

Abstract
We present a comprehensive electronic structure analysis of structurally simple BN heterocycles using a combined UV-photoelectron spectroscopy (UV-PES) / computational chemistry approach. Gas-phase He I photoelectron spectra of 1,2-dihydro-1,2-azaborine 1, N-Me-1,2-BN-toluene 2, and N-Me-1,3-BN-toluene 3 have been recorded, assessed by density functional theory calculations, and compared with their corresponding carbonaceous analogues benzene and toluene. The first ionization energies of these BN heterocycles are in the order N-Me-1,3-BN-toluene 3 (8.0 eV) < N-Me-1,2-BN-toluene 2 (8.45 eV) < 1,2-dihydro-1,2-azaborine 1 (8.6 eV) < toluene (8.83 eV) < benzene (9.25 eV). The computationally determined molecular dipole moments are in the order 3 (4.577 Debye) > 2 (2.209 Debye) > 1 (2.154 Debye) > toluene (0.349 Debye) > benzene (0 Debye) and are consistent with experimental observations. The λmax in the UV-Vis absorption spectra are in the order 3 (297 nm) > 2 (278 nm) > 1 (269 nm) > toluene (262 nm) > benzene (255 nm). We also establish that the measured anodic peak potentials and electrophilic aromatic substitution (EAS) reactivity of BN heterocycles 1–3 are consistent with the electronic structure description determined by the combined UV-PES/computational chemistry approach.
1. Introduction
Boron(B)-nitrogen(N)-containing heteroaromatic compounds (BN arenes) have recently emerged as a new structural motif relevant to biomedical research and materials science.1 They are isoelectronic and isostructural to the versatile family of arenes. Thus, their development (i.e., 1,2-, 1,3-, and 1,4-azaborines; Scheme 1) significantly expands the structural diversity and potential utility of aromatic compounds. Dewar’s early work on ring-fused polycyclic and monocyclic 1,2-BN heterocycles pioneered the field in the late 1950s and early 1960s.2,3 Since the turn of the millennium, contributions by the groups of Ashe, Piers, Kawashima, Yamaguchi, Perepichka, as well as our group have further enriched the chemistry of these compounds. Ashe developed new synthetic methods for the preparation of monocyclic 1,2-azaborines4 and demonstrated that 1,2-azaborines readily undergo electrophilic aromatic substitution reactions.5 Piers introduced new BN internalized ring-fused polycyclic compounds and investigated their optoelectronic properties.6 Kawashima made extended BN-acenes involving a 1,4-azaborine.7 More recently, Yamaguchi,8 Perepichka,9 and Nakamura10 synthesized new 1,2-azaborine structures for potential materials science applications.
Scheme 1.

BN isosteres of benzene.
Our group developed a general method for B-substituted 1,2-azaborines,11 which ultimately led to the isolation of its parent compound 1 (Scheme 1).12 We have also provided experimental and computational evidence describing the aromatic character of 1,2-azaborines.12,13 Very recently, we established the first synthesis of a 1,3-azaborine derivative A (Scheme 1).14
To improve our understanding of the electronic structure of the heterocyclic core of aromatic BN heterocycles, we have been focusing on the investigation of monocyclic BN heterocycles that minimize the influence of substituent effects. The experimental access to these simple structures allows a direct comparison between BN arenes and their classic organic counterparts (e.g., benzene and toluene). In this work, we provide a comprehensive electronic structure analysis of BN heterocycles 1, 2, and 3 shown in Scheme 215 in direct comparison with their carbonaceous derivatives using a combined UV-photoelectron spectroscopy (UV-PES)/computational chemistry approach.
Scheme 2.

The UV-PES technique allows the determination of accurate values of ionization energies for electronic structure characterization of molecules and ions. Since the pioneering work by Turner and Baker16 in the early 1960’s, UV-PES has been developed into a well-established method that provides ionization band patterns as “molecular fingerprints” of occupied molecular orbitals of organic and organometallic compounds in the gas phase.17 Simply put, UV-PES is the experimental technique for determining the energies of occupied molecular orbitals. To the best of our knowledge, the only examples of UV-PES analysis of heteroaromatic carbon-boron-nitrogen-containing organic molecules are those of diazaborolidines, diazaboroles, and benzodiazaboroles.18
For a reliable assignment of UV photoelectron spectroscopic bands and for the interpretation of spectra, a combined UV-PES / theoretical approach is necessary. The Chrostowska group has calibrated different computational methods (e.g., OVGF (the standard outer valence green function), DFT (density functional theory), ΔSCF / TD-DFT (self-consistent field / time-dependent density functional theory (TD-DFT), CASPT2 (complete active space 2nd order perturbation theory), and SAOP XC (statistical average of different orbital model potential exchange correlation functional)) against the experimentally determined UV-PES ionization energies (IE).19 The combined UV-PES / computational modeling approach developed by Chrostowska and coworkers are used to investigate the electronic structure of the compounds illustrated in Scheme 2.
2. Experimental and Computational Methods
Coupled UV-Photoelectron Spectroscopy – Mass Spectrometry Measurements
The UV-PES spectra were recorded on a home-built (IPREM/ECP), three-part spectrometer equipped with a main body device, He-I radiation source (21.21 eV and/or 48 eV) and a 127° cylindrical analyzer. The spectrometer works at constant analyzer energy under 5×10−6 Torr working pressure and ≤10−7 Torr for channeltron (X914L) pressure. The monitoring is done by a microcomputer supplemented by a digital–analogue converter (AEI spectrum). The spectra resulting from a single scan are built from 2048 points and are accurate within 0.05 eV. Spectra are calibrated with lines of xenon (12.13 and 13.44 eV) and of argon (15.76 and 15.94 eV). The accuracy of the ionization potentials is ± 0.03 eV for sharp peaks and ± 0.05 eV for broad and overlapping signals. Mass spectra were recorded on a modified quadrupole mass spectrometer (PFEIFFER Prisma QMS200) with an electron-impact at 50 eV (mass range: 200 amu; detection limit: ≤ 10−14 Torr; working pressure: 2×10−7 Torr; operating temperature: 200 °C; electronic amplifier in working conditions: 10−10 A, QUAD STAR422 software for recording and treatment of MS data). The samples were slowly vaporized under low pressure (10−6 Torr) inside a handmade three-valve injector (3/4 inch diameter; 10 cm length; working temperature: −190 °C ≤ T ≤ +300 °C), and the gaseous flow was then continuously and simultaneously analyzed by both UV-photoelectron and mass spectrometers.
Computational Methods
All calculations were performed using the Gaussian 0920 program package with the 6-311G(d,p) basis set. DFT has been shown to predict various molecular properties of similar compounds successfully.21 All geometry optimizations were carried out with the CAM-B3LYP22 functionals and were followed by frequency calculations in order to verify that the stationary points obtained were true energy minima. Ionization energies (IE) were calculated with ΔSCF-DFT, which means that separate SCF calculations were performed to optimize the orbitals of the ground state and the appropriate ionic state (IE = Ecation − Eneutral). The advantages of the most frequently employed ΔSCF-DFT method of calculations of the first ionization energies have been demonstrated previously.23 The TD-DFT19,24 approach provides a first-principal method for the calculation of excitation energies within a density functional context taking into account the low-lying ion calculated by the ΔSCF method. The vertical ionization energies were also calculated at the ab initio level according to OVGF25 (in this case the effects of electron correlation and reorganization are included beyond the Hartree-Fock approximation and the self-energy part was expanded up to third order) and SAC-CI26 (Symmetry Adapted Cluster / Configuration Interaction methods of Nakatsuji and coworkers which describes accurately and efficiently the electronic structures of the excited, ionized and electron-attached states of molecules) methods. Finally, the so-called “corrected” IEs19 were evaluated applying a uniform shift, x = −IEvexp − εKSHOMO, where εKSHOMO is the CAM-B3LYP/6-311G(d,p)) Kohn-Sham energy of the highest occupied MO of the molecule in the ground state, and IEvexp is the lowest experimental ionization energy of this species, as was suggested previously by Stowasser and Hoffmann27 and in our studies on different methods for the calculation of IEs.17 MOLEKEL28 was used as a visualization tool for all MOs.
3. Results and Discussion
Synthesis
We recently disclosed the first synthetic example of a 1,3-azaborine, i.e., compound A (Scheme 1).14 For electronic structure characterization focusing on the aromatic heterocyclic core, compound A is a relatively complex di-substituted molecule. To reduce the complexity of heterocycle A, we thought to convert the NiPr2 group on boron into a simple hydrogen substituent. Gratifyingly, treatment of A with acetic acid followed by addition of LiAlH4 afforded the desired BN toluene 3 (Scheme 3, top sequence). Compound 3 is the most simple derivative of the 1,3-azaborine family reported to date. In order to provide a direct electronic structure comparison with 3, which we could not further simplify synthetically,29 we prepared the corresponding N-Me-1,2-BN-toluene 2 (Scheme 3, bottom sequence).11 Condensation of the in situ-generated allylboron dichloride with methylallyl amine produced diene 4 in 66% yield. Ring closing metathesis of compound 4 catalyzed by Grubbs 1st generation catalyst yielded the ring-closed BN heterocycle 5. Dehydrogenation of 5 with catalytic amounts of Pd/C followed by treatment with LiAlH4 ultimately furnished the desired N-Me-1,2-BN-toluene 2.
Scheme 3.

Synthesis of N-Me-1,3-BN-Toluene 3 and N-Me-1,2-BN-Toluene 2.
UV-PES Analysis
Having BN arenes 1–3 and the commercially available benzene and toluene in our hands (see Scheme 2), we subjected these five arenes to UV-PES conditions with simultaneous monitoring via mass spectrometry. The concomitant use of mass spectrometry ensures the correct identity and purity of the gas-phase molecule being analyzed. The UV-PE spectra for benzene and toluene have been reported about half a decade ago.30 For sake of consistency and to allow a direct comparison between the compounds under current investigation, we have recollected the UV-PES data for benzene and toluene at a higher resolution. For the reliable assignment of PE bands, density functional theory [ΔSCF/TD-DFT (CAM-B3LYP)] and ab initio (OVGF and SAC-CI) calculations of ionization energies using the 6-311G(d,p) basis set have been carried out on optimized geometrical parameters of benzene, toluene, and BN arenes 1–3. In addition, the experimental PE data were compared to “corrected” IEs derived from a “shifting” of calculated Kohn–Sham energies by an experimentally determined correction value. The comparison of the theoretically predicted IEs and experimental data are summarized in Tables S1–S5 for benzene, 1,2-dihydro-1,2-azaborine 1, toluene, N-Me-1,2-BN-toluene 2, and N-Me-1,3-BN-toluene 3, respectively.31 The chosen computational models agree well with the experimentally determined IEs.
Figure 1a–e illustrates the UV-PE spectra and the highest occupied molecular orbitals corresponding to the experimentally determined IEs of each of the five molecules. As can be seen from Figure 1a, the photoelectron spectrum of benzene exhibits a low-energy band at 9.25 eV, which corresponds to its degenerate HOMOs of π symmetry (e1g, π1 and π2). This is followed by a band at 11.53 eV, which corresponds to a set of degenerate orbitals of σ symmetry (e2g). The symmetrical fifth MO of π symmetry (a2u, π3) is attributed to the PE band at 12.38 eV. The first two bands of the UV-PES of benzene are relatively broad and exhibit vibrational structure. This is due to the Jahn-Teller splitting that lifts the degeneracy of these IE levels.32
Figure 1.

UV-PE spectra of (a) benzene and (b) 1,2-dihydro-1,2-azaborine 1, (c) toluene, (d) N-Me-1,2-BN-toluene 2, and (e) N-Me-1,3-BN-toluene 3. The y-axis is defined as the negative value of the experimentally determined ionization energy (-IE). The top seven occupied molecular orbitals associated with the corresponding energy levels for each of the molecules are also depicted
For 1,2-dihydro-1,2-azaborine 1, the first PE band at 8.6 eV is attributed to the ejection of an electron from the HOMO (Figure 1b). The nature of this MO can be described as A” symmetry (πCBN - πC=C) and is comparable with the π1 HOMO of benzene. The second IE located at 10.3 eV (HOMO-1) is attributed to the MO featuring the nitrogen lone pair and π-bonding interactions between the C3-C4-C5 atoms, also of A” symmetry (πC=C - nNπ). This orbital correlates with the π2 of benzene. The third and fourth PE bands at 11.1 and 12.0 eV, respectively, reflect the ionizations of different σ-symmetry orbitals. The fifth ionization band of the 1,2-dihydro-1,2-azaborine spectrum at 12.7 eV is due to a MO corresponding to the π bonding interaction between all carbons in benzene (π3).
As can be seen from the comparison between Figure 1a and Figure 1b, the replacement of two adjacent carbon atoms in benzene by nitrogen and boron as in 1,2-dihydro-1,2-azaborine 1 causes a significant change in the electronic structure. In heterocycle 1, the degeneracy found in benzene is lifted and results in a 0.65 eV destabilization of the HOMO and 1.05 eV stabilization of the HOMO-1 (vs. benzene’s degenerate HOMO level at −9.25 eV). This lifting of degeneracy is also observed for HOMO-2 and HOMO-3 of 1,2-azaborine 1, which leads to a 0.43 eV destabilization and a 0.47 eV stabilization vs. benzene’s degenerate σ MOs at −11.53 eV, respectively. The fifth MO in 1 is stabilized by 0.32 eV compared to the corresponding π3 MO of the benzene molecule.
A comparison of Figure 1c (toluene) vs. Figure 1a (benzene) shows that the presence of a methyl substituent on the six-membered ring leads to a more electron rich system than the parent benzene molecule. A lifting of degeneracy of orbitals is also observed. The HOMO is found at −8.83 eV, 0.42 eV higher than the corresponding HOMO π2 in benzene at −9.25 eV, whereas the HOMO-1 (corresponding to π1 in benzene) is stabilized by 0.11 eV. The toluene MO associated with the totally symmetrical π3 MO of benzene is also destabilized by 0.4 eV.
Figure 1c–e illustrates the changes in the electronic structure associated with the two different variants of BN/CC isosterism of toluene, i.e., 1,2- and the 1,3-isosteres. In N-Me-1,2-BN-toluene 2, the HOMO is destabilized by 0.38 eV compared to toluene. In N-Me-1,3-BN-toluene 3, the destabilization of HOMO is even more pronounced, with the HOMO energy level difference between 3 and toluene being 0.83 eV. On the other hand, a stabilization of the HOMO-1 and π3-type π-symmetry orbitals is observed for the BN toluenes. Relative to toluene, the HOMO-1 is stabilized by 0.49 eV and 0.39 eV for 2 and 3, respectively. Similarly, the π3-type orbitals for BN toluenes 2 and 3 are lowered in energy compared to toluene by 0.52 and 0.72 eV, respectively. Interestingly, the HOMO is of π1-type symmetry for 1,2-azaborines (see Figure 1b and Figure 1d) whereas it is of π2-type symmetry for toluene and 1,3-azaborines (see Figure 1c and Figure 1e).
Additional Computational Results
We have also computationally investigated the ground-state dipole moments of the five arenes. As can be seen from Table 1, CAM-B3LYP/6-311G(d,p) calculations predict the following trend in molecular dipole moments in the order of increasing strength: benzene (0 Debye), toluene (0.349 Debye), 1,2-dihydro-1,2-azaborine 1 (2.154 Debye), N-Me-1,2-BN-toluene 2 (2.209 Debye), and N-Me-1,3-BN-toluene 3 (4.577 Debye). Thus, BN/CC isosterism leads to molecules with stronger molecular dipole moments.33 Noteworthy is also the prediction that the 1,3-BN toluene isostere is significantly more polar than the 1,2-BN toluene isostere (4.577 Debye vs. 2.209 Debye, respectively). Furthermore, TD-DFT calculations suggest that the direction and magnitude of the dipole moments do not significantly change upon excitation of the ground state to the first excited state for benzene, toluene, and N-Me-1,3-BN-toluene 3. On the other hand, an increase of ~1 Debye is predicted for 1,2-azaborine structures 1 and 2 upon excitation to the first excited state.
Table 1.
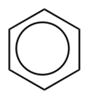
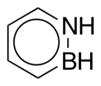
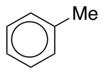
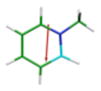
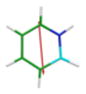
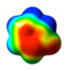
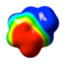
CAM-B3LYP/6-311G(d,p) Ground and First Excited State Dipole Moments (Debye) and MP2/6-31+G(d,p) Electrostatic Potential Surface Map at the 0.001 electron a.u.−3 density iso-contour level (from +12.55 to −12.55 kcal/mol), ΔSCF Ionization Energy (IE), HOMO, LUMO energies and HOMO-LUMO gap, Electron Affinity (EA, calculated with 6-311++G(d,p)), Δ(EA+IE), first HOMO → LUMO UV transition (calculated with 6-311++G(d,p)) for benzene, 1,2-dihydro-1,2-azaborine 1, toluene, N-Me-1,2-BN-toluene 2, and N-Me-1,3-BN-toluene 3
|
|
|

|

|
|
|---|---|---|---|---|---|
| ground state dipole moment (Debye) | 0.0 |
 2.154 |
 0.349 |
 2.209 |
 4.577 |
| first excited state dipole moment (Debye) | 0.0 |
 3.106 |
 0.423 |
 2.897 |
 4.251 |
| electrostatic potential surface map |

|
|

|

|
|
| HOMO (eV) | −8.444 | −7.678 | −8.092 | −7.581 | −7.175 |
| LUMO (eV) | 1.096 | 0.561 | 1.130 | 0.511 | 0.180 |
| HOMO-LUMO gap (eV) | 9.540 | 8.239 | 9.222 | 8.093 | 7.355 |
| ΔSCF/TD-DFT (ionization energy IE) (eV) | 9.298 | 8.514 | 8.851 | 8.336 | 7.899 |
| electron affinity (EA) (eV) | −0.792 | −0.964 | −0.750 | −0.801 | −0.480 |
| Δ(IE+EA) (eV) | 8.506 | 7.550 | 8.101 | 7.535 | 7.419 |
| first HOMO → LUMO transition (eV) | 5.54 | 5.14 | 5.43 | 5.00 | 4.56 |
The direction and magnitude of the dipole moments are consistent with the calculated electrostatic potential surface maps for the five arenes at the 0.001 electron a.u.−3 density iso-contour level. The color red indicates negative charge whereas the color blue represents positive charge. The symmetrical nature of the charge distribution in benzene and toluene is consistent with resulting low dipole moments. For the 1,2-BN isostere, the negative charge is more localized at the C(3), C(5), and at the B–H hydride positions while the positive charge is located at the N-substituent. For the 1,3-BN isostere, the negative charge is concentrated at the vicinity of the boron atom whereas the positive charge is again localized at the N-substituent. The concentration of negative charge at the boron atom in the 1,3-BN system is somewhat surprising as boron is the least electronegative element in the molecule. This may suggest that in addition of simple inductive arguments, strong resonance / π electron delocalization effects are at play for the 1,3-BN system that provides the boron atom with extra negative charge. As can be seen from Figure 2, both limiting resonance structures for N-Me-1,2-BN-toluene 3 have a formal negative charge on the boron atom and a formal positive charge at the nitrogen atom.
Figure 2.
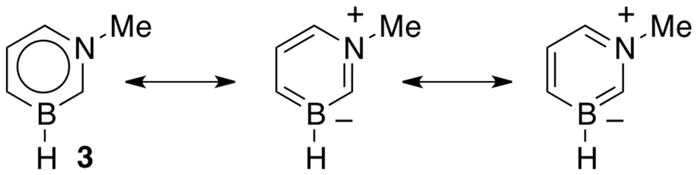
Limiting resonance structures for 3 highlighting the localization of formal charge.
Noteworthy is also the predicted electron affinity of the different arenes. A comparison of the electron affinities of benzene, 1, toluene, N-Me-1,2-BN-toluene 2, and N-Me-1,3-BN-toluene 3 indicates that the 1,2-azaborine system exhibits the most negative electron affinity followed by the carbonaceous arenes and then the 1,3-azaborine system.
Correlation of Experimental Characterization with UV-PES/Computational Electronic Structure Data
1) Thin-Layer Chromatography (TLC)
In order to evaluate the relative polarity of toluene, N-Me-1,2-BN-toluene 2, N-Me-1,3-BN-toluene 3, we have conducted simple TLC experiments using silica gel as the stationary phase and the non-polar pentane as the eluent. The resulting Rf values for toluene, 2, and 3 are >0.9, 0.7–0.8, and < 0.1, respectively. This experimental observation is consistent with the computationally determined molecular dipole moments for toluene (0.349 Debye), 2 (2.209 Debye), and 3 (4.577 Debye).
2) UV-Vis Absorption Spectra
The experimental UV-Vis absorption spectra and the calculated absorption maxima of benzene, 1,2-dihydro-1,2-azaborine 1, toluene, N-Me-1,2-BN-toluene 2, and N-Me-1,3-BN-toluene 3 are displayed in Figure 3. The λmax for 1 is calculated to be at 241 nm and corresponds to the energy of the HOMO → LUMO transition. This band is observed in the UV/Vis spectrum at λmax= 269 nm (Figure 3, entry 2). In the case of heterocycle 2, the λmax is calculated to be at 248 nm and is experimentally observed at λmax = 278 nm (Figure 3, entry 4). For compound 3, this lowest-energy absorption band is calculated to be at 272 nm and is observed at λmax = 297 nm (Figure 3, entry 5). In comparison to the theoretically derived gas-phase values, the experimentally observed λmax values are bathochromically shifted by ~30 nm, showing some limitations of the TD-DFT method in predicting absorbance spectra.
Figure 3.

Comparison of TD-DFT calculations (CAM-B3LYP/6-311++G(d,p)) and observed UV-Vis absorption spectra for benzene, 1,2-dihydro-1,2-azaborine 1, toluene, N-Me-1,2-BN-toluene 2, and N-Me-1,3-BN-toluene 3.
3) Electrochemistry
Cyclic voltametry reveals electronic differences between carbonaceous arenes and BN arenes. As established by UV-PES experiments, BN/CC isosterism of benzene and toluene leads to heterocycles with higher HOMO levels (Figure 1a vs. 1b and Figure 1c vs. 1d and 1e). Figure 4 illustrates all oxidations are irreversible and that 1,2-dihydro-1,2-azaborine 1 has an oxidation wave peaking at ~1.4 V vs. Ag/Ag+. N-Me-1,2-BN-toluene 2 exhibits an oxidation peak at a slightly lower potential at 1.31 V. This is in stark contrast to the cyclic voltammogram for N-Me-1,3-BN-toluene 3, which shows a peak at a much lower potential (0.94 V). The lower oxidation potential measured for the 1,3- vs. the 1,2-BN isostere is consistent with a higher energy HOMO for 3 vs. 2 determined by UV-PES (Figure 1d vs 1e). Under the same conditions, benzene and toluene show anodic peak potentials at significantly higher potentials than their BN isosteres (2.35 V and ~2.1 V, respectively).34
Figure 4.

Cyclicvoltammograms of (a) benzene and (b) 1,2-dihydro-1,2-azaborine 1, (c) toluene, (d) N-Me-1,2-BN-toluene 2, and (e) N-Me-1,3-BN-toluene 3 (0.1M TBABF4 in CH3CN; scan rate, 50 mV/s).
4) Chemical Reactivity
We have previously determined that 1,3-azaborine A undergoes electrophilic aromatic substitution (EAS) reactions with dimethyl(methylene)ammonium chloride preferencially at the 6-position (eq 1).14 The regioselectivity is consistent with the π-orbital coefficient distribution in the HOMO, which indicates a large orbital coefficient at the 6-position in addition to the 2- and the 4-positions (Figure 5(a)). The preference for the EAS reaction of compound A at the 6-position over the 2- or the 4-positions is consistent with simple steric arguments, i.e., the diisopropylamino group blocks the electrophile from attacking the 2- and the 4-positions.
Figure 5.

(a) HOMO of A with corresponding p orbital coefficients. (b) Electrostatic potential surface of A at the 0.001 electron a.u.−3 density iso-contour level (+12.55 to −12.55 kcal mol−1).
4. Conclusion
In summary, we described a comprehensive electronic structure analysis of structurally simple BN heterocycles using a combined UV-photoelectron spectroscopy (UV-PES) / computational chemistry approach. This analysis provided an in-depth look into the electronic structure changes associated with BN/CC isosterism of the classic arenes benzene and toluene. As part of this study we prepared the most simple 1,3-dihydro-1,3-azaborine derivative to date, i.e., N-Me-1,3-BN-toluene 3. UV-PES / computational chemistry revealed the character of the highest occupied molecular orbitals of the compounds investigated. We determined the following trends in 1) HOMO energies (via UV-PES; highest to lowest): N-Me-1,3-BN-toluene 3 (−8.0 eV) > N-Me-1,2-BN-toluene 2 (−8.45 eV) > 1,2-dihydro-1,2-azaborine 1 (−8.6 eV) > toluene (−8.83 eV) > benzene (−9.25 eV); 2) molecular ground state dipole moment (strongest to weakest): 3 (4.577 Debye) > 2 (2.209 Debye) > 1 (2.154 Debye) > toluene (0.349 Debye) > benzene (0 Debye); 3) λmax in the UV-Vis absorption spectrum (longest to shortest wavelength): 3 (297 nm) > 2 (278 nm) > 1 (269 nm) > toluene (262 nm) > benzene (255 nm); anodic peak potential in CV (lowest to highest): 3 (0.94 V) < 2 (1.31 V) < 1 (~1.4 V) < toluene (~ 2.1 V) < benzene (2.35 V). We also established that polarity measurements via TLC and EAS reactivity of BN heterocycles 1–3 are consistent with the electronic structure description determined by the UV-PES / computational chemistry approach. The data provided by this work should provide the foundation for further development of BN arenes for potential applications in biomedical research and materials science.
Supplementary Material
Acknowledgments
Support has been provided by the National Institutes of Health (National Institute of General Medical Sciences, Grant R01-GM094541). Funding for the University of Oregon Chemistry Research and Instrumentation Services has been furnished in part by the NSF (CHE–0923589). AC and AM are grateful to the Communauté de Communes de Lacq (France) for financial support.
Footnotes
Supporting Information. Experimental procedures, spectroscopic data, complete reference 20, and additional computational data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For an overview of azaborine chemistry, see: Bosdet MJD, Piers WE. Can J Chem. 2009;87:8–29.Campbell PG, Marwitz AJV, Liu SY. Angew Chem Int Ed. 2012;51 doi: 10.1002/anie.201200063.
- 2.For an overview of the early work on 1,2-azaborine, see: Fritsch AJ. Chem Heterocycl Compd. 1977;30:381–440.
- 3.For pioneering work by Dewar, see: Dewar MJS, Kubba VP, Pettit R. J Chem Soc. 1958:3073–3076.Dewar MJS, Dietz R. J Chem Soc. 1959:2728–2730.Dewar MJS, Marr PA. J Am Chem Soc. 1962;84:3782.Davies KM, Dewar MJS, Rona P. J Am Chem Soc. 1967;89:6294–6297.
- 4.(a) Ashe AJ, III, Fang X. Org Lett. 2000;2:2089–2091. doi: 10.1021/ol0001113. [DOI] [PubMed] [Google Scholar]; (b) Ashe AJ, III, Fang X, Fang X, Kampf JW. Organometallics. 2001;20:5413–5418. [Google Scholar]; (c) Ashe AJ., III Organometallics. 2009;28:4236–4248. [Google Scholar]
- 5.Pan J, Kampf JW, Ashe AJ., III Org Lett. 2007;9:679–681. doi: 10.1021/ol062968r. [DOI] [PubMed] [Google Scholar]
- 6.For leading references, see: Jaska CA, Emslie DJ, Bosdet MJ, Piers WE, Sorensen TS, Parvez M. J Am Chem Soc. 2006;128:10885–10896. doi: 10.1021/ja063519p.Bosdet MJ, Piers WE, Sorensen TS, Parvez M. Angew Chem Int Ed. 2007;46:4940–4943. doi: 10.1002/anie.200700591.Bosdet MJ, Jaska CA, Piers WE, Sorensen TS, Parvez M. Org Lett. 2007;9:1395–1398. doi: 10.1021/ol070328y.
- 7.For leading references, see: Agou T, Arai H, Kawashima T. Chem Lett. 2010;39:612–613.Agou T, Kojima T, Kobayashi J, Kawashima T. Org Lett. 2009;11:3534–3537. doi: 10.1021/ol901035d.Agou T, Sekine M, Kobayashi J, Kawashima T. Chem Commun. 2009:1894–1896. doi: 10.1039/b818505k.Agou T, Kobayashi J, Kawashima T. Org Lett. 2006;8:2241–2244. doi: 10.1021/ol060539n.
- 8.Taniguchi T, Yamaguchi S. Organometallics. 2010;29:5732–5735. [Google Scholar]
- 9.Lepeltier M, Lukoyanova O, Jacobson A, Jeeva S, Perepichka DF. Chem Commun. 2010;46:7007–7009. doi: 10.1039/c0cc01963a. [DOI] [PubMed] [Google Scholar]
- 10.Hatakeyama T, Hashimoto S, Seki S, Nakamura M. J Am Chem Soc. 2011;133:18614–18617. doi: 10.1021/ja208950c. [DOI] [PubMed] [Google Scholar]
- 11.Marwitz AJV, Abbey ER, Jenkins JT, Zakharov LN, Liu SY. Org Lett. 2007;9:4905–4908. doi: 10.1021/ol702383u. [DOI] [PubMed] [Google Scholar]
- 12.Marwitz AJV, Matus MH, Zakharov LN, Dixon DA, Liu SY. Angew Chem Int Ed. 2009;48:973–977. doi: 10.1002/anie.200805554. [DOI] [PubMed] [Google Scholar]
- 13.(a) Abbey ER, Zakharov LN, Liu SY. J Am Chem Soc. 2008;130:7250–7252. doi: 10.1021/ja8024966. [DOI] [PubMed] [Google Scholar]; (b) Campbell PG, Abbey ER, Neiner D, Grant DJ, Dixon DA, Liu SY. J Am Chem Soc. 2010;132:18048–18050. doi: 10.1021/ja109596m. [DOI] [PubMed] [Google Scholar]; (c) Lamm AN, Garner EB, Dixon DA, Liu SY. Angew Chem Int Ed. 2011;50:8157–8160. doi: 10.1002/anie.201103192. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Matus MH, Liu SY, Dixon DA. J Phys Chem A. 2010;114:2644–2654. doi: 10.1021/jp9102838. [DOI] [PubMed] [Google Scholar]
- 14.Xu S, Zakharov LN, Liu SY. J Am Chem Soc. 2011;133:20152–20155. doi: 10.1021/ja2097089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.A simple monocyclic 1,4-azaborine still remains elusive.
- 16.Turner DW, Baker C, Baker AD, Brundle CR. Molecular Photoelectron Spectroscopy. Wiley-Interscience; New York: 1970. [Google Scholar]
- 17.For representative studies from the laboratory at Pau, see: Guillemin JC, Chrostowska A, Dargelos A, Nguyen TXM, Guenot P. Chem Commun. 2008:4204–4206. doi: 10.1039/b806771f.Chrostowska A, Dargelos A, Graciaa A, Beylère P, Lee VYa, Nakamoto M, Sekiguchi A. Organometallics. 2008;27:2915–2917.Chrostowska A, Lemierre V, Dargelos A, Baylère P, Leigh WJ, Rima G, Weber L, Schimmel M. J Organomet Chem. 2009;694:43–51.Chrostowska A, Nguyen TXM, Dargelos A, Khayar S, Graciaa A, Guillemin JC. J Phys Chem A. 2009;113:2387–2396. doi: 10.1021/jp8087447.Chrostowska A, Dargelos A, Graciaa A. Austr J Chem. 2010;63:1608–1614.Chrostowska A, Matrane A, Maki D, Khayar S, Ushiki H, Graciaa A, Belachemi L, Guillemin JC. Chem Phys Chem. 2011;12:226–236. doi: 10.1002/cphc.201100672.
- 18.(a) Weber L, Domke I, Greschner W, Miqueu K, Chrostowska A, Baylère P. Organometallics. 2005;24:5455–5463. [Google Scholar]; (b) Chrostowska A, Maciejczyk M, Dargelos A, Baylère P, Weber L, Werner V, Eickhoff D, Stammler HG, Neumann B. Organometallics. 2010;29:5192–5198. [Google Scholar]
- 19.Lemierre V, Chrostowska A, Dargelos A, Chermette H. J Phys Chem A. 2005;109:8348–8355. doi: 10.1021/jp050254c. [DOI] [PubMed] [Google Scholar]
- 20.Frisch MJ, et al. Gaussian 09, revision B.01. Gaussian, Inc; Wallingford, CT: 2009. [Google Scholar]
- 21.(a) Parr RG, Yang W. Functional Theory of Atoms and Molecules. Oxford University Press; New York: 1989. [Google Scholar]; (b) Frisch MJ, Trucks GW, Cheeseman JR. In: In Recent Development and Applications of Modern Density Functional Theory, Theoretical and Computational Chemistry. Semminario JM, editor. Vol. 4. Elsevier; Amsterdam – Lausann - New York - Oxford-Shannon - Tokyo: 1996. pp. 679–707. [Google Scholar]; (c) Limacher PA, Mikkelsen KV, Lüthi HP. J Chem Phys. 2009;130:194114. doi: 10.1063/1.3139023. [DOI] [PubMed] [Google Scholar]; (d) Kobayashi R, Amos RD. Chem Phys Lett. 2006;420:106–109. [Google Scholar]; (e) Jacquemin D, Perpète EA, Scalmani G, Frisch MJ, Kobayashi R, Adamo C. J Chem Phys. 2007;126:144105. doi: 10.1063/1.2715573. [DOI] [PubMed] [Google Scholar]
- 22.(a) Becke AD. Phys Rev. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]; (b) Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]; (c) Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; (d) Yanai T, Tew D, Handy N. Chem Phys Lett. 2004;393:51–57. [Google Scholar]
- 23.(a) Joantéguy S, Pfister-Guillouzo G, Chermette H. J Phys Chem. 1999;103:3505–3511. [Google Scholar]; (b) Chrostowska A, Miqueu K, Pfister-Guillouzo G, Briard E, Levillain J, Ripoll JL. J Mol Spectrosc. 2001;205:323–330. doi: 10.1006/jmsp.2000.8273. [DOI] [PubMed] [Google Scholar]; (c) Bartnik R, Baylère P, Chrostowska A, Galindo A, Lesniak S, Pfister-Guillouzo G. Eur J Org Chem. 2003:2475–2479. [Google Scholar]
- 24.(a) Stratmann RE, Scuseria GE, Frisch MJ. J Chem Phys. 1998;109:8218–8224. [Google Scholar]; (b) Casida ME, Jamorski C, Casida KC, Salahub DR. J Chem Phys. 1998;108:4439–4449. [Google Scholar]
- 25.(a) von Niessen W, Schirmer J, Cederbaum LS. Comput Phys Rep. 1984;1:57–125. [Google Scholar]; (b) Ortiz JV. J Chem Phys. 1988;89:6348–6352. [Google Scholar]
- 26.Nakatsuji H, Hirao K. J Chem Phys. 1978;68:2053–2065. [Google Scholar]
- 27.Stowasser R, Hoffmann R. J Am Chem Soc. 1999;121:3414–3420. [Google Scholar]
- 28.Portmann Stefan, Lüthi Hans Peter. MOLEKEL 4.3. Vol. 54. CHIMIA; 2000. pp. 766–770. [Google Scholar]
- 29.Efforts to prepare the parent 1,3-dihydro-1,3-azaborine have thus far met with failure.
- 30.Dewar MJS, Worley SD. J Chem Phys. 1969;50:654–667. [Google Scholar]
- 31.See Supporting Information for details.
- 32.Jahn H, Teller E. Proceedings of the Royal Society of London. Series A, Mathematical and Physical Sciences. 1937;161:220–235. [Google Scholar]
- 33.Daly AM, Tanjaroon C, Marwitz AJV, Liu SY, Kukolich SG. J Am Chem Soc. 2010;132:5501–5506. doi: 10.1021/ja1005338. [DOI] [PubMed] [Google Scholar]
- 34.(a) Pleskov Y, Krotova M, Elkin V, Varnin V, Teremetskaya I. Int J Electrochem. 2012;2012 doi: 10.1155/2012/437063. [DOI] [PubMed] [Google Scholar]; (b) Li E, Shi G, Hong X, Wu P. J Appl Polym Sci. 2004;93:189–195. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.