

Abstract
Y-family DNA-polymerases have larger active sites that can accommodate bulky DNA adducts allowing them to bypass these lesions during replication. One member, polymerase eta (pol eta), is specialized for the bypass of UV-induced thymidine-thymidine dimers, correctly inserting two adenines. Loss of pol eta function is the molecular basis for xeroderma pigmentosum (XP) variant where the accumulation of mutations results in a dramatic increase in UV-induced skin cancers. Less is known about the role of pol eta in the bypass of other DNA adducts. A commonly encountered DNA adduct is that caused by benzo[a]pyrene diol epoxide (BPDE), the ultimate carcinogenic metabolite of the environmental chemical benzo[a]pyrene. Here, treatment of pol eta-deficient fibroblasts from humans and mice with BPDE resulted in a significant decrease in Hprt gene mutations. These studies in mammalian cells support a number of in vitro reports that purified pol eta has error-prone activity on plasmids with site-directed BPDE adducts. Sequencing the Hprt gene from this work shows that the majority of mutations are G>T transversions. These data suggest that pol eta has error-prone activity when bypassing BPDE-adducts. Understanding the basis of environmental carcinogen-derived mutations may enable prevention strategies to reduce such mutations with the intent to reduce the number of environmentally relevant cancers.
Introduction
Environmental mutagens such as the ultraviolet (UV) component of sunlight and chemical mutagens in food and cigarette smoke are well-established human carcinogens. One such compound is benzo[a]pyrene (B[a]P), which is a polycyclic aromatic hydrocarbon present in cigarette smoke, diesel exhaust and well-cooked meat. The carcinogenic effects of B[a]P are largely due to its metabolism to the highly reactive product benzo[a]pyrene diol epoxide (BPDE), which principally binds to the exocyclic amine of guanine [1]. Nucleotide excision repair recognizes and removes adducts induced by BPDE using transcription-coupled and global genomic repair mechanisms [2], [3]. However, if a cell enters S-phase with persistent damage, helix-distorting lesions halt the progression of the replication apparatus. Data indicate that specialized DNA polymerases that have relaxed base-pairing requirements may facilitate bypass of bulky DNA adducts in a process called translesion DNA synthesis (TLS), but with potentially mutagenic consequences. A subset of these proteins has been classified as Y-family polymerases based on structural similarities [4].
There are four members of the Y-family: REV1, pol eta, pol iota, and pol kappa. The role of these proteins in human disease is best characterized for pol eta. Loss of pol eta activity is the molecular defect underlying the XP variant syndrome [5], [6]. These patients have a very high incidence of sunlight-induced skin cancer but are nucleotide-excision repair proficient unlike the classic XP complementation groups A–G [7]. Cells from these patients are extremely hypermutable after exposure to UV due to the deficiency of pol eta which normally bypasses the most common UV-induced photoproducts, thymine-thymine cyclobutane pyrimidine dimers, in an error-free manner [8], [9]. In the absence of pol eta, the highly error-prone pol iota assumes this bypass function resulting in the accumulation of UV-induced mutations and an increased susceptibility to skin cancer [10]–[12].
The role of the Y-family proteins in the bypass of BPDE-induced adducts is considerably less clear. Pol kappa has been shown to participate in error-free bypass of lesions induced by BPDE in vitro using purified mouse and human enzymes [13]–[15]. Similar conclusions were reported using mouse cells lacking pol kappa activity [15], [16]. While there are some data concerning the role of pol eta in TLS of BPDE-induced DNA adducts, the data are somewhat contradictory. Purified pol eta has been shown to be capable of performing error-prone bypass of BPDE-adducted plasmids in vitro [13], [17], [18] and in mammalian cells using shuttle vectors to measure TLS efficiency and fidelity [19]. However, measurements of mutations at the endogenous hypoxanthine-guanine phosphoribosyltransferase (HPRT) locus in pol eta-deficient human cells treated with BPDE suggest that pol eta is error-free in bypassing BPDE-induced DNA adducts [20].
The B family member pol zeta is also implicated in the bypass of BPDE lesions. Purified pol zeta has alternatively been shown to have error-free [21] and error-prone [22], [23] bypass activity on BPDE-adducted DNA templates in vitro. In addition, pol zeta deficiency has been shown to affect TLS of BPDE lesions in mammalian cells [24], [25].
Here, BPDE treatment of both cells derived from pol eta-knockout mice and pol eta-deficient cells from an XP variant patient resulted in lower induced mutant frequencies. Our results suggest that pol eta is involved in error-prone bypass of BPDE-induced lesions and may be partially responsible for the mutagenic effects of this carcinogen. Understanding the mechanisms by which environmental chemicals induce mutations in DNA increases our understanding of how cancers can be linked to the environment and will increase our ability to develop strategies to prevent such cancers, for example through tissue-specific selective inhibition of one or more of these enzymes.
Results
Cytotoxicity from BPDE exposure is independent of pol eta status
The survival of mouse primary fibroblasts after treatment with 150 nM BPDE was assessed using clonogenic assays. Wild-type and pol eta knockout cells exhibited similar levels of cytotoxicity when exposed to 150 nM BPDE (41±5% and 37±8% survival, respectively, p = 0.67) (Figure 1). Human cells with wild-type pol eta and XP variant cells without functional pol eta exhibited similar sensitivities to 150 nM BPDE (46±9% and 36±10%, respectively, p = 0.51) (Figure 1). Mutagenic responses could therefore be assessed in comparable numbers of surviving cells after treatment.
Figure 1. Polymerase eta does not affect cell survival after 150nM BPDE treatment.
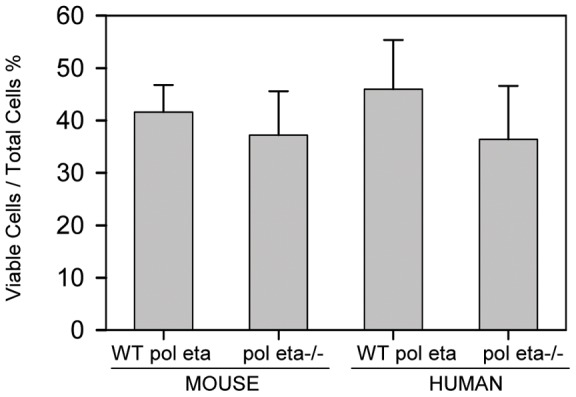
Viable cell calculations were made based on the number of clones present relative to the number of cells plated. This value was adjusted based on the number of clones observed in vehicle-only treated cells and expressed as average ± SEM.
Reduced BPDE-induced mutant frequency was observed in the absence of pol eta
Fibroblasts deficient for pol eta were treated with 150 nM BPDE and mutant frequency was assessed by the formation of 6-thioguanine-(TG) resistant clones. The induced mutant frequency of wild type mouse fibroblasts in response to 150 nM BPDE was 213±48 mutants per 1×106 cells. Relative to wild-type, the BPDE-induced mutant frequency in pol eta null mouse fibroblasts was significantly decreased, 34±10 mutants per 1×106cells (p<0.05) (Figure 2).
Figure 2. Induced mutant frequency is reduced in polymerase eta-deficient cells after BPDE treatment.
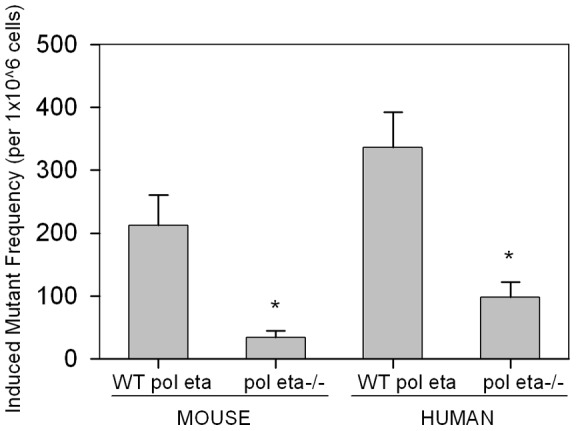
Mutant frequency induced by 150 nM BPDE was calculated relative to the frequency of vehicle treated cells and expressed as average ± SEM. *p<0.05.
Similarly, wild type human fibroblasts had a much higher induced mutant frequency relative to XP variant fibroblasts containing a non-functional pol eta. Normal human fibroblasts had an induced mutant frequency of 337±56 mutants per 1×106cells while the induced mutant frequency in XP variant fibroblasts was 96±24 mutants per 1×106cells (p<0.05) (Figure 2). Both human and mouse cells had low background mutation frequency which was not affected by the loss of pol eta (data not shown).
Sequencing of Hprt mutant clones
After two weeks of growth in selective medium, macroscopic colonies of murine fibroblasts were isolated and lysed for RT-PCR amplification of Hprt cDNA and subsequent sequence analysis. Overall, the majority of the mutations in these mutant clones were guanine to thymine transversions which accounted for 50% and 70% of the mutations in wild-type and pol eta-null fibroblasts, respectively (Table 1).
Table 1. Kinds of Mutations Induced in the Hprt Gene of Murine Dermal Fibroblasts.
| Pol eta+/+ | Pol eta−/− | |||
| No. | % | No. | % | |
| Deletion of one Purine | 1 | 4.55 | 1 | 4.35 |
| Tandems | 2 | 9.09 | 0 | - |
| Transitions | ||||
| G•C → A•T | 4 | 18.18 | 3 | 13.04 |
| A•T → G•C | 1 | 4.55 | 2 | 8.70 |
| Transversions | ||||
| G•C → T•A | 11 | 50.00 | 16 | 69.57 |
| G•C → C•G | 0 | - | 1 | 4.35 |
| A•T → T•A | 1 | 4.55 | 0 | - |
| A•T → C•G | 2 | 9.09 | 0 | - |
| Total | 22 | 100 | 23 | 100 |
Discussion
The somatic mutation hypothesis of cancer asserts that changes arising from mutations in the genome can result in cells with a growth advantage and thus tumorigenic potential. While it is currently estimated that the spontaneous mutation rate in human cells is around 1–2×10−8 per nucleotide per generation [26], the overwhelming evidence for environmental carcinogen-induced cancers is attributed to the ability of these agents to cause DNA damage that results in the increased accumulation of mutations far exceeding those that occur spontaneously. The ability of a cell to bypass DNA-damage during replication prevents prolonged stalling of replication fork complexes that could signal cell death. However, depending on both the lesion encountered and the polymerase recruited, this bypass could result in replication errors that become mutations upon subsequent cell divisions. Polymerases of the Y-family are well known to participate in bypass replication and research has been focused on determining the accuracy of each enzyme when a particular DNA lesion is encountered. While polymerase kappa is likely specialized for protection against endogenously produced DNA adducts including those resulting from reactive oxygen species, this enzyme has also been implicated in the error-free bypass of BPDE lesions [13]–[15]. Other members of this polymerase family however, namely pol eta and pol iota, appear to have lower fidelity when they encounter BPDE-adducted bases. It was previously reported that pol eta may be involved in error-prone bypass of BPDE adducts using in vitro and yeast systems [18], [22], [27]. The only data examining pol eta in mammalian cells showed that human fibroblasts without a functional copy of pol eta were equally mutable relative to their wild-type counterparts after exposure to BPDE, indicating that pol eta does not participate in BPDE adduct bypass [20]. However, in this investigation, the eta-null XP variant cells and the normal human fibroblasts were not treated simultaneously, rather the normal fibroblast results were obtained from a different study in which a modified treatment protocol was used [28]. In the current study both murine and human fibroblasts without a functional pol eta accumulated fewer BPDE-induced mutations indicating an error-prone role for this enzyme in the bypass of BPDE-adducted DNA. Additionally, the current study used an asynchronous population of cells to determine mutant frequency while the previous studies used synchronized cells. Another possible reason for discrepancies in the induced mutant frequency in pol eta-deficient human cells between these two studies may be due to differences in the cell lines used as they were derived from different XP variant patients. XP4BE cells used by Watanabe et al [20] express a truncated version of pol eta that is 27 amino acids long due to a deletion of four nucleotides in the gene, while the XP115LO cells used in this study express a 127 amino acid truncated form as a result of a nonsense mutation. However, neither enzyme has been shown to have measurable enzyme activity. Data presented here from mouse cells support the low mutant frequency seen in human XP variant cells in a second species. In addition, pol eta was disrupted in these mice by removal of exon 4 and the generation of a frameshift mutation 30 bp downstream. The resultant mutant transcript would therefore encode a 92 amino acid truncated form of pol eta. This deletion is similar to that found in XP4BE cells and supports our hypothesis that loss of functional pol eta reduces BPDE-induced mutagenesis.
The observation of a reduced mutant frequency in pol eta-null mouse fibroblasts and in human fibroblasts deficient for pol eta relative to their pol eta wild-type counterparts in response to BPDE exposure indicates that this enzyme is involved in error-prone bypass of BPDE-adducts, in contrast to its error-free role in the bypass of UV-induced lesions. Thus, while it would be catastrophic to target pol eta in the body as a whole as an anti-cancer strategy, increasing the risk of UV-induced mutations and cancers, it may be possible to selectively inhibit this enzyme in tissues other than the skin that accumulate bulky lesions like those induced by BPDE. It was recently shown that ribozyme-mediated knockdown of the Y-family polymerase REV1 in the lung via aerosol delivery reduced the multiplicity of lung tumors in B[a]P-treated mice [29]. Lung-directed targeting of pol eta may be useful for those with increased exposure to environmental BPDE, as is the case for smokers. Determining the role of pol eta in the bypass of BPDE-adducts will further our knowledge of environmental carcinogen-linked cancers with the intent to determine possible prevention strategies.
Materials and Methods
Ethics Statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee at the University of Louisville protocol number 09059. All efforts were made to minimize animal suffering.
Cells and Cell Culture
Pol eta knockout mice were a gift from the Kucherlapati laboratory [30]. These mice were backcrossed with C57BL/6 mice and were congenic in the C57BL/6 genetic background. Mice were tested for mutations in pol eta and pol iota using PCR-based protocols [31], [32]. All mice used were proficient for pol iota. Primary murine fibroblasts were established from 8- to 9- week-old mice using standard techniques as published [33]. Briefly, using an autoclaved ear punch, small pieces of ear from anesthetized mice were removed and plated in MEM-a (Life Technologies) supplemented with 10% fetal calf serum (Hyclone), 2mmol/L glutamine, nonessential amino acids (Life Technologies), penicillin (100 units/mL), streptomycin (100 ug/mL), and fungizone (Life Technologies; 1 ug/mL). Fibroblasts were grown at 37°C under hypoxic conditions (2–3% O2, 5% CO2) previously reported to increase the number of population doublings and lengthen the time before senescence of primary murine cells [34]. The medium was changed within 48 h, and the fungizone was omitted.
NF1604 human lung embryonic fibroblasts human fibroblasts were a gift from Dr. Lisa McDaniels [35] at the University of Texas Southwestern Medical Branch under the terms of an MTA between Greon Corporation and WGM. XP115LO fibroblasts were a gift from Dr. Veronica Maher [36]. Human fibroblasts were grown in DMEM (Gibco) supplemented with 10% supplemented calf serum (Atlanta Biologicals), penicillin (100 units/mL), and streptomycin (100ug/mL) (Sigma) at 37°C in 5% CO2.
Exposure to BPDE
BPDE was purchased from the National Cancer Institute Chemical Carcinogen Repository. Powdered BPDE was dissolved in anhydrous tetrahydrofuran (THF) (Sigma). For cytotoxic and mutagenic studies the culture medium was aspirated and the cells were washed with sterile PBS (pH 7.4) and replaced with serum-free medium. BPDE was added to the culture medium to a final concentration of 150nM. The control cells were exposed to solvent (THF) only. After 1 hour incubation at 37°C, the cells were washed twice with PBS and the medium was replaced with complete medium.
Cytotoxic and Mutagenic Assays
Primary mouse fibroblasts were assayed for cytotoxic and mutagenic responses to BPDE at early passages (six to eight population doublings) after primary cultures were established. A series of independent populations (1.5×106 cells each, plated on 150-mm-diameter plates) were allowed to attach overnight before exposure to BPDE. Cytotoxic responses to BPDE were established by exposing the cells at the same density as used for the mutagenesis experiments, namely 1×104 cells/cm2. After exposure, the cells were plated at cloning density to measure colony-forming ability. Fresh media was added 7 days after treatment and cells were stained with 1% crystal violet (Fisher Scientific) at 14 days. For mutagenesis, cells were allowed 3–5 days of growth before trypsinization and passage of 1.5×106cells. After 8–9 days of post-treatment growth, cells were selected in 20uM 6-thioguanine (TG) at a density of 450 cells/cm2. Colony-forming ability was also determined at the time of TG selection by plating the cells at cloning density in nonselective medium and was used to correct for the mutant frequency. After 14–20 days, TG resistant clones were isolated for Hprt coding region amplification. After colony isolation, plates were stained, and mutant frequency was determined, defined as the number of observed TG clones per 106 clonable cells (corrected for cloning efficiency). Data were analyzed using a student's t-test assuming unequal variance.
Amplification and Sequencing of Hprt cDNA
Isolation of thioguanine clones, reverse transcriptase-PCR of the Hprt cDNA, and sequence analysis of the PCR products were performed as previously described [10], [33].
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was partially supported by USPHS Grant T32-ES011564, by a predoctoral fellowship to LJS, F30-ES017730 to ACK, and by R01-CA112197 and R03-CA112664 to WGM. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Cosman M, de los Santos C, Fiala R, Hingerty BE, Singh SB, et al. Solution conformation of the major adduct between the carcinogen (+)-anti-benzo[a]pyrene diol epoxide and DNA. Proc Natl Acad Sci U S A. 1992;89:1914–1918. doi: 10.1073/pnas.89.5.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang LL, Maher VM, McCormick JJ. Error-free excision of the cytotoxic, mutagenic N2-deoxyguanosine DNA adduct formed in human fibroblasts by (+/−)-7 beta, 8 alpha-dihydroxy-9 alpha, 10 alpha-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene. Proc Natl Acad Sci U S A. 1980;77:5933–5937. doi: 10.1073/pnas.77.10.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dreij K, Seidel A, Jernstrom B. Differential removal of DNA adducts derived from anti-diol epoxides of dibenzo[a, l]pyrene and benzo[a]pyrene in human cells. Chem Res Toxicol. 2005;18:655–664. doi: 10.1021/tx0497090. [DOI] [PubMed] [Google Scholar]
- 4.Ohmori H, Friedberg EC, Fuchs RP, Goodman MF, Hanaoka F, et al. The Y-family of DNA polymerases. Mol Cell. 2001;8:7–8. doi: 10.1016/s1097-2765(01)00278-7. [DOI] [PubMed] [Google Scholar]
- 5.Johnson RE, Kondratick CM, Prakash S, Prakash L. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science. 1999;285:263–265. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]
- 6.Broughton BC, Cordonnier A, Kleijer WJ, Jaspers NG, Fawcett H, et al. Molecular analysis of mutations in DNA polymerase eta in xeroderma pigmentosum-variant patients. Proc Natl Acad Sci U S A. 2002;99:815–820. doi: 10.1073/pnas.022473899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tung BS, McGregor WG, Wang YC, Maher VM, McCormick JJ. Comparison of the rate of excision of major UV photoproducts in the strands of the human HPRT gene of normal and xeroderma pigmentosum variant cells. Mutat Res. 1996;362:65–74. doi: 10.1016/0921-8777(95)00034-8. [DOI] [PubMed] [Google Scholar]
- 8.Maher VM, Ouellette LM, Curren RD, McCormick JJ. Frequency of ultraviolet light-induced mutations is higher in xeroderma pigmentosum variant cells than in normal human cells. Nature. 1976;261:593–595. doi: 10.1038/261593a0. [DOI] [PubMed] [Google Scholar]
- 9.Washington MT, Johnson RE, Prakash L, Prakash S. Accuracy of lesion bypass by yeast and human DNA polymerase eta. Proc Natl Acad Sci U S A. 2001;98:8355–8360. doi: 10.1073/pnas.121007298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dumstorf CA, Clark AB, Lin Q, Kissling GE, Yuan T, et al. Participation of mouse DNA polymerase iota in strand-biased mutagenic bypass of UV photoproducts and suppression of skin cancer. Proc Natl Acad Sci U S A. 2006;103:18083–18088. doi: 10.1073/pnas.0605247103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gueranger Q, Stary A, Aoufouchi S, Faili A, Sarasin A, et al. Role of DNA polymerases eta, iota and zeta in UV resistance and UV-induced mutagenesis in a human cell line. DNA Repair (Amst) 2008;7:1551–1562. doi: 10.1016/j.dnarep.2008.05.012. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Woodgate R, McManus TP, Mead S, McCormick JJ, et al. Evidence that in xeroderma pigmentosum variant cells, which lack DNA polymerase eta, DNA polymerase iota causes the very high frequency and unique spectrum of UV-induced mutations. Cancer Res. 2007;67:3018–3026. doi: 10.1158/0008-5472.CAN-06-3073. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Wu X, Guo D, Rechkoblit O, Wang Z. Activities of human DNA polymerase kappa in response to the major benzo[a]pyrene DNA adduct: error-free lesion bypass and extension synthesis from opposite the lesion. DNA Repair (Amst) 2002;1:559–569. doi: 10.1016/s1568-7864(02)00055-1. [DOI] [PubMed] [Google Scholar]
- 14.Ogi T, Mimura J, Hikida M, Fujimoto H, Fujii-Kuriyama Y, et al. Expression of human and mouse genes encoding polkappa: testis-specific developmental regulation and AhR-dependent inducible transcription. Genes Cells. 2001;6:943–953. doi: 10.1046/j.1365-2443.2001.00478.x. [DOI] [PubMed] [Google Scholar]
- 15.Avkin S, Goldsmith M, Velasco-Miguel S, Geacintov N, Friedberg EC, et al. Quantitative analysis of translesion DNA synthesis across a benzo[a]pyrene-guanine adduct in mammalian cells: the role of DNA polymerase kappa. J Biol Chem. 2004;279:53298–53305. doi: 10.1074/jbc.M409155200. [DOI] [PubMed] [Google Scholar]
- 16.Ogi T, Shinkai Y, Tanaka K, Ohmori H. Polkappa protects mammalian cells against the lethal and mutagenic effects of benzo[a]pyrene. Proc Natl Acad Sci U S A. 2002;99:15548–15553. doi: 10.1073/pnas.222377899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rechkoblit O, Zhang Y, Guo D, Wang Z, Amin S, et al. trans-Lesion synthesis past bulky benzo[a]pyrene diol epoxide N2-dG and N6-dA lesions catalyzed by DNA bypass polymerases. J Biol Chem. 2002;277:30488–30494. doi: 10.1074/jbc.M201167200. [DOI] [PubMed] [Google Scholar]
- 18.Chiapperino D, Kroth H, Kramarczuk IH, Sayer JM, Masutani C, et al. Preferential misincorporation of purine nucleotides by human DNA polymerase eta opposite benzo[a]pyrene 7,8-diol 9,10-epoxide deoxyguanosine adducts. J Biol Chem. 2002;277:11765–11771. doi: 10.1074/jbc.M112139200. [DOI] [PubMed] [Google Scholar]
- 19.Shachar S, Ziv O, Avkin S, Adar S, Wittschieben J, et al. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009;28:383–393. doi: 10.1038/emboj.2008.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watanabe M, Maher VM, McCormick JJ. Excision repair of UV- or benzo[a]pyrene diol epoxide-induced lesions in xeroderma pigmentosum variant cells is ‘error free’. Mutat Res. 1985;146:285–294. doi: 10.1016/0167-8817(85)90070-7. [DOI] [PubMed] [Google Scholar]
- 21.Simhadri S, Kramata P, Zajc B, Sayer JM, Jerina DM, et al. Benzo[a]pyrene diol epoxide-deoxyguanosine adducts are accurately bypassed by yeast DNA polymerase zeta in vitro. Mutat Res. 2002;508:137–145. doi: 10.1016/s0027-5107(02)00211-7. [DOI] [PubMed] [Google Scholar]
- 22.Xie Z, Braithwaite E, Guo D, Zhao B, Geacintov NE, et al. Mutagenesis of benzo[a]pyrene diol epoxide in yeast: requirement for DNA polymerase zeta and involvement of DNA polymerase eta. Biochemistry. 2003;42:11253–11262. doi: 10.1021/bi0346704. [DOI] [PubMed] [Google Scholar]
- 23.Zhao B, Wang J, Geacintov NE, Wang Z. Poleta, Polzeta and Rev1 together are required for G to T transversion mutations induced by the (+)- and (−)-trans-anti-BPDE-N2-dG DNA adducts in yeast cells. Nucleic Acids Res. 2006;34:417–425. doi: 10.1093/nar/gkj446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neal JA, Fletcher KL, McCormick JJ, Maher VM. The role of hRev7, the accessory subunit of hPolzeta, in translesion synthesis past DNA damage induced by benzo[a]pyrene diol epoxide (BPDE). BMC Cell Biol. 2010;11:97. doi: 10.1186/1471-2121-11-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hashimoto K, Cho Y, Yang IY, Akagi J, Ohashi E, et al. The Vital Role of Polymerase ζ and REV1 in Mutagenic, but Not Correct, DNA Synthesis across Benzo[a]pyrene-dG and Recruitment of Polymerase ζ by REV1 to Replication-stalled Site. J Biol Chem. 2012;287:9613–9622. doi: 10.1074/jbc.M111.331728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crow JF. Spontaneous mutation as a risk factor. Exp Clin Immunogenet. 1995;12:121–128. doi: 10.1159/000424865. [DOI] [PubMed] [Google Scholar]
- 27.Chiapperino D, Cai M, Sayer JM, Yagi H, Kroth H, et al. Error-prone translesion synthesis by human DNA polymerase eta on DNA-containing deoxyadenosine adducts of 7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene. J Biol Chem. 2005;280:39684–39692. doi: 10.1074/jbc.M508008200. [DOI] [PubMed] [Google Scholar]
- 28.Yang LL, Maher VM, McCormick JJ. Relationship between excision repair and the cytotoxic and mutagenic effect of the ‘anti’ 7,8-diol-9,10-epoxide of benzo[a]pyrene in human cells. Mutat Res. 1982;94:435–447. doi: 10.1016/0027-5107(82)90306-2. [DOI] [PubMed] [Google Scholar]
- 29.Dumstorf CA, Mukhopadhyay S, Krishnan E, Haribabu B, McGregor WG. REV1 is implicated in the development of carcinogen-induced lung cancer. Mol Cancer Res. 2009;7:247–254. doi: 10.1158/1541-7786.MCR-08-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin Q, Clark AB, McCulloch SD, Yuan T, Bronson RT, et al. Increased susceptibility to UV-induced skin carcinogenesis in polymerase eta-deficient mice. Cancer Res. 2006;66:87–94. doi: 10.1158/0008-5472.CAN-05-1862. [DOI] [PubMed] [Google Scholar]
- 31.Lin Q, Clark AB, McCulloch SD, Yuan T, Bronson RT, et al. Increased Susceptibility to UV-Induced Skin Carcinogenesis in Polymerase {eta}-deficient Mice. Cancer Research. 2006;66:87–94. doi: 10.1158/0008-5472.CAN-05-1862. [DOI] [PubMed] [Google Scholar]
- 32.McDonald JP, Frank EG, Plosky BS, Rogozin IB, Masutani C, et al. 129-derived strains of mice are deficient in DNA polymerase iota and have normal immunoglobulin hypermutation. The Journal of Experimental Medicine. 2003;198:635–643. doi: 10.1084/jem.20030767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diaz M, Watson NB, Turkington G, Verkoczy LK, Klinman NR, et al. Decreased frequency and highly aberrant spectrum of ultraviolet-induced mutations in the hprt gene of mouse fibroblasts expressing antisense RNA to DNA polymerase zeta. Mol Cancer Res. 2003;1:836–847. [PubMed] [Google Scholar]
- 34.Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, et al. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5:741–747. doi: 10.1038/ncb1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ouellette MM, McDaniel LD, Wright WE, Shay JW, Schultz RA. The establishment of telomerase-immortalized cell lines representing human chromosome instability syndromes. Human molecular genetics. 2000;9:403–411. doi: 10.1093/hmg/9.3.403. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y, Woodgate R, McManus TP, Mead S, McCormick JJ, et al. Evidence that in Xeroderma Pigmentosum Variant Cells, which Lack DNA Polymerase {eta}, DNA Polymerase {iota} Causes the Very High Frequency and Unique Spectrum of UV-Induced Mutations. Cancer Research. 2007;67:3018–3026. doi: 10.1158/0008-5472.CAN-06-3073. [DOI] [PubMed] [Google Scholar]