

Abstract
Objectives
Essential fatty acids are important for growth, development, and physiologic function. Alpha-linolenic acid and linoleic acid are the precursors of docosahexaenoic and arachidonic acid, respectively, and have traditionally been considered the essential fatty acids. However, we hypothesized that docosahexaenoic acid and arachidonic acid can function as the essential fatty acids.
Methods
Using a murine model of essential fatty acid deficiency and consequent hepatic steatosis, we provided mice with varying amounts of docosahexaenoic and arachidonic acids to determine whether exclusive supplementation of docosahexaenoic and arachidonic acids could prevent essential fatty acid deficiency and inhibit or attenuate hepatic steatosis.
Results
Mice supplemented with docosahexaenoic and arachidonic acids at 2.1% or 4.2% of their calories for 19 days had normal liver histology and no biochemical evidence of essential fatty acid deficiency, which persisted when observed after 9 weeks.
Conclusion
Supplementation of sufficient amounts of docosahexaenoic and arachidonic acids alone without alpha-linolenic and linoleic acids meets essential fatty acid requirements and prevents hepatic steatosis in a murine model.
Supplementary key words: alpha-linolenic acid, linoleic acid, omega-3 fatty acid, omega-6, fatty liver, polyunsaturated fatty acids
INTRODUCTION
Essential fatty acids (EFAs) are polyunsaturated fatty acids (PUFAs) that are required nutrients for growth, development, and other important physiologic functions. EFAs are termed as such because they must be acquired through dietary sources, as mammals cannot synthesize them from simple carbon precursors or interconvert other fatty acids. When less than 1–2% of total calories are provided from EFAs [1], essential fatty acid deficiency (EFAD) typically occurs.
There are three major families of PUFAs: omega-3, omega-6, and omega-9, all of which are metabolized using the same group of enzymes. Traditionally, α-linolenic acid (ALA, omega-3) and linoleic acid (LA, omega-6) are considered the only EFAs [2, 3]. Their downstream products are the highly physiologically relevant eicosapentaenoic acid (EPA, omega-3), docosahexaenoic acid (DHA, omega-3), and arachidonic acid (AA, omega-6). These are considered critical metabolites, as EPA and AA are important precursors to eicosanoids including prostaglandins, thromboxanes, prostacyclins and leukotrienes, which mediate numerous physiological and biochemical processes. EPA and DHA are important for optimal visual and cognitive function [4] in addition to providing the source of anti-inflammatory metabolites and promoting resolution of the inflammatory process [5].
A fish oil-based lipid emulsion provided as the sole source of fat has been reported to be effective in treating EFAD and reversing cholestasis [6]. It has also been shown that a fish oil-based diet with more than 13% of the total calories from fish oil will prevent EFAD and maintain growth in a murine model [7]. Since DHA and AA represent the principally active omega-3 and omega-6 fatty acids in fish oil, we hypothesize that DHA and AA will prevent EFAD and the resultant hepatic steatosis as well as provide ample amounts of EPA in a murine model of hepatic steatosis.
EXPERIMENTAL PROCEDURES
1. Animal model
Hepatic steatosis and biochemical model of EFAD
Experiments were performed on 4–6 week-old C57Bl6 mice (Jackson Laboratories, Bar Harbor, ME). The animals were housed in a barrier room with a 12 hour light cycle and were acclimated to their environment for at least 72 hours prior to initiation of the experiment. Experimental mice were housed in groups of five and fed an ad libitum liquid, fat-free, high carbohydrate diet (HCD), similar to the parenteral nutrition used clinically at Children’s Hospital Boston. The HCD contained 20% dextrose, a mixture of 2% essential and nonessential amino acids (TrophAmine, B. Braun Medical, Irvine, CA), 30 mEq/L sodium, 20 mEq/L potassium, 15 mEq/L calcium (as gluconate), 10 mEq/L magnesium, 10 mM phosphate, 5 mEq/L acetate, 30 mEq/L chloride, 0.2% pediatric trace elements (American Reagent, Shirley, NY), and 0.5% pediatric multivitamins (MaynePharma USA, Paramus, NJ). The mice were kept on this diet for 19 days, a duration known to cause severe hepatic steatosis [8]. In the long-term study, animals received this diet for 9 weeks. Control animals were fed the standard AIN-93M purified rodent diet (Dyets Inc., Bethlehem, PA) containing 140 g/kg casein, 1.8 g/kg L-cystine, 100 g/kg sucrose, 465.9 g/kg cornstarch, 155 g/kg dextrose, 40 g/kg soybean oil, 0.8 mg/kg t-butylhydroquinone, 50 g/kg cellulose, 35 g/kg mineral mix, 10 g/kg vitamin mix, and 2.5 g/kg choline bitartrate. The fatty acid composition of the soybean oil included: 6.42% ALA (18:3n-3), 51.3% LA (18:2n-6), 21.9% oleic acid (18:1n-9), 10% palmitic acid (16:0), 3.9% stearic acid (18:0), 0.29% arachidic acid (20:0), and 0.32% behenic acid (22:0). Animals also had access to water ad libitum.
Animal protocols complied with the NIH Animal Research Advisory Committee guidelines and were approved by the Children’s Hospital Boston Animal Care and Use Committee.
2. Treatment
Hydrogenated coconut oil (HCO) and AA (98% grade) were purchased from Cayman Chemical (Ann Arbor, MI). DHA (87.4% DHA, 12.6% sterols) was provided by Martek (Columbia, MD). A 19-day experiment was conducted on six groups of mice (n=5 in each), and another three groups (n=5 in each) were used in a 9-week experiment to test the long-term effects. In the 19-day experiment, mice were fed with both a fat-free liquid diet (HCD-only) and an EFA-deficient liquid diet (HCD-0) to create EFAD. In both experiments, mice on the HCD (except HCD-only) also received a daily lipid mixture equivalent to 5% of their daily caloric intake via orogastric gavage. The groups are outlined in Table 1. The lipid mixture was comprised of HCO, DHA and AA. In the 19-day experiment, the supplemented lipid mixture had a DHA:AA ratio of 20:1 and provided 0% (HCD-0), 0.21% (HCD-0.21), 2.1% (HCD-2.1) or 4.2% (HCD-4.2) of daily caloric intake. The amount of PUFAs in the fish oil-based lipid emulsion previously used fluctuates in its composition at approximately 14.4–30.9% DHA, 12.5–28.2% EPA, 1–4% AA, 0.1–0.7% LA, and <0.2% ALA [9]. Based on these data, the ratio of DHA:AA could range from 3.6:1 to 30.9:1. We chose the DHA:AA ratio of 20:1 for this experiment. HCO, which contains only trace amounts of PUFAs, was added to bring the total fat calories of the mixtures to 5% in the HCD-lipid containing diets. The 9-week experiment was comprised of a control group, a HCD-0L group, and a HCD-2.1L group. The HCD-2.1L group was selected based on the results of the 19-day experiment.
Table 1.
Percentage of HCO, DHA and AA in each lipid mixture
| Group | HCO (%) | DHA (%) | AA (%) |
|---|---|---|---|
| Control | - | - | - |
| HCD-only | 0.00 | 0.00 | 0.00 |
| HCD-0, HCD-0L | 5.00 | 0.00 | 0.00 |
| HCD-0.21 | 4.79 | 0.20 | 0.01 |
| HCD-2.1, HCD-2.1L | 2.90 | 2.00 | 0.10 |
| HCD-4.2 | 0.80 | 4.00 | 0.20 |
The 19-day experiment had 6 groups: Control, HCD-only, HCD-0, HCD-0.21, HCD-2.1 and HCD-4.2. The 9-week experiment had 3 groups: Control, HCD-0L and HCD-2.1L. The total fat in each lipid mixture provided 5% of the daily caloric intake.
3. Measurement of serum liver enzymes
At the end of each experiment, mice were fasted for 4 hours and then anesthetized with 300 μl of 2.5% Tribromoethanol (Sigma-Aldrich Corporation, St. Louis, MO) via intraperitoneal injection. Approximately 600 μl of blood was then collected from each mouse via retro-orbital puncture and centrifuged at 4°C at 1,000 g for 10 minutes. The serum was aspirated and about 100 μl was brought to the Clinical Laboratory at Children’s Hospital for measurement of alanine aminotransferase (ALT). The remaining serum was stored at −80°C and used for serum fatty acid analysis.
4. Histological evaluation and degree of steatosis
Mice were euthanized and their livers were excised. A designated lobe of the liver was fixed in 10% formalin overnight, washed with chilled PBS solution, and embedded in paraffin. The specimens were then sliced and stained with hematoxylin and eosin (H&E). The remaining portion of liver was immediately snap-frozen in liquid nitrogen and used for liver fatty acid analysis.
Histological analysis was performed on H&E sections by an experienced pathologist in a blinded fashion. The degree of steatosis was graded based on an established system [10]. Macrovesicular steatosis was graded on a scale 0–3 based on the percentage of hepatocytes affected in each specimen. The score was 0 for none; 1 for up to 33%; 2 for 33–66%; and 3 for more than 66%. Data were expressed as median with interquartile range (IQR).
5. Analysis of Serum and Liver Fatty Acid Composition
Serum and liver samples were prepared and the total lipids were extracted based on the methods of Folch [11]. Briefly, chloroform and methanol (Fisher, Fair Lawn, NJ) were added at a ratio of 2:1 followed by a potassium chloride (Aldrich, Milwaukee, WI) salt wash to isolate the total lipid fraction. Plasma total lipids were extracted from 40 to 150 μl of plasma. Tricosanoic free fatty acid (Sigma, St. Louis, MO) was added to each sample as an internal standard. The plasma and liver total lipids were saponified with 0.5 N methanolic sodium hydroxide (Sigma, St. Louis, MO) and the fatty acids were converted to methyl esters with 14% BF3/methanol (Sigma-Aldrich, St. Louis, MO) at 100°C for 30 minutes (Morrison and Smith, 1964). Butylatedhydroxytoluene (Sigma-Aldrich, St. Louis, MO) was added before saponification and all samples were purged with N2 throughout the process to minimize oxidation.
Fatty acid methyl esters were analyzed by gas liquid chromatography using a Hewlett Packard 6890 equipped with a flame ionization detector. Peaks were identified by comparison of retention times with external fatty acid methyl ester standard mixtures from NuCheck Prep (Elysian, MN). The fatty acid profiles were expressed as percentage of the total fatty acid (weight percent).
6. Determination of EFAD
The triene-tetraene (T:T) ratio is a biochemical marker used to characterize EFAD, as determined when the ratio of serum Mead acid (20:3 n-9) to AA (20:4 n-6) is greater than 0.2. Another marker of EFAD is the EFA index, defined as the (n-3 + n-6) to (n-7 + n-9) ratio. These ratios are widely accepted biochemical markers to diagnose EFAD in both animals [7, 12, 13] and humans [6, 14].
7. Statistical analysis
The group means were compared using analysis of variance (ANOVA) for differences with post hoc analysis using Tukey’s test. For nonparametric data, group medians were compared using the Kruskal-Wallis test. P< 0.05 was considered statistically significant. All statistical tests were performed using SPSS version 17.0 (SPSS Inc. Chicago, IL) and figures were created using Prism 5.01v Software (GraphPad Software Inc, La Jolla, CA). Steatosis scores are reported as medians (IQR). Other data are expressed as mean ± standard deviation (SD).
RESULTS
1. Animal outcome
All animals survived to completion of the study. The growth curves of both the 19-day and 9-week experiments are shown in Figure 1. In the 19-day experiment (Figure 1A), HCD-4.2 mice experienced weight loss in the first week, although animals had appropriate weight gain after 19 days. This effect was thought to be due to toxicity of AA [15, 16]. There were no statistically significant differences in body weight gain between groups during the study. Since mice in the HCD-4.2 group experience weight loss in the first week, we decided to eliminate this group from the long term (9-week) experiment. In the 9-week experiment, there were no statistically significant differences in percentage body weight gain among the three groups (Figure 1B). Mice in the control, HCD-0L and HCD-2.1L groups gained 51%, 50% and 52% of their weight, respectively. None of the mice developed dermatitis or alopecia, the hallmark clinical signs of EFAD.
Figure 1. Growth curves of mice in the 19-day and 9-week experiments.
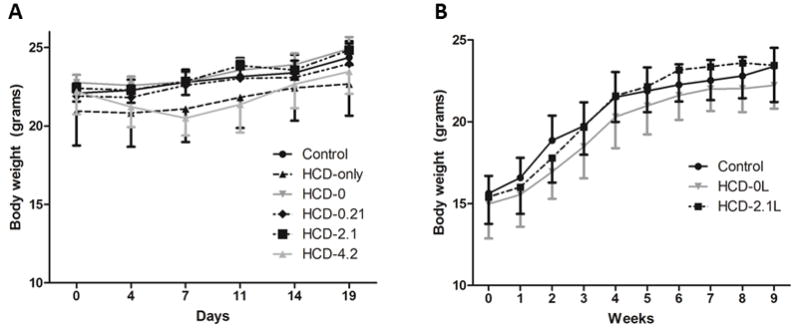
(A) In the 19-day experiment, mice received a supplemented lipid mixture with a DHA:AA ratio of 20:1 which provided 0% (HCD-0), 0.21% (HCD-0.21), 2.1% (HCD-2.1) or 4.2% (HCD-4.2) of their daily caloric intake. Control mice received the standard AIN-93M purified rodent diet. (B) In the 9-week experiment, mice received a lipid mixture that provided 0% (HCD-0L), 2.1% (HCD-2.1L) of their daily caloric intake. Mice in HCD-4.2 group experienced weight loss in the first week of treatment but caught up by day 14. There was no difference in the mean body weight or the percentage of body weight gain among the groups by the end of the experiment. Data are shown as mean body weight ± SD.
2. Fatty acid analysis
Alpha-linolenic acid and Linoleic acid
ALA (18:3 n-3) and LA (18:2 n-6) are considered to be the two EFAs. Their levels in all the treated groups were significantly lower compared to the control group in both serum and liver (Tables 2 and 3). Interestingly, serum LA levels were also significantly lower in HCD-2.1 and HCD-4.2 groups compared to the EFA-deficient mice (HCD-0 group).
Table 2.
Fatty acid composition of serum and liver in the 19-day experiment
| Mole % | Control | HCD-only | HCD-0 | HCD-0.21 | HCD-2.1 | HCD-4.2 |
|---|---|---|---|---|---|---|
| Serum | ||||||
| 16:0 | 17.54 ± 1.51 | 19.77 ± 0.65 | 19.88 ± 0.78 | 21.86 ± 1.60§ | 23.55 ± 2.10§,¶ | 23.10 ± 0.47§,¶ |
| 16:1 n-7 | 2.17 ± 0.24 | 4.99 ± 0.46§ | 5.71 ± 0.37§ | 5.95 ± 0.88§ | 5.05 ± 0.51§ | 4.32 ± 0.36§,¶ |
| 18:0 | 11.75 ± 1.06 | 10.51 ± 0.61 | 10.28 ± 1.04 | 11.31 ± 0.96 | 11.19 ± 1.94 | 11.67 ± 0.91 |
| 18:1 n-9 | 15.79 ± 2.37 | 24.54 ± 1.17§ | 26.33 ± 3.85§ | 26.41 ± 2.39§ | 22.70 ± 5.16§ | 16.49 ± 3.93 |
| 18:1 n-7 | 3.28 ± 0.34 | 5.26 ± 0.84§ | 5.17 ± 1.00§ | 5.11 ± 0.47§ | 2.67 ± 1.19¶ | 1.42 ± 0.37§,¶ |
| 18:2 n-6 | 24.25 ± 1.03 | 4.70 ± 0.50§ | 5.73 ± 2.08§ | 5.98 ± 0.83§ | 2.96 ± 0.72§,¶ | 2.54 ± 0.80§,¶ |
| 18:3 n-6 | 0.18 ± 0.02 | 0.20 ± 0.03 | 0.23 ± 0.07 | 0.14 ± 0.01¶ | 0.03 ± 0.03§,¶ | 0.03 ± 0.02§,¶ |
| 18:3 n-3 | 0.32 ± 0.06 | 0.05 ± 0.01§ | 0.07 ± 0.03§ | 0.06 ± 0.03§ | 0.09 ± 0.03§ | 0.10 ± 0.01§ |
| 20:3 n-9 | 0.41 ± 0.21 | 8.53 ± 1.58§ | 6.92 ± 1.82§ | 2.73 ± 0.99§,¶ | 0.18 ± 0.20¶ | 0.01 ± 0.02¶ |
| 20:3 n-6 | 1.64 ± 0.25 | 1.68 ± 0.19 | 1.77 ± 0.36 | 1.29 ± 0.15 | 0.30 ± 0.14§,¶ | 0.17 ± 0.04§,¶ |
| 20:4 n-6 | 12.06 ± 1.28 | 10.54 ± 1.51§ | 8.90 ± 2.09§ | 6.47 ± 1.56§ | 7.51 ± 1.62§ | 8.16 ± 0.98§ |
| 20:5 n-3 | 1.31 ± 0.27 | 0.20 ± 0.02 | 0.32 ± 0.12 | 0.98 ± 0.13 | 4.58 ± 1.66§,¶ | 7.20 ± 1.57§,¶ |
| 22:4 n-6 | 0.08 ± 0.02 | 0.11 ± 0.05 | 0.10 ± 0.01 | 0.04 ± 0.04 | 0.04 ± 0.04 | 0.00 ± 0.00§,¶ |
| 22:5 n-6 | 0.13 ± 0.04 | 0.86 ± 0.06§,¶ | 0.55 ± 0.07§ | 0.15 ± 0.04,¶ | 0.02 ± 0.03§,¶ | 0.00 ± 0.00§,¶ |
| 22:5 n-3 | 0.30 ± 0.02 | 0.04 ± 0.05§ | 0.11 ± 0.03§ | 0.15 ± 0.02§ | 0.54 ± 0.14§,¶ | 0.70 ± 0.06§,¶ |
| 22:6 n-3 | 7.11 ± 1.14 | 6.62 ± 0.52 | 6.29 ± 1.00 | 9.50 ± 3.01 | 16.65 ± 3.13§,¶ | 22.13 ± 3.37§,¶ |
| Liver | ||||||
| 16:0 | 19.49 ± 0.59 | 22.01 ± 0.36§,¶ | 23.74 ± 0.41§ | 24.96 ± 0.43§ | 26.03 ± 0.86§,¶ | 25.12 ± 1.21§ |
| 16:1 n-7 | 2.82 ± 0.84 | 6.67 ± 0.91§ | 6.34 ± 0.50§ | 7.33 ± 0.69§ | 6.51 ± 0.64§ | 5.08 ± 0.67§ |
| 18:0 | 10.81 ± 2.16 | 3.15 ± 0.12§ | 4.04 ± 0.53§ | 4.46 ± 0.79§ | 6.85 ± 1.32§,¶ | 9.86 ± 1.22¶ |
| 18:1 n-9 | 23.63 ± 6.91 | 49.67 ± 1.76§ | 46.12 ± 1.37§ | 42.62 ± 2.91§ | 28.85 ± 9.65¶ | 17.81 ± 3.29¶ |
| 18:1 n-7 | 4.72 ± 0.91 | 9.56 ± 0.85§ | 9.31 ± 0.65§ | 8.37 ± 0.65§ | 3.33 ± 1.74¶ | 1.49 ± 0.25§,¶ |
| 18:2 n-6 | 13.44 ± 1.21 | 1.45 ± 0.26§ | 1.91 ± 0.70§ | 2.28 ± 0.58§ | 2.47 ± 0.77§ | 2.23 ± 0.50§ |
| 18:3 n-6 | 0.14 ± 0.02 | 0.06 ± 0.02§ | 0.05 ± 0.02§ | 0.03 ± 0.01§ | 0.02 ± 0.01§ | 0.02 ± 0.00§,¶ |
| 18:3 n-3 | 0.35 ± 0.09 | 0.03 ± 0.01§ | 0.03 ± 0.02§ | 0.05 ± 0.02§ | 0.12 ± 0.06§,¶ | 0.10 ± 0.02§ |
| 20:3 n-9 | 0.39 ± 0.05 | 1.44 ± 0.13§ | 1.25 ± 0.28§ | 0.63 ± 0.13¶ | 0.08 ± 0.07§,¶ | 0.02 ± 0.01§,¶ |
| 20:3 n-6 | 1.42 ± 0.20 | 0.24 ± 0.02§ | 0.32 ± 0.09§ | 0.30 ± 0.07§ | 0.15 ± 0.05§ | 0.14 ± 0.03§ |
| 20:4 n-6 | 10.25 ± 3.20 | 1.43 ± 0.15§ | 1.95 ± 0.45§ | 2.02 ± 0.61§ | 3.36 ± 0.48§ | 4.99 ± 0.74§,¶ |
| 20:5 n-3 | 0.63 ± 0.02 | 0.04 ± 0.00 | 0.05 ± 0.02 | 0.20 ± 0.07 | 1.99 ± 1.08§,¶ | 3.72 ± 0.28§,¶ |
| 22:4 n-6 | 0.18 ± 0.06 | 0.04 ± 0.01§ | 0.05 ± 0.01§ | 0.04 ± 0.02§ | 0.08 ± 0.02§ | 0.07 ± 0.01§ |
| 22:5 n-6 | 0.15 ± 0.06 | 0.18 ± 0.03 | 0.14 ± 0.02 | 0.06 ± 0.01§,¶ | 0.01 ± 0.02§,¶ | 0.00 ± 0.00§,¶ |
| 22:5 n-3 | 0.41 ± 0.02 | 0.02 ± 0.01§ | 0.04 ± 0.03§ | 0.10 ± 0.03 | 0.87 ± 0.40§,¶ | 1.05 ± 0.24§,¶ |
| 22:6 n-3 | 9.01 ± 1.65 | 1.41 ± 0.16§ | 1.75 ± 0.30§ | 3.82 ± 0.96 | 16.94 ± 8.47§,¶ | 26.60 ± 4.41§,¶ |
The group means were compared using analysis of variance (ANOVA) for difference with post hoc analysis using Tukey’s test.
indicate statistically significant differences with P<0.05 compared to the control and the EFA-deficient HCD-0 groups, respectively.
Table 3.
Fatty acid composition of serum and liver in the 9-week experiment
| Serum | Liver | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Mole % | Control | HCD-0L | HCD-2.1L | Control | HCD-0L | HCD-2.1L |
| 16:0 | 21.99 ± 1.26 | 20.38 ± 0.52 | 26.11 ± 1.56§,¶ | 22.88 ± 1.21 | 21.40 ± 0.46 | 23.15 ± 1.01¶ |
| 16:1 n-7 | 4.97 ± 1.07 | 10.82 ± 1.05§,¶ | 7.16 ± 1.68§,¶ | 6.76 ± 0.68 | 11.34 ± 0.62§ | 12.43 ± 0.93§ |
| 18:0 | 11.10 ± 0.56 | 10.37 ± 0.59 | 12.84 ± 1.70¶ | 8.30 ± 0.40 | 3.08 ± 0.32§ | 6.85 ± 0.55§,¶ |
| 18:1 n-9 | 15.97 ± 1.12 | 25.10 ± 1.42§ | 14.67 ± 1.86¶ | 26.45 ± 4.13 | 43.88 ± 0.96§ | 28.03 ± 2.24¶ |
| 18:1 n-7 | 4.08 ± 0.62 | 6.97 ± 0.46§ | 2.40 ± 0.53§,¶ | 5.58 ± 0.51 | 9.58 ± 0.86§ | 4.33 ± 0.22§,¶ |
| 18:2 n-6 | 16.08 ± 0.82 | 2.67 ± 0.66§ | 2.27 ± 0.56§ | 10.91 ± 1.43 | 0.54 ± 0.13§ | 1.54 ± 0.36§ |
| 18:3 n-6 | 0.16 ± 0.09 | 0.00 ± 0.00§ | 0.00 ± 0.00§ | 0.03 ± 0.03 | 0.00 ± 0.00§ | 0.00 ± 0.00§ |
| 18:3 n-3 | 0.25 ± 0.07 | 0.00 ± 0.00§ | 0.00 ± 0.00§ | 0.25 ± 0.04 | 0.00 ± 0.00§ | 0.05 ± 0.01§,¶ |
| 20:3 n-9 | 0.92 ± 0.19 | 13.02 ± 0.73§ | 0.47 ± 0.38¶ | 0.59 ± 0.13 | 2.20 ± 0.34§ | 0.13 ± 0.06§,¶ |
| 20:3 n-6 | 2.11 ± 0.11 | 0.73 ± 0.13§ | 0.26 ± 0.04§,¶ | 1.42 ± 0.24 | 0.13 ± 0.03§ | 0.14 ± 0.03§ |
| 20:4 n-6 | 14.55 ± 1.69 | 4.29 ± 0.46§ | 8.74 ± 1.12§,¶ | 9.33 ± 1.12 | 1.11 ± 0.21§ | 2.83 ± 0.51§,¶ |
| 20:5 n-3 | 0.39 ± 0.07 | 0.00 ± 0.00 | 7.90 ± 0.99§,¶ | 0.18 ± 0.06 | 0.00 ± 0.00 | 2.87 ± 0.43§,¶ |
| 22:4 n-6 | 0.12 ± 0.02 | 0.03 ± 0.02§ | 0.01 ± 0.01§ | 0.21 ± 0.04 | 0.00 ± 0.00§ | 0.04 ± 0.01§,¶ |
| 22:5 n-6 | 0.51 ± 0.10 | 0.50 ± 0.29 | 0.00 ± 0.00§,¶ | 0.42 ± 0.12 | 0.13 ± 0.03§ | 0.00 ± 0.00§,¶ |
| 22:5 n-3 | 0.13 ± 0.03 | 0.11 ± 0.25 | 0.34 ± 0.02 | 0.12 ± 0.03 | 0.00 ± 0.00 | 0.97 ± 0.12§,¶ |
| 22:6 n-3 | 4.63 ± 0.29 | 2.00 ± 0.28§ | 14.77 ± 0.93§,¶ | 3.75 ± 0.51 | 0.51 ± 0.11§ | 13.59 ± 1.98§,¶ |
The group means were compared using analysis of variance (ANOVA) for difference with post hoc analysis using Tukey’s test.
indicate statistically significant differences with P<0.05 compared to the control and the EFA-deficient HCD-0L groups, respectively.
Eicosapentaenoic acid
EPA (20:5 n-3) is an important downstream product of ALA. EPA is not present in the control soybean-based diet and is synthesized in vivo from ALA. Its mean percentage in the serum and liver at 19-days was 1.31% and 0.63%, respectively (Table 2). After 9 weeks, the mean percentage decreased to 0.39% and 0.18% in the serum and liver, respectively (Table 3). Mice in the HCD-only and HCD-0 groups, both with expected EFAD, had lower percentages of EPA in the serum and liver compared to the control mice. When mice in the 19-day experiment were provided increasing amounts of DHA, the percentage of serum and liver EPA increased steadily compared to the control group. After 9 weeks, mice in the HCD-0 group had an essentially undetectable level of EPA in both the serum and liver, whereas the percentage of EPA in the HCD-2.1 mice was increased.
Docosahexaenoic acid
Like EPA, DHA (22:6 n-3) is an important downstream product of ALA and is not present in the control diet. By increasing the dietary amounts of pure DHA, the mean percentage of DHA in serum increased from 6.29% in HCD-0 group to 9.50%, 16.65% and 22.13% in HCD-0.21, HCD-2.1 and HCD-4.2 groups, respectively (Table 2). The percentage of liver DHA also increased from 1.75% in the HCD-0 group to 3.82%, 16.94% and 26.60% in HCD-0.21, HCD-2.1 and HCD-4.2 groups, respectively (Table 3). The 9-week experiment also showed a significant increase in the percentage of serum and liver DHA in the HCD-2.1 mice compared to HCD-0 mice.
Arachidonic acid
The mean percentage of AA (20:4 n-6) was 12.06% in the serum and 10.25% in control mouse livers after 19 days (Table 2). These values remained relatively unchanged after 9 weeks (Table 3). The mean percentage of AA remained relatively constant in the serum of mice in the HCD-only and HCD-0 groups, whereas the mean percentage in the liver decreased. After 9 weeks, the mean percentage of serum AA in the HCD-0 group decreased to 4.29% while the liver levels continued to be low at 1.11%. Mice in the HCD-2.1 and HCD-4.2 groups had an intermediate mean percentage of serum AA at 7.51% and 8.16%, respectively. It was also noted that the mean percentage of serum and liver AA remained relatively unchanged at 8.74% after 9 weeks in the HCD-2.1 group. The percentage of liver AA also remained stable (3.36% after 19 days and 2.83% after 9 weeks).
3. Evidence of EFAD
T:T ratio
As expected, control mice in the 19-day and 9-week experiments did not have EFAD as their serum and liver T:T ratios were well below 0.2 (Table 4,5). Fat-free mice (HCD-only) and EFA-free mice (HCD-0) all had severe EFAD with elevated serum and liver T:T ratios after 19 days (Table 4). The T:T ratio of HCD-0 mice became even more elevated after 9 weeks (Table 5) suggesting more severe EFAD. HCD-0.21 mice had mean serum and liver T:T ratios surpassing the value of 0.2 indicating EFAD. HCD-2.1 mice had serum and liver T:T ratios well below the value 0.2 after 19 days and remained similar to the control group after 9 weeks. HCD-4.2 mouse serum and liver T:T ratios were well below 0.2.
Table 4.
Triene:tetraene ratio and EFA index of serum and liver in the 19-day experiment
| Group | Triene:tetraene ratio | EFA Index | ||
|---|---|---|---|---|
|
| ||||
| Serum | Liver | Serum | Liver | |
| Control | 0.03 ± 0.02 | 0.04 ± 0.01 | 2.20 ± 0.46 | 1.25 ± 0.58 |
| HCD-only | 0.84 ± 0.27§ | 1.02 ± 0.14§ | 0.58 ± 0.10§ | 0.07 ± 0.01§ |
| HCD-0 | 0.84 ± 0.35§ | 0.66 ± 0.18§ | 0.56 ± 0.21§ | 0.10 ± 0.02§ |
| HCD-0.21 | 0.42 ± 0.10§,¶ | 0.34 ± 0.13§,¶ | 0.72 ± 0.15§ | 0.15 ± 0.04§ |
| HCD-2.1 | 0.02 ± 0.03¶ | 0.03 ± 0.02§,¶ | 1.12 ± 0.46§ | 0.79 ± 0.53 |
| HCD-4.2 | 0.00 ± 0.00¶ | 0.00 ± 0.00§,¶ | 1.87 ± 0.52¶ | 1.63 ± 0.44¶ |
The group means were compared using analysis of variance (ANOVA) for difference with post hoc analysis using Tukey’s test.
indicate statistically significant differences with P<0.05 compared to the control and the EFA-deficient HCD-0 groups, respectively.
Table 5.
Triene:tetraene ratio and EFA index of serum and liver in the 9-week experiment
| Group | Triene:tetraene ratio | EFA Index | ||
|---|---|---|---|---|
|
| ||||
| Serum | Liver | Serum | Liver | |
| Control | 0.06 ± 0.02 | 0.06 ± 0.01 | 1.49 ± 0.23 | 0.67 ± 0.14 |
| HCD-0L | 3.07 ± 0.44§ | 2.02 ± 0.29§ | 0.19 ± 0.04§ | 0.01 ± 0.00§ |
| HCD-2.1L | 0.05 ± 0.03¶ | 0.05 ± 0.02¶ | 1.40 ± 0.22¶ | 0.48 ± 0.08§,¶ |
The group means were compared using analysis of variance (ANOVA) for difference with post hoc analysis using Tukey’s test.
indicate statistically significant differences with P<0.05 compared to the control and the EFA-deficient HCD-0L groups, respectively.
EFA index
EFA index, another biochemical marker of EFAD, was also used to evaluate these mice for evidence of biochemical EFAD. The results showed that both the fat-free and EFA-deficient mice had suppressed serum and liver EFA indices compared to control mice (Table 4) in the 19-day experiment. Mice that received 2.1% of their calories from DHA and AA (HCD-2.1) had significantly lower serum EFA index after 19 days. However, their serum EFA indices after 9 weeks were equivalent to those of control mice.
Mead Acid
ALA>LA>oleic acid is the order of affinity for the elongation and desaturation enzymes. In the state of EFAD, Mead acid (20:3 n-9) is synthesized from oleic acid (18:1 n-9) which serves to partially compensate for the reduction in double bonds in tissue membranes due to the lack of precursor omega-3 and omega-6 fatty acids. Therefore, an increased Mead acid level is another important indicator of EFAD. In the 19-day experiment, the mean percentage of Mead acid was 0.41% in the serum and 0.39% in the liver of the control mice (Table 2). As expected, mean percentages of Mead acid increased significantly in the serum and livers of mice in the HCD-only and HCD-0 groups compared to the control group. Mice in the HCD-0.21 group also had a significant, but smaller increase in the percentage of serum Mead acid compared to control mice. Mice in the HCD-2.1 and HCD-4.2 groups had even lower percentages of Mead acid in their livers compared to controls. Similarly, results from the 9-week experiment also showed a significant increase in serum and liver Mead acid percentages in the HCD-0 group (Table 3). There was no significant difference in serum and liver Mead acid percentage in the HCD-2.1 group compared to the control group.
4. Histological evaluation and degree of steatosis
In both experiments, there were no inflammatory cells present on liver sections, suggesting that there was no evidence of steatohepatitis in any group (Figures 2, 3). Control mice showed normal hepatic architecture with no hepatic steatosis. In contrast, liver sections from the HCD-only and the HCD-0 mice exhibited diffuse macro- and microvesicular steatosis in the periportal and midzone areas. Liver sections from mice in the HCD-0.21 group showed moderately preserved hepatic architecture, less macrovesicular steatosis, and more sparing of the midzone. In both the 19-day and 9-week experiments, livers of mice receiving 2% DHA and 0.1% AA (HCD-2.1 and HCD-2.1L) had well-preserved hepatic architecture with rare focal microvacuoles in the cytoplasm of the midzone hepatocytes. No macro- or microvesicular steatosis was observed in liver sections of HCD-4.2 mice in the 19-day experiment.
Figure 2. Liver histology and degree of hepatic steatosis after 19 days.
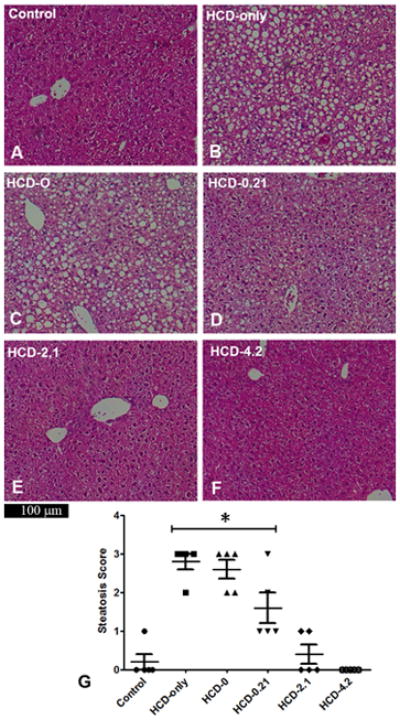
Hematoxylin and eosin stained liver sections of mice with different lipid supplementation. Liver sections from control mice showed normal hepatic architecture (A). HCD-only livers exhibited diffuse macro- and microvesicular steatosis in the periportal and midzone hepatocytes (B). HCD-0 livers were similar to HCD-only livers (C). HCD-0.21 livers showed a moderate reduction of steatosis in the periportal and midzone regions (D). HCD-2.1 and HCD-4.2 livers had well-preserved hepatic architecture with no focal microvacuoles (E–F). The results were reflected in the steatosis scores of these groups (G) in which mice in the HCD-only, HCD-0 and HCD-0.21 groups had higher median scores than control animals (*P<0.05). Data are expressed as median ± IQR. Group medians were compared using the Kruskal-Wallis test.
Figure 3. Liver histology and degree of steatosis after 9 weeks.
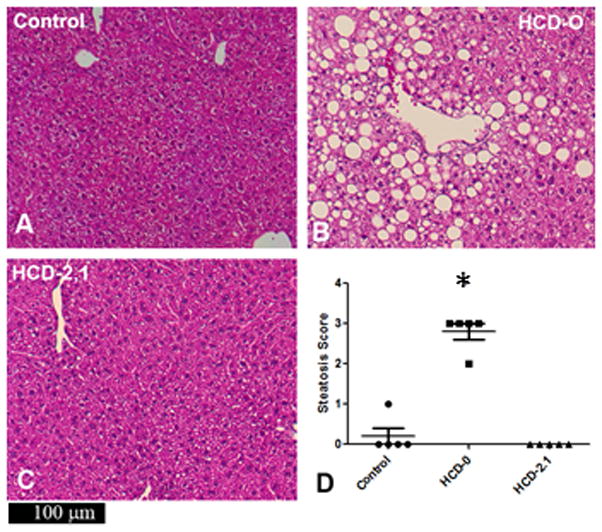
Livers of the HCD-0L mice had severe macrovesicular steatosis (B). Livers of the HCD-2.1L mice retained normal architecture (C). Livers of the HCD-0L mice had a significantly higher steatosis score compared to both the control and HCD-2.1L mice (*P<0.05). Data are expressed as median ± IQR. Group medians were compared using the Kruskal-Wallis test.
5. Serum liver enzymes
ALT levels were obtained to assess the degree of liver injury. Values from the control mice that were maintained on the standard rodent diet were used as a reference. In the 19-day experiment, the ALT value in the HCD-only group was higher but not statistically significant compared to the control group (Figure 4A). However, the ALT level of the HCD-4.2 group was significantly lower than the HCD-only group. In the 9-week experiment, HCD-0 mice had a higher ALT value than both the control and HCD-2.1 mice (Figure 4B).
Figure 4. Serum ALT in the 19-day and 9-week experiments.
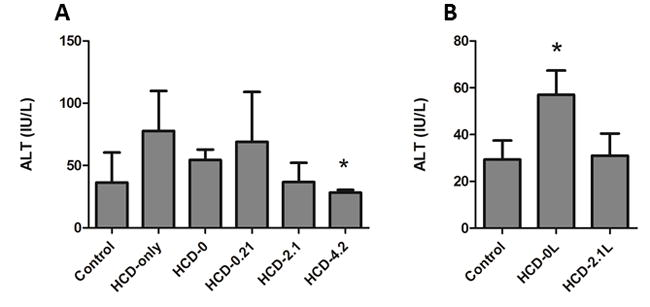
After 19 days (A), the mean ALT value in control mice was 36.3 ± 13.9 IU/L. HCD-only, HCD-0 and HCD-0.21 values were higher than the control value but not significant (77.8 ± 16.0 IU/L, 54.4 ± 3.8 IU/L and 69.0 ± 23.1 IU/L, respectively). The ALT value of the HCD-2.1 mice was 35.8 ± 6.9 IU/L. The ALT value of the HCD-4.2 mice (28.3 ± 1.1 IU/L) was significantly lower than ALT values of HCD-only and HCD-0 mice (*P<0.05). After 9 weeks (B), the mean ALT value of the HCD-0 mice was 57.0± 4.6 IU/L, which was significantly higher than the ALT values of both control mice (29.4 ± 3.6 IU/L) and the HCD-2.1 mice (31.0 ± 4.2 IU/L) (*P<0.05). Data are expressed as mean ± SD. The group means were compared using the analysis of variance (ANOVA) for difference with post hoc analysis using Tukey’s test.
DISCUSSION
ALA and LA are considered to be the only EFAs. EFAs are termed as such because they are required for important body functions but cannot be synthesized in the body [17]. Clinical signs of EFAD typically include growth retardation and dermatitis [18]. However, these clinical signs are now rarely seen in adults with the wide availability of PN and lipid emulsions containing EFA; therefore, biochemical markers such as T:T ratio and EFA index were derived to identify the development of biochemical EFAD [14]. In animals, clinical evidence of EFAD is often not reported [12] and generally requires a long duration of feeding an EFAD diet and/or feeding beginning early in infancy. In our experiments, adult mice fed both fat-deficient and EFA-deficient diets exhibited biochemical EFAD with accompanying hepatic steatosis without any clinical signs of EFAD.
Our results demonstrate that mice provided with at least 2% of their calories from DHA and at least 0.1% of calories from AA did not develop clinical or biochemical evidence of EFAD. The prevention of biochemical evidence of EFAD was demonstrated using a combination of T:T ratio, EFA index, and Mead acid level as well as the prevention of hepatic steatosis. In fact, the mean Mead acid levels were lower in the HCD-2.1 and HCD-4.2 groups as compared to the control group. This presumably reflects the impact of DHA and/or EPA to reduce elongation and desaturation of oleic acid. In the animals that received DHA and AA yet did not develop evidence of EFAD, the percentage of the traditional EFA, ALA and LA remained significantly lower than in control animals. This presumably reflects their absence in the diets containing DHA and AA and the lack of appreciable retro-conversion of DHA beyond the level of EPA (i.e. to ALA) or of AA to LA. Furthermore, ALA and LA, which are present in the control diet but not in the other diets, would competitively inhibit the elongation of oleic acid to Mead acid and thus lead to lower levels of Mead acid. However, the level of Mead acid and the T:T ratio were maintained in the higher DHA and AA-containing diets as in the control diets reflecting equivalent EFA status, whereas the AA level was lower. This is thought to reflect the ability of EPA to substitute for AA in tissue membranes [19]. Since a lower AA level in membranes produced by EPA supplementation is associated with an anti-inflammatory effect [20], this suggests a potential benefit for the use of DHA and AA to meet EFA requirements. While ALA and LA can meet all the needs for EPA, DHA, and AA, it would appear that DHA and AA can alternatively serve as the EFAs.
The lipid mixture used in our experiments had a 20:1 ratio of DHA:AA. This ratio was used to mimic the ratio of n-3 to n-6 fatty acids present in the oil of cold water fish. HCO was added to increase the non-EFA fat content. Since coconut oil contains approximately 0.1% ALA and 1.9% LA, it was hydrogenated to ensure that only trace LA was present. Although there have been reports of retarded growth in infants and growing animals on high omega-3 to omega-6 ratio diets [21, 22], the effect is most likely due to lack of AA [23] since EPA can so profoundly lower AA levels. In addition, we have previously shown that a diet containing 13% of calories from fish oil which contains EPA, DHA, and AA prevented EFAD and maintained growth in a long-term 9-week study despite inadequate amounts of LA in the fish oil [7]. In this study, we show that a 20:1 ratio of DHA:AA results in an increase in serum and liver DHA levels while keeping AA at levels significantly lower than with the control diet despite similar EFA status. The data also show that when mice receive increasing amounts of exogenous DHA, both serum and liver EPA increase. Since these mice received no exogenous EPA, this is likely to be the result of retro-conversion of DHA to EPA, as previously described [24]. When mice received DHA:AA at 4.2% of their daily calories, they developed weight loss in the first week but eventually caught up with the control group by day 14. This effect was confirmed on repeated experiments. In the HCD-4.2 group, each mouse received approximately 200mg/kg/day of AA, which is lethal if administered alone in intravenous form [15, 16]. However, when given with DHA in oral form, the level of toxicity is often diminished [25]. The results suggest that the amount of AA administered to the HCD-4.2 group was relatively toxic which resulted in weight loss likely due to an increased inflammatory response as a result of increased inflammatory cytokine production. Continued exposure to high dose AA might have enabled these mice to adapt and metabolize AA more efficiently to avoid a toxic effect. Due to the toxic effect of AA, this group was not included in the 9-week study.
We also demonstrate that DHA and AA, with the background intake of other fat as HCO, are able to prevent hepatic steatosis in our model. Although it is known that enhanced de novo lipogenesis and resultant hepatic steatosis occur secondary to EFAD, we have shown that the route of administration and EFAD may act independently when fat is provided parenterally [26]. However, in this study all feeding occurred by the enteral route. It is also known that fat intake of any type can impair de novo lipogenesis, albeit to different degrees [27]. Nonetheless, this study showed that the degree of steatosis was similar in the fat-free diet and with added HCO only, suggesting that EFAD was the principal factor. One explanation for the prevention of hepatic steatosis is the lack of or major reduction in de novo lipogenesis in mice receiving DHA/AA. One of the markers of de novo lipogenesis is the “desaturation index”, which is the serum ratio of 18:1/18:0 [28]. Results from this study showed that “desaturation indices” were higher in both the fat-free and HCO-only groups (2.33 and 2.56, respectively) compared to the control value of 1.34. This ratio decreased to 2.34, 2.03 and 1.41 in the HCD-0.21, HCD-2.1 and HCD-4.2 groups, respectively. This reflects decreased de novo lipogenesis with increasing DHA. These results are consistent with the histological findings.
Although this is beyond the scope of this study, one limitation is that the lipid mixtures employed do not reflect the normal dietary intake of PUFA nor provide the usual PUFA tissue levels in either humans or rodents. The basal diet used was based on our previous experimentation where mice fed with this diet developed EFAD and hepatic steatosis [8], the latter which we viewed as an excellent metabolic measure of EFAD. The lipid mixtures were created to ensure that DHA and AA were the sole sources of PUFA. However, by eliminating other sources of PUFA, we can confidently conclude that the DHA and AA are the only two responsible dietary PUFAs in this study, although their effect on other PUFAs such as EPA in vivo would have a major impact on metabolism. These results therefore can serve as the basis for a future study of other diets using this expanded definition of EFAs particularly where EPA is included in the dietary mix.
These results should be interpreted with caution as they were derived from murine models. Moreover, the optimal amount of DHA and AA to prevent EFAD and hepatic steatosis is yet to be determined. Further studies need to be performed to determine the optimal ratio of DHA:AA, as well as the minimal requirement of each fatty acid in order to design novel parenteral lipid emulsions that provide adequate amounts of EFA to prevent and treat EFAD while limiting hepatic steatosis and reducing AA due to its putative anti-inflammatory effects.
Overall, this is the first demonstration that the provision of DHA and AA can prevent biochemical and clinical evidence of EFAD in both a short and long-term murine model. DHA and AA supplementation can also prevent hepatic steatosis as an important metabolic consequence of EFAD. These changes occurred despite dietary absence of ALA and LA and while their serum and tissue levels diminished. This suggests that the paradigm that ALA and LA are the only EFA should at least be reconsidered.
CLINICAL RELEVANCY.
We found that docosahexaenoic acid and arachidonic acid alone without the traditional essential fatty acids alpha-linolenic and linoleic acid meet essential fatty acid requirements and prevent hepatic steatosis. This suggests that docosahexaenoic acid and arachidonic acid can be considered essential fatty acids. Traditional thinking is that patients have to receive adequate alpha-linolenic and linoleic acid to meet essential fatty acid requirements. With this finding, new parenteral and enteral formulas can be developed based on these two fatty acids to take advantage of their metabolically active properties without fear of clinical or biochemical essential fatty deficiency.
Acknowledgments
An abstract describing these findings was presented at the 40th American Pediatric Surgery Association annual meeting. HDL and EMF were recipients of the Joshua Ryan Rappaport Fellowship. EMF was supported by the National Institute of Diabetes And Digestive and Kidney Diseases (grant F32DK083880). MP was supported by the National Institutes of Health (grant DK069621-05). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Diabetes And Digestive and Kidney Diseases or the National Institutes of Health. HDL, JAM, VEM, EMF and MP were supported by the Children’s Hospital Surgical Foundation and The Vascular Biology Program (Boston, MA).
References
- 1.Aaes-Jorgensen E, Leppik EE, Hayes HW, Holman RT. Essential fatty acid deficiency. II. In adult rats. J Nutr. 1958;66(2):245–259. doi: 10.1093/jn/66.2.245. [DOI] [PubMed] [Google Scholar]
- 2.Das UN. Essential fatty acids: biochemistry, physiology and pathology. Biotechnology journal. 2006;1(4):420–439. doi: 10.1002/biot.200600012. [DOI] [PubMed] [Google Scholar]
- 3.Voet DV, Voet JG. Biochemistry. 3. New York: J. Wiley & Sons; 2004. [Google Scholar]
- 4.Carlson SE, Werkman SH, Tolley EA. Effect of long-chain n-3 fatty acid supplementation on visual acuity and growth of preterm infants with and without bronchopulmonary dysplasia. The American journal of clinical nutrition. 1996;63(5):687–697. doi: 10.1093/ajcn/63.5.687. [DOI] [PubMed] [Google Scholar]
- 5.Bannenberg G, Serhan CN. Specialized pro-resolving lipid mediators in the inflammatory response: An update. Biochim Biophys Acta. 2010;1801(12):1260–1273. doi: 10.1016/j.bbalip.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Puder M, Valim C, Meisel JA, Le HD, de Meijer VE, Robinson EM, Zhou J, Duggan C, Gura KM. Parenteral Fish Oil Improves Outcomes in Patients With Parenteral Nutrition-Associated Liver Injury. Annals of surgery. 2009 doi: 10.1097/SLA.0b013e3181b36657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strijbosch RA, Lee S, Arsenault DA, Andersson C, Gura KM, Bistrian BR, Puder M. Fish oil prevents essential fatty acid deficiency and enhances growth: clinical and biochemical implications. Metabolism. 2008;57(5):698–707. doi: 10.1016/j.metabol.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alwayn IP, Javid PJ, Gura KM, Nose V, Ollero M, Puder M. Do polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREPB-1 suppression or by correcting essential fatty acid deficiency. Hepatology. 2004;39(4):1176–1177. doi: 10.1002/hep.20189. author reply 1177–1178. [DOI] [PubMed] [Google Scholar]
- 9.de Meijer VE, Gura KM, Le HD, Meisel JA, Puder M. Fish Oil-Based Lipid Emulsions Prevent and Reverse Parenteral Nutrition-Associated Liver Disease: The Boston Experience. Jpen. 2009 doi: 10.1177/0148607109332773. [DOI] [PubMed] [Google Scholar]
- 10.Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. The American journal of gastroenterology. 1999;94(9):2467–2474. doi: 10.1111/j.1572-0241.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 11.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. The Journal of biological chemistry. 1957;226(1):497–509. [PubMed] [Google Scholar]
- 12.Lukovac S, Los EL, Stellaard F, Rings EH, Verkade HJ. Effects of essential fatty acid deficiency on enterohepatic circulation of bile salts in mice. American journal of physiology. 2009 doi: 10.1152/ajpgi.00091.2009. [DOI] [PubMed] [Google Scholar]
- 13.Saether T, Tran TN, Rootwelt H, Grav HJ, Christophersen BO, Haugen TB. Essential fatty acid deficiency induces fatty acid desaturase expression in rat epididymis, but not in testis. Reproduction (Cambridge, England) 2007;133(2):467–477. doi: 10.1530/REP-06-00294. [DOI] [PubMed] [Google Scholar]
- 14.Aldamiz-Echevarria L, Vallo A, Aguirre M, Sanjurjo P, Gonzalez-Lamuno D, Elorz J, Prieto JA, Andrade F, Rodriguez-Soriano J. Essential fatty acid deficiency profile in patients with nephrotic-range proteinuria. Pediatric nephrology (Berlin, Germany) 2007;22(4):533–540. doi: 10.1007/s00467-006-0366-1. [DOI] [PubMed] [Google Scholar]
- 15.Tsurumi K, Fujimura H. Effect of anti-inflammatory drugs on endotoxin-induced diarrhea in mice. Jpn J Pharmacol. 1983;33(1):165–173. doi: 10.1254/jjp.33.165. [DOI] [PubMed] [Google Scholar]
- 16.Myers A, Papadopoulos A, O’Day D, Ramey E, Ramwell P, Penhos J. Sexual differentiation of arachidonate toxicity in mice. J Pharmacol Exp Ther. 1982;222(2):315–318. [PubMed] [Google Scholar]
- 17.Burr GO, Burr MM. A new deficiency disease produced by the rigid exclusion of fat from the diet. The Journal of Biological Chemistry. 1929;82(2):23. doi: 10.1111/j.1753-4887.1973.tb06008.x. [DOI] [PubMed] [Google Scholar]
- 18.Alfin-Slater RB, Aftergood L. Essential fatty acids reinvestigated. Physiol Rev. 1968;48(4):758–784. doi: 10.1152/physrev.1968.48.4.758. [DOI] [PubMed] [Google Scholar]
- 19.Calder PC. Dietary modification of inflammation with lipids. Proc Nutr Soc. 2002;61(3):345–358. doi: 10.1079/pns2002166. [DOI] [PubMed] [Google Scholar]
- 20.Ling PR, Boyce P, Bistrian BR. Role of arachidonic acid in the regulation of the inflammatory response in TNF-alpha-treated rats. JPEN J Parenter Enteral Nutr. 1998;22(5):268–275. doi: 10.1177/0148607198022005268. [DOI] [PubMed] [Google Scholar]
- 21.Jensen CL, Prager TC, Fraley JK, Chen H, Anderson RE, Heird WC. Effect of dietary linoleic/alpha-linolenic acid ratio on growth and visual function of term infants. The Journal of pediatrics. 1997;131(2):200–209. doi: 10.1016/s0022-3476(97)70154-9. [DOI] [PubMed] [Google Scholar]
- 22.Wainwright PE, Jalali E, Mutsaers LM, Bell R, Cvitkovic S. An imbalance of dietary essential fatty acids retards behavioral development in mice. Physiology & behavior. 1999;66(5):833–839. doi: 10.1016/s0031-9384(99)00028-1. [DOI] [PubMed] [Google Scholar]
- 23.Wainwright PE, Xing HC, Mutsaers L, McCutcheon D, Kyle D. Arachidonic acid offsets the effects on mouse brain and behavior of a diet with a low (n-6):(n-3) ratio and very high levels of docosahexaenoic acid. The Journal of nutrition. 1997;127(1):184–193. doi: 10.1093/jn/127.1.184. [DOI] [PubMed] [Google Scholar]
- 24.Brossard N, Croset M, Pachiaudi C, Riou JP, Tayot JL, Lagarde M. Retroconversion and metabolism of [13C]22:6n-3 in humans and rats after intake of a single dose of [13C]22:6n-3-triacylglycerols. The American journal of clinical nutrition. 1996;64(4):577–586. doi: 10.1093/ajcn/64.4.577. [DOI] [PubMed] [Google Scholar]
- 25.Hempenius RA, Van Delft JM, Prinsen M, Lina BA. Preliminary safety assessment of an arachidonic acid-enriched oil derived from Mortierella alpina: summary of toxicological data. Food Chem Toxicol. 1997;35(6):573–581. doi: 10.1016/s0278-6915(97)00025-2. [DOI] [PubMed] [Google Scholar]
- 26.Javid PJ, Greene AK, Garza J, Gura K, Alwayn IP, Voss S, Nose V, Satchi-Fainaro R, Zausche B, Mulkern RV, et al. The route of lipid administration affects parenteral nutrition-induced hepatic steatosis in a mouse model. J Pediatr Surg. 2005;40(9):1446–1453. doi: 10.1016/j.jpedsurg.2005.05.045. [DOI] [PubMed] [Google Scholar]
- 27.Wilson MD, Blake WL, Salati LM, Clarke SD. Potency of polyunsaturated and saturated fats as short-term inhibitors of hepatic lipogenesis in rats. The Journal of nutrition. 1990;120(6):544–552. doi: 10.1093/jn/120.6.544. [DOI] [PubMed] [Google Scholar]
- 28.Attie AD, Krauss RM, Gray-Keller MP, Brownlie A, Miyazaki M, Kastelein JJ, Lusis AJ, Stalenhoef AF, Stoehr JP, Hayden MR, et al. Relationship between stearoyl-CoA desaturase activity and plasma triglycerides in human and mouse hypertriglyceridemia. Journal of lipid research. 2002;43(11):1899–1907. doi: 10.1194/jlr.m200189-jlr200. [DOI] [PubMed] [Google Scholar]