

Abstract
MK-7246, an antagonist of the chemoattractant receptor on T helper type 2 (Th2) cells, is being developed for the treatment of respiratory diseases. In a first-in-human study, we investigated whether genetic polymorphisms contributed to the marked intersubject variability in the pharmacokinetics of MK-7246 and its glucuronide metabolite M3. Results from in vitro enzyme kinetic studies suggested that UGT2B17 is probably the major enzyme responsible for MK-7246 metabolism in both the liver and the intestine. As compared with those with the UGT2B17*1/*1 wild-type genotype, UGT2B17*2/*2 carriers, who possess no UGT2B17 protein, had 25- and 82-fold greater mean dose-normalized values of area under the plasma concentration–time curve (AUC) and peak concentration of MK-7246, respectively, and a 24-fold lower M3-to-MK-7246 AUC ratio. The apparent half-life of MK-7246 was not as variable between these two genotypes. Therefore, the highly variable pharmacokinetics of MK-7246 is attributable primarily to the impact of UGT2B17 genetic polymorphisms and extensive first-pass metabolism of MK-7246.
MK-7246, an antagonist of a chemoattractant receptor on T helper type 2 (Th2) cells, is being developed for the treatment of respiratory diseases.1,2 The metabolism of MK-7246 was studied in both preclinical species and in hepatocytes. After oral administration of [3H]MK-7246 in bile-duct-cannulated rats, ~84% of the administered radioactivity was recovered, with 77% excreted in the bile. Biotransformation was the major route of elimination of MK-7246. Less than 3% of the radioactive dose was excreted as unchanged parent drug in rat bile, and the acyl glucuronide M3 accounted for ~90% of the radioactivity in the bile. These data suggest that direct glucuronidation is the major elimination route for MK-7246 in rats. In human hepatocytes, M3 was the major metabolite observed; minor metabolites included an oxidative metabolite and a glucuronide of an oxidative metabolite. The metabolites observed in rat hepatocytes were qualitatively similar to those observed in human hepatocytes; M3 was also the major metabolite observed in rat hepatocytes (Merck, unpublished data, data on file). Taken together, these data suggest that glucuronidation is probably the major metabolic pathway in humans.
Uridine 5′-diphosphate-glucuronosyltransferases (UGTs) catalyze glucuronidation of various endogenous substances and xenobiotics. The UGT family of phase II enzymes consists of the subfamilies UGT1A, UGT2A, UGT2B, UGT3A, and UGT8. Some of the UGT genes are highly polymorphic and are associated with interindividual variability in pharmacokinetics of certain therapeutic agents.3 For example, patients with the UGT1A1*28 mutation were associated with a 30–50% lower glucuronidation ratio of SN-38 glucuronide to SN-38, an active metabolite of irinotecan; the ratio is correlated with irinotecan-induced diarrhea.4,5,6 Oral clearance of zidovudine was shown to be approximately twofold higher in UGT2B7*1c carriers than in noncarriers.7 The UGT2B15 D85Y polymorphism has been identified as a major determinant of oxazepam clearance; median oxazepam oral clearance was ~50% lower in subjects with 85YY than in those with 85DD.8 Overall, the effects of genetic polymorphisms of UGT enzymes on the pharmacokinetics of drugs have generally been found to be less than twofold.
The first-in-human study of the pharmacokinetics of MK-7246 showed high variability. The objective of this study was to identify the UGT responsible for MK-7246 glucuronidation and to investigate whether UGT genetic polymorphisms are responsible for the large variability observed in MK-7246 pharmacokinetics.
Results
Variability of MK-7246 pharmacokinetics as observed in the first-in-human study
In response to oral administration of single doses of MK-7246, large intersubject variability in MK-7246 plasma concentrations was observed; the coefficient of variation (CV) of area under the plasma concentration–time curve from time 0 to infinity (AUC0→∞) of MK-7246 ranged from 74 to 177%. At doses <20 mg, only two subjects had detectable concentrations of MK-7246 in plasma samples collected at 24 h after administration. Individual plasma concentration–time profiles of MK-7246 after a single 40-mg dose are shown in Figure 1a,b. Study subjects could be categorized into high- and low-exposure groups based on their MK-7246 plasma concentrations, and intersubject variability within these groups was <20%. Plasma drug concentrations of subjects in the high- and low-exposure groups remained high and low, respectively, throughout the entire dose range tested. The values of the M3-to-MK-7246 AUC ratio were ~0.5 and ~12 in subjects with high and low MK-7246 plasma concentrations, respectively.
Figure 1.

Plasma concentration–time profiles of MK-7246 after administration of a single 40-mg dose of MK-7246 in the fasted state in individuals in panels (a) A and (b) B of the first-in-human study. Eight subjects, including two who received the placebo, participated in each panel. Each symbol and line represents a separate individual who received MK-7246 within each panel.
MK-7246 glucuronidation
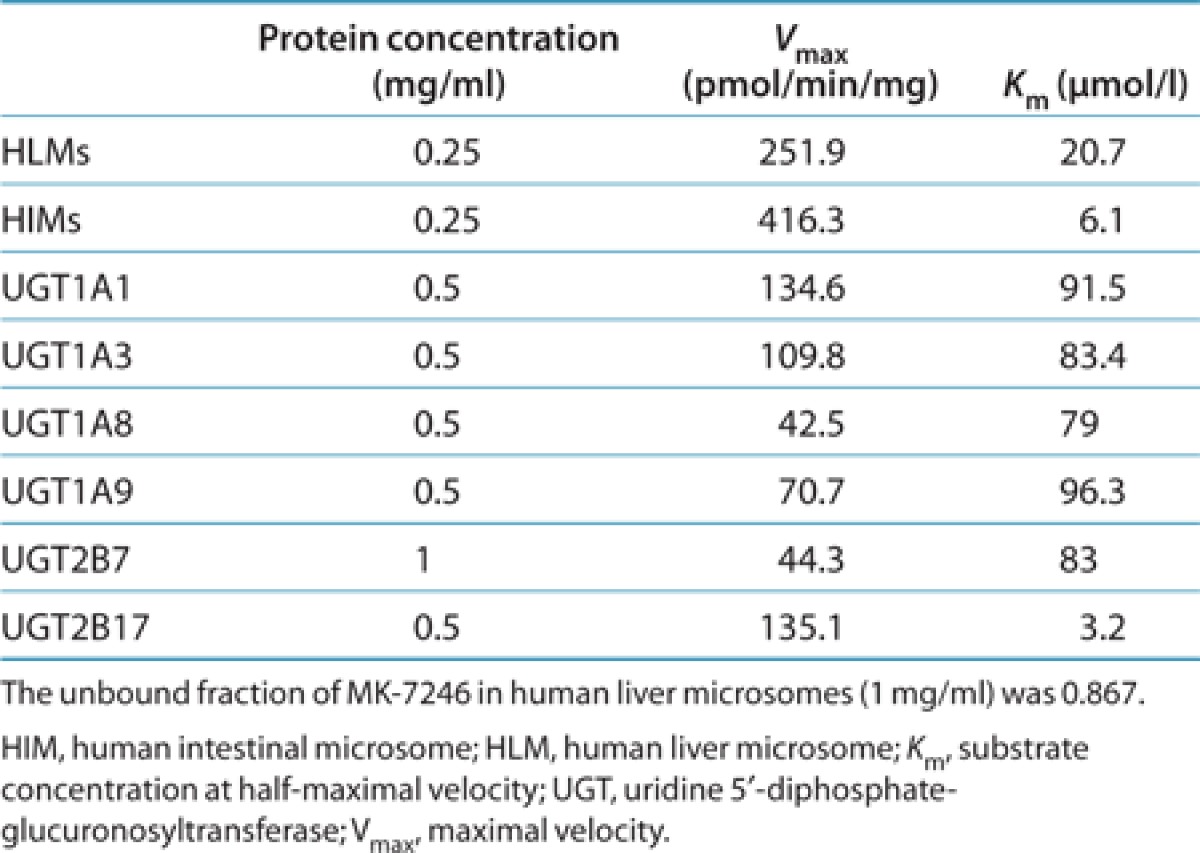
The apparent difference in metabolite-to-parent AUC ratio between the low- and high-exposure groups indicated that the pharmacokinetic variability was probably related to glucuronidation of MK-7246. To determine which of the UGTs were capable of metabolizing MK-7246 to M3, we screened 12 recombinant human UGTs for MK-7246 glucuronidation activity at 5 and 50 µmol/l MK-7246. All UGTs were capable of forming the acyl glucuronide M3 (Figure 2). Among the 12 enzymes, UGT1A4, UGT1A6, UGT1A7, UGT1A10, UGT2B4, and UGT2B15 were the least active, and UGT1A1, UGT1A3, UGT2B17, UGT1A8, UGT1A9, and UGT2B7 were the most active, based on the peak area ratios. Subsequently, enzyme kinetics studies were performed for the six most active isoforms. MK-7246 glucuronidation by the six isoforms, human liver microsomes (HLMs), and human intestinal microsomes (HIMs) exhibited hyperbolic Michaelis–Menten kinetics. The kinetic parameters substrate concentration at half-maximal velocity (Km) and maximal velocity (Vmax) are summarized in Table 1. Recombinant UGT2B17 exhibited high affinity for MK-7246, with a Km value of 3.2 µmol/l, which was comparable to the Km value generated in HIMs. Km values of other recombinant UGTs were ~25- to 30-fold greater as compared with recombinant UGT2B17. In addition, recombinant UGT2B17 was also the only enzyme that had a lower Km value for MK-7246 than the value generated in HLMs. The Km values of other recombinant UGTs were more than fourfold greater than the value generated in HLMs.
Figure 2.

Formation of acyl glucuronide M3 by recombinant human UGTs. MK-7246 at 5 and 50 µmol/l MK-7246 were incubated with UGTs in the presence of UDPGA for 90 min. Each bar and error bar represents the mean and standard deviation, respectively, of determinations carried out in triplicate. HLM, human liver microsome; UDPGA, uridine 5′-diphospho-glucuronic acid; UGT, uridine 5′-diphosphate-glucuronosyltransferase.
Table 1. Kinetic parameters for MK-7246 glucuronidation by human liver microsomes, human intestinal microsomes, and recombinant UGTs.
Effect of UGT2B17 polymorphism on the pharmacokinetics of MK-7246 and M3
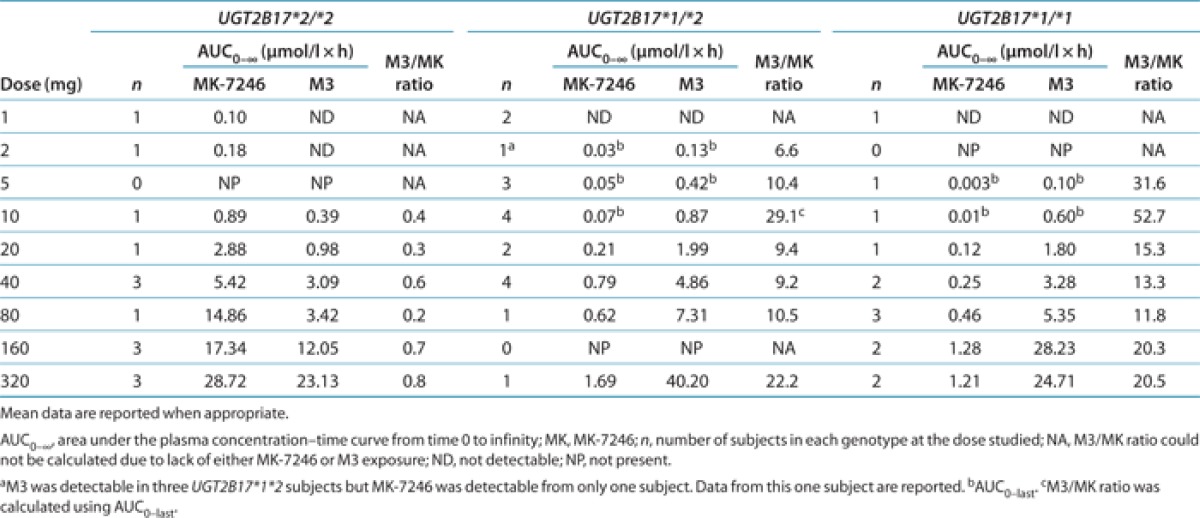
The results of the in vitro glucuronidation studies indicate that UGT2B17 could be the major UGT responsible for acyl glucuronidation of MK-7246, in both the liver and the intestine. To investigate whether UGT2B17 genetic polymorphisms are associated with the high variability observed in MK-7246 pharmacokinetics, we conducted a genetic analysis. Of the 16 subjects who participated in the first-in-human study, 13 consented to be genotyped. These subjects were allocated to one of the three groups according to their UGT2B17 genotypes (Table 2). There was no statistically significant difference in mean age, height, or weight between subjects with at least one UGT2B17*1 allele and subjects with the UGT2B17*2/*2 genotype (recessive model; P < 0.2). The effects of UGT2B17 polymorphisms on the exposure levels of MK-7246 and M3 at different doses are shown in Table 3. As compared with subjects with at least one UGT2B17*1 allele, those with the UGT2B17*2/*2 genotype had much higher MK-7246 concentrations. In response to administration of doses ranging from 1 to 160 mg, the plasma concentrations of M3 were generally lower in the subjects with the UGT2B17*2/*2 genotype than in those with at least one UGT2B17*1 allele. M3 concentration levels were below the limit of detection in subjects with the UGT2B17*2/*2 genotype after administration of doses <10 mg. In addition, the M3-to-MK-7246 AUC ratios in subjects with the UGT2B17*2/*2 genotype were consistently <1 throughout the entire dose range.
Table 2. Demographics of all study subjects and subjects genotyped for UGT2B17 genotype.
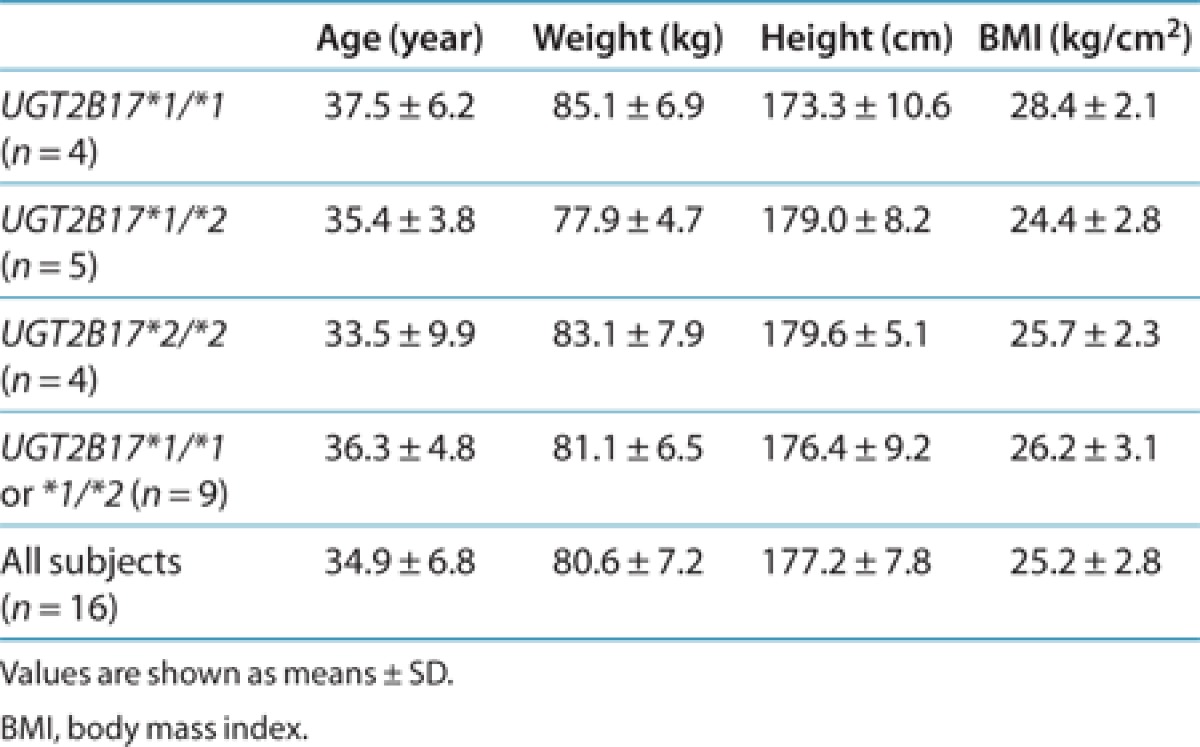
Table 3. Mean MK-7246 and M3 exposure following single doses of MK-7246 in relation to UGT2B17 genotype.
The 13 subjects who were genotyped participated in the first-in-human study and received different dose levels. To compare the effects of UGT2B17 polymorphisms among genotype groups, the plasma concentrations and pharmacokinetic parameters of MK-7246 and M3 were normalized to the 40-mg dose for nine subjects, to the 20-mg dose for two subjects, and to the 80-mg dose for two subjects. The mean dose-normalized AUC0-∞ of MK-7246 was 25-fold greater in subjects with the UGT2B17*2/*2 genotype than in those with the UGT2B17*1/*1 genotype and 10-fold higher than in heterozygous subjects (Figure 3, Table 4). The intersubject variability in dose-normalized AUC0-∞ of MK-7246 within the heterozygous genotype group was high, with a CV of 93%; the CVs with respect to other genotype groups were <20%. The mean dose-normalized peak concentration in plasma (Cmax) of MK-7246 was ~82-fold greater in subjects with the UGT2B17*2/*2 genotype than in those with the UGT2B17*1/*1 genotype and ~19-fold greater than in heterozygous subjects. The intersubject variability in dose-normalized Cmax of MK-7246 within the heterozygous genotype group was high, with a CV of 103%; the CVs with respect to other genotype groups were <35%. The effects of the UGT2B17 genotype on time to Cmax (tmax) and terminal half-life (t1/2) of MK-7246 were less dramatic. There was a tendency toward shorter tmax and t1/2 in subjects with the UGT2B17*2/*2 genotype. The tmax and t1/2 of MK-7246 varied in the range of ~2- and ~2.5-fold between individual subjects within each of the three genotype groups.
Figure 3.

Dose-normalized individual (N = 13) and mean plasma concentration–time profiles of MK-7246 and its acyl glucuronide metabolite, M3, in UGT2B17 genotype groups of healthy volunteers after a single dose of MK-7246.
Table 4. Dose-normalized pharmacokinetic variables of MK-7246 in 13 healthy subjects in relation to UGT2B17 genotype.
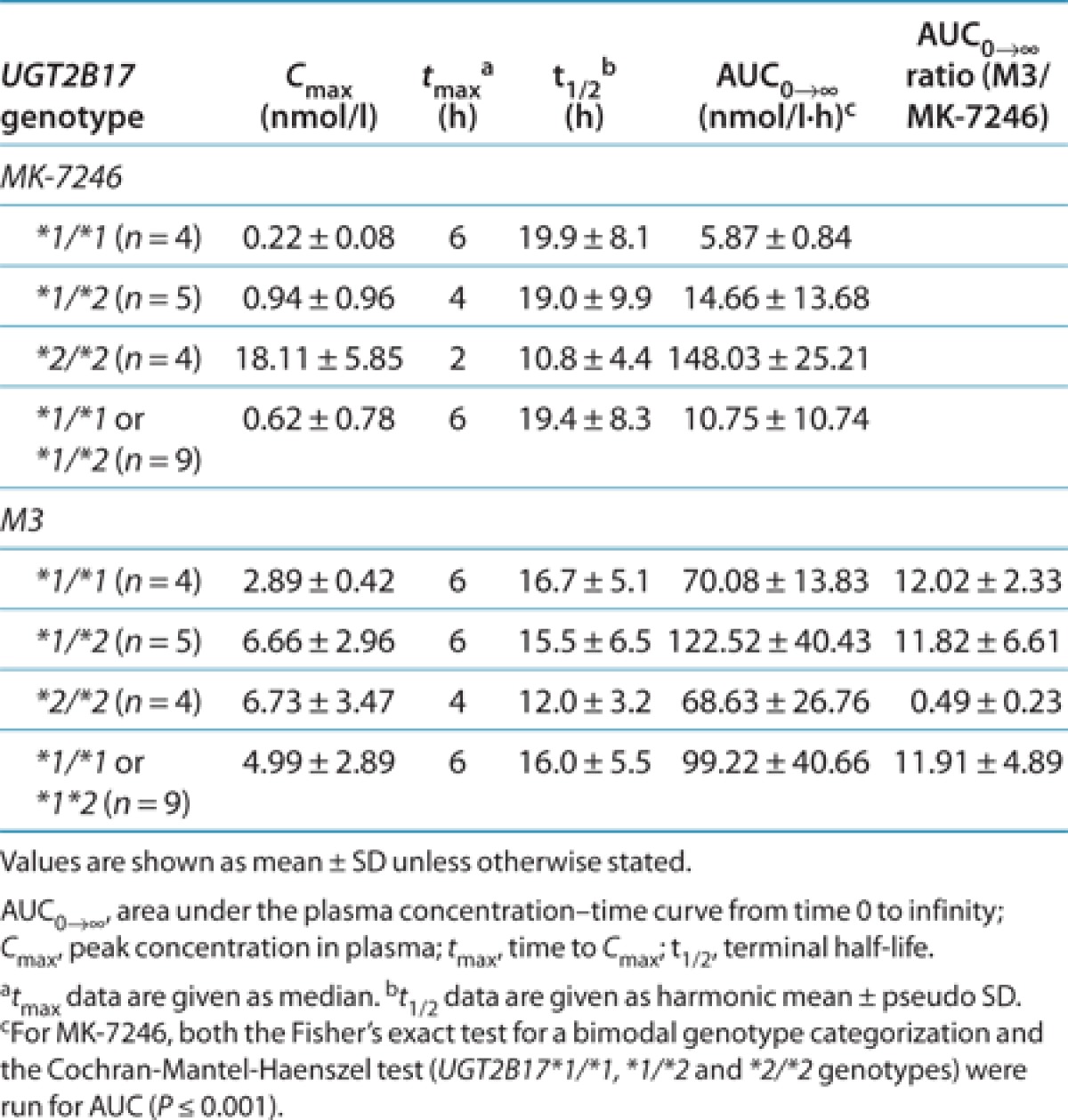
The associations between UGT2B17 genotypes and the pharmacokinetics of M3 at the dose levels studied were not statistically significant (Figure 3; Table 4). Although greater AUC0-∞ values were observed in heterozygous subjects, the mean dose-normalized AUC0-∞ of the acyl glucuronide M3 was similar in subjects with the UGT2B17*2/*2 genotype and those with the UGT2B17*1/*1 genotype. The mean Cmax of M3 was lower in subjects with the UGT2B17*1/*1 genotype than in those in other genotype groups. The dose-normalized AUC0-∞ and Cmax of M3 were less variable in subjects with the UGT2B17*1/*1 genotype as compared with subjects in other genotype groups. The CVs of AUC0–∞ of M3 in the UGT2B17*1/*1, UGT2B17*1/*2, and UGT2B17*2/*2 genotype groups were 20, 44, and 39%, respectively, while the corresponding values of CVs with respect to Cmax were 15, 33, and 52%, respectively. The mean t1/2 of M3 in all genotypes was similar to the corresponding mean t1/2 of MK-7246. The median tmax of M3 was delayed relative to the median tmax of MK-7246 in subjects with the UGT2B17*1/*1 and heterozygous genotypes.
Discussion
The present study shows that the UGT2B17 enzyme is the major UGT enzyme responsible for the glucuronidation of MK-7246. UGT2B17 genetic polymorphism is associated with the highly variable pharmacokinetics observed in the MK-7246 first-in-human study. This study is the first to report that genetic polymorphisms of a UGT enzyme can have such a profound effect on the pharmacokinetics of a compound in drug development.
The UGT2B17 enzyme is involved in androgen metabolism and is capable of metabolizing a variety of xenobiotics including several coumarins, anthraquinones, and flavonoids.9 UGT2B17 messenger RNA (mRNA) is expressed in both the liver and the intestine.10 There are two common alleles of the UGT2B17 gene. The UGT2B17*1 allele is considered the wild-type allele. The UGT2B17*2 allele has a deletion of 150 kb spanning the entire UGT2B17 gene11 and resulting in complete absence of UGT2B17 protein. The frequency of the UGT2B17*2/*2 genotype varies significantly among ethnic groups. For example, the frequency of the UGT2B17*2/*2 genotype was reported to be ~9–15% in Caucasians,12 67% in a Korean population,13 and >80% in a Japanese population.14 The UGT2B17*2/*2 genotype is strongly associated with urinary testosterone levels; the urinary testosterone/epitestosterone ratio was ~16-fold lower in subjects with the UGT2B17*2/*2 genotype than in those with the UGT2B17*1/*1 genotype.15
In the MK-7246 first-in-human study, a 20-fold difference in mean exposure of MK-7246 was observed between subjects carrying at least one UGT2B17*1 allele and those with the UGT2B17*2/*2 genotype. This is probably attributable to the complete absence of UGT2B17 enzyme activity in the subjects with the UGT2B17*2/*2 genotype. In subjects with this genotype, Cmax values of MK-7246 were ~80-fold greater than in subjects with at least one UGT2B17*1 allele, but the values of apparent t1/2 were similar. This indicates that the UGT2B17*2 polymorphism affects mainly first-pass metabolism of MK-7246. These findings may be due to potentially high expression of UGT2B17 in the intestine relative to other UGTs. A recent study showed that UGT2B17 mRNA expression in the intestine was ~13-fold higher than in the liver and that UGT2B17 mRNA had the highest expression in the intestine among all UGT mRNAs.10 In addition, UGT2B17 mRNA was not highly expressed in the liver.10 Furthermore, MK-7246 is a specific substrate for UGT2B17, as shown by in vitro UGT studies. The high affinity of MK-7246 for UGT2B17 relative to the other UGTs tested, and the potentially high expression of UGT2B17 in the intestine, may result in extensive metabolism of MK-7246 in the intestine in subjects with at least one UGT2B17*1 allele, thereby contributing to the large difference in MK-7246 concentrations observed among the genotype groups.
Interestingly, the mean apparent t1/2 in subjects with at least one UGT2B17*1 allele tends to be longer than in subjects with the UGT2B17*2/*2 genotype. This finding appears to be inconsistent with the extensive metabolism of MK-7246 but may be explained by possible enterohepatic recirculation of MK-7246. For example, in subjects with at least one UGT2B17*1 allele, MK-7246 was extensively metabolized, thereby forming more acyl glucuronide M3; the conversion of M3 to MK-7246 in the intestine could lead to reabsorption of MK-7246, thereby prolonging the duration for which MK-7246 remains in the body. Extensive metabolism of MK-7246 in the intestine in these subjects may also contribute to the absence of multiple peaks (a signature of enterohepatic circulation) in their plasma concentration–time profiles. In subjects with the UGT2B17*2/*2 genotype, less M3 was formed because of lack of the UGT2B17 enzyme, and therefore less M3 was available for the recirculation process. Consequently, in subjects with the UGT2B17*2/*2 genotype, M3 appeared to have a shorter t1/2.
The effects of UGT2B17*2 polymorphism on M3 pharmacokinetics were less dramatic. In subjects with the UGT2B17*2/*2 genotype, M3 concentrations were undetectable for doses <10 mg. As the dose increased, M3 concentrations in these subjects increased, but the exposure levels remained at approximately half of those observed in subjects with at least one UGT2B17*1 allele. This may indicate that other UGT enzymes could be involved in MK-7246 glucuronidation; this is likely to occur when MK-7246 concentrations in the intestine and the liver exceed their Km values. For example, on the basis of its known solubility characteristics, we could assume that the 10-mg dose of MK-7246 was completely dissolved in 250 ml of fluid. Under these conditions, the concentration of MK-7246 in the intestine could reach 76.8 µmol/l, which would be close to the Km values of UGT1A8 and UGT2B7. At the 20-mg dose, MK-7246 concentration in the intestine could exceed the Km values of all six UGT isoenzymes that are capable of metabolizing MK-7246. It is difficult to estimate the extent to which these UGT enzymes are involved in MK-7246 glucuronidation because the protein expressions of these UGTs in the tissues and in the recombinant enzyme systems used in this study are not known. The difference in M3 concentrations between subjects with the UGT2B17*2/*2 genotype and those with at least one UGT2B17*1 allele was much less than the difference observed in MK-7246 concentrations (Table 4). This could be due to formation-limited elimination of M3, as indicated by the similar values of apparent t1/2 for M3 and MK-7246; M3 concentrations in plasma may not reflect the amount of M3 actually formed in these subjects because the metabolite was eliminated as soon as it was formed.
MK-7246 concentrations in the heterozygous subjects were similar to those in subjects with the UGT2B17*1/*1 genotype, suggesting that a gene-dose effect was not present. Similar observations were reported in a doping test study15 and a study that linked the UGT2B17 polymorphism with androgen expression in pubertal boys.16 The urinary testosterone glucuronide concentrations and urinary testosterone/epitestosterone ratios in the heterozygous subjects were similar to those in subjects with the UGT2B17*1/*1 genotype.15,16 Consistent with these findings, HLMs taken from heterozygous subjects had similar relative UGT2B17 mRNA expression17 and testosterone glucuronidation activity13 as compared with microsomes taken from subjects with the UGT2B17*1/*1 genotype. In our study, higher interindividual variability was observed within the heterozygous subjects as compared with other genotype groups. Two subjects had relatively higher MK-7246 concentrations than the rest of the subjects in the heterozygous genotype group. The reason for high variability in this group is not known. Other factors (such as the influence of genetic variants) may be involved in the elimination of MK-7246. However, the small sample size in this study hinders further investigation.
In summary, we report that genetic variations in the UGT2B17 gene dramatically affect the pharmacokinetics of MK-7246 in healthy subjects. We found that the large variability in MK-7246 plasma concentration–time profiles is probably due to the extensive first-pass metabolism in subjects with at least one UGT2B17*1 allele and the complete absence of the UGT2B17 enzyme in subjects homozygous for the UGT2B17*2 allele. In recent years, substantial efforts have been made in reducing the risk of drug–drug interactions related to cytochrome P450 enzymes and variability caused by polymorphic expression of cytochrome P450 enzymes (e.g., CYP2D6). According to study reports, drug interactions and genetic polymorphisms of UGT enzymes generally cause less than twofold alterations in the pharmacokinetic parameters of drugs18; this is probably attributable to the overlapping substrate specificity of UGT enzymes. As a result, there is currently a move toward developing drugs that are cleared through alternative pathways such as glucuronidation. Our study results demonstrate that marked intersubject variability in healthy subjects could occur when a compound is primarily metabolized by glucuronidation, especially when the compound is a specific substrate of UGT2B17. The effects of UGT2B17 polymorphisms on the pharmacokinetics of MK-7246 suggest that the potential effects of a potent inhibitor of UGT2B17 on a concomitant specific UGT2B17 substrate cannot be ignored. Therefore, caution should be exercised when a compound is primarily eliminated by glucuronidation, and UGT phenotyping should be performed to rule out potentially serious drug interactions or large variability in pharmacokinetics.
Methods
Clinical study design. This first-in-human study was a double-blind, randomized, placebo-controlled, serial-panel, rising-single-oral-dose study in healthy male subjects. The study was designed to evaluate the safety, pharmacokinetics, and pharmacodynamics of MK-7246. The study and the subsequent pharmacogenetic sample collection were approved by the institutional review board of Ziekenhuis Netwerk Antwerpen (Antwerp, Belgium). The subjects gave informed consent separately for the study and for pharmacogenetic sample collection. All procedures were performed in accordance with the principles of the Declaration of Helsinki. The study consisted of two panels (A and B); each panel had eight subjects. All 16 subjects reported their ethnicity as Caucasian. Each subject received single rising doses of 1, 2, 5, 10, 20, and 40 mg (panel A) or 40 (with or without food), 80, 160, and 320 mg (panel B) of MK-7246 or a matching placebo. All doses were administered after an 8-h overnight fast, except that subjects in panel B who received 40 mg in the fasted state were administered the 40-mg dose again later during the study after a standard high-fat meal. During each treatment period (or dose) in each panel, six subjects received MK-7246 and two subjects received placebo in a randomized, balanced, blinded fashion. After administration of MK-7246, there was a minimum washout period of 7 days before readministration of MK-7246 in the subsequent period, for any individual subject. Dosing in panel B did not commence until completion of dosing in panel A. Pharmacokinetic sampling was collected immediately before dosing and at 0.5, 1, 2, 4, 6, 8, 12, 16, 24, 48, and 72 h after the dose.
Determination of MK-7246 and M3 concentrations in plasma. An analytical method for the simultaneous determination of MK-7246 (definitive assay) and acyl glucuronide (M3) metabolite (exploratory assay) in human plasma was developed and validated over the concentration ranges of 0.5–500 ng/ml and 5.0–500 ng/ml, respectively. The analytes were isolated through protein precipitation extraction in a 96-well format with a 100 µl acidified plasma sample fortified with 50 µl of stable isotope-labeled internal standard. An aliquot of the supernatant was analyzed using reverse-phase high-performance liquid chromatography (Acquity UPLC; Waters, Milford, MA) with tandem mass spectrometric (API4000; Applied Biosystems Sciex, Concord, Ontario, Canada) detection fitted with a turbo ionspray interface. Multiple-reaction monitoring of the precursor to product ion pairs was used for quantification. The mean correlation coefficients (R2) were 0.9972 for MK-7246 and 0.9964 for M3. The intraday and interday coefficients of variation were ≤7.0% for MK-7246 and for M3 across all standard concentrations.
Pharmacokinetics. The pharmacokinetics of MK-7246 and its metabolite M3 were estimated by means of noncompartmental analysis using WinNonlin 5.0.1 (Pharsight, Mountain View, CA). Concentrations of the two analytes in plasma and nominal sampling times were used to estimate pharmacokinetic parameters in each subject. AUC0–∞ was calculated using the linear trapezoidal method for ascending concentrations and the log trapezoidal method for descending concentrations. The extrapolated portion of the AUC from the last time point of sample collection (tlast) to infinity was determined by dividing the observed concentration at tlast (Clast) by the terminal rate constant (k). AUC0–∞ was then calculated from the sum of the AUC through the last measured time point (AUClast) and the extrapolated area. The value of k was determined from the slope of a least-squares fit to the terminal phase of the log-transformed concentration–time curve. The t1/2 was then calculated from the equation t1/2 = ln (2)/k. The Cmax and tmax were obtained by inspection of the plasma concentration–time data.
Reagents. MK-7246 and its acyl glucuronide metabolite were synthesized by Merck Research Laboratories (Rahway, NJ). Baculovirus-expressed human UGTs 1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B15, and 2B17, and control supersomes, and pooled HLMs were obtained from BD Gentest (Woburn, MA). Pooled HIMs were purchased from XenoTech (Lenexa, KS).
Quantification of the acyl glucuronide M3. To determine the UGT isoforms capable of metabolizing MK-7246 to M3, the formation of M3 was measured by incubating HLMs and UGT supersomes (1 mg/ml) in 50 mmol/l phosphate buffer (pH 7.4) with 1 mmol/l magnesium chloride, alamethicin (0.025 mg/ml), D-saccharic acid-1,4-lactone (2.5 mmol/l), and MK-7246 (5 or 50 µmol/l). The reactions were initiated by adding 4 mmol/l uridine 5′-diphosphate-glucuronic acid and were then terminated after 90 min at 37 °C by adding two volumes of acetonitrile containing internal standard (labetalol) and 0.1% formic acid. The samples were centrifuged and analyzed using liquid chromatography–tandem mass spectrometry. High-performance liquid chromatography separation was performed on a Fluophase PFP column (50 × 3 mm, 5 µm; Thermo-Electron, Waltham, MA) using aqueous 0.1% formic acid and acetonitrile containing 0.1% formic acid as the mobile phase and operated at a flow rate of 0.5 ml/min for 9.5 min. Tandem mass spectrometry analysis was performed on a TSQ Quantum mass spectrometer (Thermo-Electron) using electrospray ionization in the positive ion mode. The transitions monitored were: m/z 417.1 → 228.1 for MK-7246, m/z 593.0 → 371.0 for MK-7246 glucuronide, and m/z 329.0 → 162.0 for labetalol. The range of quantification was 0.05–10 µmol/l. The relative amounts of M3 formation were assessed on the basis of the peak area ratios relative to the internal standard.
Enzyme kinetics for MK-7246 glucuronidation. MK-7246 glucuronidation kinetics with HLMs, HIMs, and recombinant human UGT1A1, 1A3, 1A8, 1A9, 2B7, and 2B17 were performed over a range of MK-7246 concentrations (0.5–400 µmol/l) using conditions similar to those described above except that linear conditions were used with respect to protein concentrations (Table 1) and incubation times. The reactions were terminated after 30 min of incubation. For determining the kinetic parameters, MK-7246 concentration and velocity data for MK-7246 glucuronidation were fitted to the standard Michaelis–Menten model to obtain the Vmax and Km. SigmaPlot version 10.0 (Systat Software, San Jose, CA) was used for the model fitting.
Genotyping. Clinical samples of DNA were extracted from 200 µl of ethylenediaminetetraacetic acid–anticoagulated blood using the Agencourt Genfind v2 DNA purification system (Beckman Coulter Genomics, Danvers, MA) in accordance with the manufacturer's instructions. Control HapMap DNA samples with known UGT2B17 copy-number variation were purchased as purified DNA from the Coriell Institute for Medical Research. The subjects were genotyped for the UGT2B17 deletion using two TaqMan copy-number assays obtained from Applied Biosystems (Foster City, CA): Hs03185327 and Hs04282679. The results from both assays were concordant for all samples and gave correct genotypes for HapMap controls.
Statistical analysis. A recessive inheritance model was assumed, based on the genetics. The UGT2B17*1/*1 genotype and the UGT2B17*1/*2 genotype were combined (≥1 copy number) and compared with the recessive UGT2B17*2/*2 genotype (0 copy number). Fisher's exact test was used to examine the high vs. low pharmacokinetic responders by genotype. Sensitivity analyses were performed by examining the genotypes as belonging to three distinct categories, pooled into two groups to represent the recessive inheritance models. One-way analysis of variance models with genotype also confirmed the nonparametric tests when the pharmacokinetic end point was dichotomized into the categories of high and low responders.
Author Contributions
Y.-H.W. wrote the manuscript, designed research, and analyzed data. M.T. designed research and analyzed data. J.J.M. performed research and analyzed data. P.H.W. analyzed data. C.M. analyzed data. C.D.T. analyzed data. T.P. analyzed data. G.C.G. designed research. R.D. designed research. K.J.W. performed research and analyzed data. R.C.S. performed research. J.A.K. performed research and analyzed data. G.J.O. designed research. E.V. performed research. J.A.T. performed research and analyzed data. C.G. performed research and analyzed data. T.L. designed research and performed research. P.P. designed research and analyzed data. M.I. analyzed data. P.M.S. designed research. J.A.W. designed research. J.C.H. designed research.
Acknowledgments
The authors thank Ronda K. Rippley from Department of Pharmacokinetics, Pharmacodynamics, and Drug Metabolism for helpful discussions during the first-in-human study.
E.V. is an employee of SGS Life Science Services, which was contracted by Merck & Co. to conduct the clinical trial. All the other authors are or were employees of Merck & Co.
References
- Gallant M.et al. Discovery of MK-7246, a selective CRTH2 antagonist for the treatment of respiratory diseases Bioorg. Med. Chem. Lett 21288–293.2011 [DOI] [PubMed] [Google Scholar]
- Gervais F.G.et al. Pharmacological characterization of MK-7246, a potent and selective CRTH2 (chemoattractant receptor-homologous molecule expressed on T-helper type 2 cells) antagonist Mol. Pharmacol 7969–76.2011 [DOI] [PubMed] [Google Scholar]
- Maruo Y., Iwai M., Mori A., Sato H., &, Takeuchi Y. Polymorphism of UDP-glucuronosyltransferase and drug metabolism. Curr. Drug Metab. 2005;6:91–99. doi: 10.2174/1389200053586064. [DOI] [PubMed] [Google Scholar]
- Ando Y., Saka H., Asai G., Sugiura S., Shimokata K., &, Kamataki T. UGT1A1 genotypes and glucuronidation of SN-38, the active metabolite of irinotecan. Ann. Oncol. 1998;9:845–847. doi: 10.1023/a:1008438109725. [DOI] [PubMed] [Google Scholar]
- Ando Y., Ueoka H., Sugiyama T., Ichiki M., Shimokata K., &, Hasegawa Y. Polymorphisms of UDP-glucuronosyltransferase and pharmacokinetics of irinotecan. Ther. Drug Monit. 2002;24:111–116. doi: 10.1097/00007691-200202000-00018. [DOI] [PubMed] [Google Scholar]
- Iyer L.et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity Pharmacogenomics J 243–47.2002 [DOI] [PubMed] [Google Scholar]
- Kwara A.et al. Interindividual variability in pharmacokinetics of generic nucleoside reverse transcriptase inhibitors in TB/HIV-coinfected Ghanaian patients: UGT2B7*1c is associated with faster zidovudine clearance and glucuronidation J. Clin. Pharmacol 491079–1090.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X.et al. Evidence for oxazepam as an in vivo probe of UGT2B15: oxazepam clearance is reduced by UGT2B15 D85Y polymorphism but unaffected by UGT2B17 deletion Br. J. Clin. Pharmacol 68721–730.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgeon D., Carrier J.S., Chouinard S., &, Bélanger A. Glucuronidation activity of the UGT2B17 enzyme toward xenobiotics. Drug Metab. Dispos. 2003;31:670–676. doi: 10.1124/dmd.31.5.670. [DOI] [PubMed] [Google Scholar]
- Ohno S., &, Nakajin S. Determination of mRNA expression of human UDP-glucuronosyltransferases and application for localization in various human tissues by real-time reverse transcriptase-polymerase chain reaction. Drug Metab. Dispos. 2009;37:32–40. doi: 10.1124/dmd.108.023598. [DOI] [PubMed] [Google Scholar]
- Wilson W. 3rd.et al. Characterization of a common deletion polymorphism of the UGT2B17 gene linked to UGT2B15 Genomics 84707–714.2004 [DOI] [PubMed] [Google Scholar]
- Park J.et al. Deletion polymorphism of UDP-glucuronosyltransferase 2B17 and risk of prostate cancer in African American and Caucasian men Cancer Epidemiol. Biomarkers Prev 151473–1478.2006 [DOI] [PubMed] [Google Scholar]
- Jakobsson J.et al. Large differences in testosterone excretion in Korean and Swedish men are strongly associated with a UDP-glucuronosyl transferase 2B17 polymorphism J. Clin. Endocrinol. Metab 91687–693.2006 [DOI] [PubMed] [Google Scholar]
- Terakura S.et al. A UGT2B17-positive donor is a risk factor for higher transplant-related mortality and lower survival after bone marrow transplantation Br. J. Haematol 129221–228.2005 [DOI] [PubMed] [Google Scholar]
- Schulze J.J., Lundmark J., Garle M., Skilving I., Ekström L., &, Rane A. Doping test results dependent on genotype of uridine diphospho-glucuronosyl transferase 2B17, the major enzyme for testosterone glucuronidation. J. Clin. Endocrinol. Metab. 2008;93:2500–2506. doi: 10.1210/jc.2008-0218. [DOI] [PubMed] [Google Scholar]
- Juul A.et al. A common deletion in the uridine diphosphate glucuronyltransferase (UGT) 2B17 gene is a strong determinant of androgen excretion in healthy pubertal boys J. Clin. Endocrinol. Metab 941005–1011.2009 [DOI] [PubMed] [Google Scholar]
- Balliet R.M., Chen G., Gallagher C.J., Dellinger R.W., Sun D., &, Lazarus P. Characterization of UGTs active against SAHA and association between SAHA glucuronidation activity phenotype with UGT genotype. Cancer Res. 2009;69:2981–2989. doi: 10.1158/0008-5472.CAN-08-4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J.A.et al. Drug-drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios Drug Metab. Dispos 321201–1208.2004 [DOI] [PubMed] [Google Scholar]