural unit comprised with two tryptophan side chains at the centre of a hydrophobic core. This specific protein can be found with the following search in the Protein database, “PACC [Gene Name] AND Aspergillus niger [ORGN]”. More than one entry is found, because these records represent copies form different source databases such as UniProt, GenBank and RefSeq. The following modified search,“PACC[Gene Name] AND Aspergillus niger [ORGN] AND “refseq”[Filter]” produces only one record. On the protein record page follow the Run BLAST link and on the BLAST page select the PHI-BLAST algorithm and enter the tandem CWCH2 pattern used in the publication. If a user is only interested in finding hits in a limited taxon set, then this can be set in the Organism search box (any taxonomic level is accepted). The PHI-BLAST results shows the list of significant alignments having a CWCH2 pattern and with an E-value better than the threshold. Scroll down to where the alignments start and click on the Multiple alignment link. The multiple alignments visualize the lack of overlap among a subset of sequences and the lack of aligned sequences in a subset of shorter proteins. At the top of the multi-alignment page, the “phylogenetic tree” link displays a guide tree using a fast minimum evolution algorithm from given distances by default (other options are also available). However, despite the wording on the link these are not phylogenetic trees but guide trees and provide a rough visualization of the similarities between sequences. This PHI-BLAST example was limited to Aspergillus species and the resulting tree produced roughly three groups. These included a PACC containing group, a group where some sequences lacked alignment on their N-terminal end and a group with shorter protein sequences (half the length of the PACC clade). Different tree rendering methods are available at the top of the page and the tree could be re-rooted at any internal node by hovering with the mouse over a node and selecting “Re-Root” from the list of actions.
The resulting PHI-BLAST alignments can also be used to run a PSI-BLAST search by clicking “Go” below the set of significant PHI-BLAST hits. The resulting search revealed the presence of a protein from Aspergillus terreus not previously found but with a 100% query coverage and an E-value equal to zero. However, when selecting this protein record and searching the fasta sequence with the tandem CWCH2 pattern (xCxWx(2,35)Cx(5,15)Hx(2,5)Hx(5,25)CxWx(1,3)Cx(5,17)Hx(2,5)Hx) no match was found. This protein could truly lack this characteristic feature or further inspection of the gene model may reveal an alternative annotation that provides a better alignment in this region.
Downloading data
Alternatively to analyze fungal data on the user’s own machine there is an option to download files via FTP sites. FTP sites exist for primary and derived data types including genomes, annotation, ESTs, microarray data, traces, SRA data, Protein Clusters, RefSeq and UniGene (http://www.ncbi.nlm.nih.gov/guide/all/#downloads_). In addition to data downloads, there are a variety of tools that are available for download that function on a standalone basis, for example Splign (Kapustin et al., 2008), BLAST, CDTree (Marchler-Bauer et al., 2010) and Genome Workbench (http://www.ncbi.nlm.nih.gov/projects/gbench/) etc. Splign is a utility for computing spliced alignments (cDNA-to-Genomic) and CDtree aid in the classification of protein sequences and investigates their evolutionary relationships. Genome Workbench can display sequence data in many ways, including graphical sequence views, various alignment views, phylogenetic tree views, and tabular views of data. It can also align your private data to data in public databases, display your data in the context of public data, and retrieve BLAST results.
Conclusion
Even though it is not possible to show all search scenarios, examples above demonstrated how search results from one database can be transferred and used as input in another database which allows for flexibility in mining data for biological significance. Examples showed that navigation between at least 10 resources (TaxMap, BLink, Protein database, Genome Project, EST database, Gene, sequence viewer, BioSystems, FLink and PubMed) was easily done to gather information.
NCBI maintain several databases, containing a wide range of information, covering the whole spectrum of life and thus feedback (info@ncbi.nlm.nih.gov) are welcome if some Fungi specific issues are not currently addressed. A request, to include a submitted genome not currently present in a pre-computed analysis, will be considered. However, by submitting an annotated complete genome, a user will benefit from most of the web tools available at NCBI.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2010 Nov 10; doi: 10.1093/nar/gkp1024. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H, Henrick K, Nakamura H, Markley JL. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007;35(Database issue):D301–3. doi: 10.1093/nar/gkl971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhagwat AA, Bhagwat M. Methods and Tools for Comparative Genomics of Foodborne Pathogens. Foodborne Pathogens and Disease. 2008;5:487–497. doi: 10.1089/fpd.2008.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R, Altman T, Dale JM, Dreher K, Fulcher CA, Gilham F, Kaipa P, Karthikeyan AS, Kothari A, Krummenacker M, Latendresse M, Mueller LA, Paley S, Popescu L, Pujar A, Shearer AG, Zhang P, Karp PD. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2010;38(Database issue):D473–9. doi: 10.1093/nar/gkp875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane G, Bates K, Apweiler R, Tateno Y, Mashima J, Kosuge T, Mizrachi IK, Schafer S, Fetchko M. Evidence standards in experimental and inferential INSDC Third Party Annotation data. OMICS. 2006;10:105–13. doi: 10.1089/omi.2006.10.105. [DOI] [PubMed] [Google Scholar]
- Cooper PS, Lipshultz D, Matten WT, McGinnis SD, Pechous S, Romiti ML, Tao T, Valjavec-Gratian M, Sayers EW. Education resources of the National Center for Biotechnology Information. Brief Bioinform. 2010;11:563–9. doi: 10.1093/bib/bbq022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft D, O’Kelly G, Wu G, Haw R, Gillespie M, Matthews L, Caudy M, Garapati P, Gopinath G, Jassal B, Jupe S, Kataskaya I, Mahajan S, May B, Ndegwa N, Schmidt E, Shamovsky V, Yung C, Birney E, Hermjakob H, D’Eustachio P, Stein L. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2010 Nov 23; doi: 10.1093/nar/gkq1018. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunesekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2010;38(Database Issue):D211–222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geer LY, Marchler-Bauer A, Geer RC, Han L, He J, He S, Liu C, Shi W, Bryant SH. The NCBI BioSystems database. Nucleic. Acids Res. 2010;38(Database Issue):D492–D496. doi: 10.1093/nar/gkp858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatayama M, Aruga J. Characterization of the tandem CWCH2 sequence motif: a hallmark of inter-zinc finger interactions. BMC Evol Biol. 2010;10:53. doi: 10.1186/1471-2148-10-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji L, Barrett T, Ayanbule O, Troup DB, Rudnev D, Muertter RN, Tomashevsky M, Soboleva A, Slotta DJ. NCBI Peptidome: a new repository for mass spectrometry proteomics data. Nucleic Acids Res. 2010;38(Database issue):D731–5. doi: 10.1093/nar/gkp1047. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminuma E, Kosuge T, Kodama Y, Aono H, Mashima J, Gojobori T, Sugawara H, Ogasawara O, Takagi T, Okubo K, Nakamura Y. DDBJ progress report. Nucleic Acids Res. 2010 Nov 9; doi: 10.1093/nar/gkq1041. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Furumichi M Kawashima, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38(Database issue):D355–60. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapustin Y, Souvorov A, Tatusova T, Lipman D. Splign: algorithms for computing spliced alignments with identification of paralogs. Biol Direct. 2008;3:20. doi: 10.1186/1745-6150-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Willger SD, Park SW, Puttikamonkul S, Grahl N, Cho Y, Mukhopadhyay B, Cramer RA, Jr, Lawrence CB. TmpL, a transmembrane protein required for intracellular redox homeostasis and virulence in a plant and an animal fungal pathogen. PLoS Pathog. 2009;5(11):e1000653. doi: 10.1371/journal.ppat.1000653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimke W, Agarwala R, Badretdin A, Chetvernin S, Ciufo S, Fedorov B, Kiryutin B, O’Neill K, Resch W, Resenchuk S, Schafer S, Tolstoy I, Tatusova T. The National Center for Biotechnology Information’s Protein Clusters Database. Nucleic Acids Res. 2009;37(Database issue):D216–23. doi: 10.1093/nar/gkn734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen R, Akhtar R, Birney E, Bower L, Cerdeno-Tárraga A, Cheng Y, Cleland I, Faruque N, Goodgame N, Gibson R, Hoad G, Jang M, Pakseresht N, Plaister S, Radhakrishnan R, Reddy K, Sobhany S, Ten Hoopen P, Vaughan R, Zalunin V, Cochrane G. The European Nucleotide Archive. Nucleic Acids Res. 2010a Oct 23; doi: 10.1093/nar/gkq967. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen R, Sugawara H, Shumway M. The sequence read archive. Nucleic Acids Res. 2010b Nov 9; doi: 10.1093/nar/gkq1019. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Doerks T, Bork P. SMART 6: recent updates and new developments. Nucleic Acids Res. 2009;37(Database issue):D229–32. doi: 10.1093/nar/gkn808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2010 Nov 28; doi: 10.1093/nar/gkq1237. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, Deweese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2010 Nov 24; doi: 10.1093/nar/gkq1189. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos JS, Agarwala R. COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics. 2007;23(9):1073–1079. doi: 10.1093/bioinformatics/btm076. (2007) [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Klimke W, Maglott DR. NCBI Reference Sequences: current status, policy and new initiatives. Nucleic Acids Res. 2009;37(Database issue):D32–6. doi: 10.1093/nar/gkn721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL, Church D, DiCuccio M, Yaschenko E, Ostell J. The Genome Assembly Archive: A New Public Resource. PLoS Biol. 2004;2(9):e285. doi: 10.1371/journal.pbio.0020285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Federhen S, Feolo M, Fingerman IM, Geer LY, Helmberg W, Kapustin Y, Landsman D, Lipman DJ, Lu Z, Madden TL, Madej T, Maglott DR, Marchler-Bauer A, Miller V, Mizrachi I, Ostell J, Panchenko A, Phan L, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Shumway M, Sirotkin K, Slotta D, Souvorov A, Starchenko G, Tatusova TA, Wagner L, Wang Y, Wilbur WJ, Yaschenko E, Ye J. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2010 Nov 21; doi: 10.1093/nar/gkq1172. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res. 2010 Nov 4; doi: 10.1093/nar/gkq1020. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]