

Abstract
AIM: To clarify the biological role of stem cell factor (SCF)-mediated wild-type KIT receptor activation in gastrointestinal stromal tumor (GIST) growth.
METHODS: The co-expression of wild-type KIT receptor and SCF was evaluated in 51 GIST samples using mutation analysis and immunohistochemistry, and the results were correlated with clinicopathological parameters, including the mitotic count, proliferative index (Ki-67 immunohistochemical staining), mitotic index (phospho-histone H3 immunohistochemical staining) and apoptotic index (terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling). Using primary cultured GIST cells, the effect of SCF-mediated wild-type KIT receptor activation was determined by western blotting, methyl thiazolyl tetrazolium (MTT), and apoptosis assays.
RESULTS: We found that wild-type KIT receptor and SCF protein were expressed in 100% and 76.5% of the 51 GIST samples, respectively, and the co-expression of wild-type KIT receptor and SCF was associated with known indicators of poor prognosis, including larger tumor size (P = 0.0118), higher mitotic count (P = 0.0058), higher proliferative index (P = 0.0012), higher mitotic index (P = 0.0282), lower apoptosis index (P = 0.0484), and increased National Institutes of Health risk level (P = 0.0012). We also found that the introduction of exogenous SCF potently increased KIT kinase activity, stimulated cell proliferation (P < 0.01) and inhibited apoptosis (P < 0.01) induced by serum starvation, while a KIT immunoblocking antibody suppressed proliferation (P = 0.01) and promoted apoptosis (P < 0.01) in cultured GIST cells.
CONCLUSION: SCF-mediated wild-type KIT receptor activation plays an important role in GIST cell growth. The inhibition of SCF-mediated wild-type KIT receptor activation may prove to be particularly important for GIST therapy.
Keywords: Gastrointestinal stromal tumor, Stem cell factor, Wild-type KIT receptor, Cell growth, In vitro
INTRODUCTION
The KIT receptor, encoded by the oncogene c-kit[1], is characterised structurally by five immunoglobulin-like extracellular domains and an intracytoplasmic domain that contains an adenosine triphosphate (ATP)-binding domain and a phosphotransferase domain, which are separated by an interkinase sequence[2-5]. Under physiological conditions, the stem cell factor (SCF) binds to KIT and induces KIT homodimerisation, resulting in the phosphorylation of its tyrosine residues. The tyrosine-phosphorylated KIT receptor subsequently becomes a new docking site for signal transduction molecules and induces substrate binding and phosphorylation[6]. Thus, the interaction between SCF and the KIT receptor is essential for normal hematopoiesis, melanogenesis, gametogenesis, and the growth and differentiation of mast cells and interstitial cells of Cajal (ICCs)[7,8]. Notably, mutations in the c-kit gene have been implicated in neoplasms arising from these cell lineages. Oncogenic mutations in c-kit cause a constitutive phosphorylation of the KIT receptor that is independent of SCF binding, leading to a cascade of intracellular signalling events that contribute to the abnormal proliferation and survival of these neoplastic cells[9,10].
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal neoplasms of the gastrointestinal tract, and they are believed to originate from ICC progenitor cells[11-13]. It has also been noted that approximately 90% of GIST cases have activating mutations in either the c-kit or platelet-derived growth factor receptor (PDGFR) A genes[14-16]. In addition, the emerging role of SCF in c-kit-mutant GISTs indicates that an autocrine-paracrine loop serves as a further mechanism of wild-type KIT receptor activation[17,18].
In this study, we demonstrated the co-existence of wild-type KIT receptor and SCF in primary GISTs by analysing the entire coding sequence of c-kit and the protein expression of KIT and SCF in these tumors, as suggested in a previous study[19]. Based on ex vivo assays, we further demonstrated that SCF-mediated wild-type KIT receptor activation affected GIST growth in a dual manner by stimulating proliferation and inhibiting the apoptosis of GIST primary cells. These data suggest that the inhibition of SCF-mediated wild-type KIT receptor activation may be particularly important for GIST therapy.
MATERIALS AND METHODS
Patients
Samples from 51 consecutive patients with GISTs who underwent surgery at Changhai Hospital (Shanghai, China) between January and October 2006 were subjected to histological analysis. In addition, GIST primary cells were isolated from three fresh GIST specimens from patients who underwent surgery at Changhai Hospital in 2009. The GIST diagnosis was confirmed as previously described[20-22], and all tumors were KIT protein (CD117)-positive. No patients had received imatinib prior to the surgical resection of the tumor. Demographic data and clinical and histological features for all of the GISTs analysed in this study are summarised in Table 1.
Table 1.
Correlations between the co-expression of wild-type KIT receptor and stem cell factor and clinicopathological factors in gastrointestinal stromal tumors
|
Co-expression of wild-type KIT receptor and stem cell factor |
|||
| Absent (n = 12) | Present (n = 39) | P value | |
| Age (yr) | 58.9 ± 13.2 | 52.8 ± 10.5 | 0.0524 |
| Sex | |||
| Male | 6 | 18 | 0.8154 |
| Female | 6 | 21 | |
| Tumor size (cm) | 3.69 ± 1.68 | 6.21 ± 3.94 | 0.0118 |
| Histological phenotype | |||
| Spindle type | 8 | 26 | 0.7262 |
| Epithelioid/mixed type | 4 | 13 | |
| Cellularity | |||
| Sparse | 1 | 6 | 0.8000 |
| Moderate/dense | 11 | 33 | |
| Tumor location | |||
| Gastric | 6 | 25 | 0.5931 |
| Non-gastric | 6 | 14 | |
| Proliferative index | 2.92 ± 2.23 | 7.21 ± 4.93 | 0.0012 |
| pHH3 | |||
| ≤ 5 | 12 | 24 | 0.0282 |
| > 5 | 0 | 15 | |
| Mitotic counts | 3 ± 2.37 | 7.36 ± 6.36 | 0.0058 |
| ≤ 5 | 10 | 14 | 0.0004 |
| > 5 | 2 | 25 | |
| Apoptosis index | 44.58 ± 19.00 | 32.59 ± 17.63 | 0.0484 |
| c-kit mutation | |||
| Presence | 5 | 27 | 0.1659 |
| Absence | 7 | 12 | |
| National Institutes of Health risk group | |||
| Very low or low | 10 | 10 | 0.0012 |
| Intermediate or high | 2 | 29 | |
The use of all human tissues was approved by the hospital’s institutional committee for human research, and informed consent was obtained from all of the subjects.
Immunohistochemistry
Immunohistochemical staining was performed using the labelled streptavidin-biotin method (DAKO LSAB-2 Kit, Peroxidase, DAKO) according to the manufacturer’s instructions. The following primary antibodies were used: CD117 (DAKO), Ki-67 (DAKO), SCF (Cell Signaling Technology, Inc.) and phospho-histone H3 (pHH3, Cell Signaling Technology). Parallel sections were used to examine the co-expression of KIT and SCF. For Ki-67 and pHH3, positive cells were counted in five randomised regions in the tumor component of each lesion, and the labelling index was calculated as follows: Labelling index (%) = (positive cell number/total cell number) × 100%.
In situ apoptosis
In situ apoptosis was assessed by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling (TUNEL, Roche Diagnostics) staining, which was performed according to the manufacturer’s instructions. The apoptotic index was calculated as follows: Apoptotic index (%) = (apoptotic cell number/total cell number) × 100%.
Polymerase chain reaction amplification and sequencing
Genomic DNA was extracted from cryopreserved (n = 51) or fresh (n = 3) specimens using a commercial kit (BBI, Canada). Next, c-kit exons 9, 11, 13, 14 and 17, as well as PDGFRA exons 12 and 18, were amplified using the following primer sequences and annealing temperatures (designed): c-kit exon 9 (5’TTTATTTTCCTAGAGTAAGCCAGGG-3’ and 5’-ATCATGACTGATA TGGTAGACAGAGC-3’, at 56 °C), c-kit exon 11 (5’-ATTATTAAAAGGTGAT CTATTTTT-3’ and 5’-ACTGTTATGTGTACCCAAAAAG-3’, at 60 °C), c-kit exon 13 (5’-CACCATCACCACTTACTTGTTGTCT-3’ and 5’-GACAGACAAT AAAAGGCAGCTTGGAC-3’, at 67 °C), c-kit exon 14 (5’-TCTCACCTTC TTTCTAACCTTTTC-3’ and 5’-AACCCTTATGACCCCATGAA-3’, at 54 °C), c-kit exon 17 (5’GAACATCATTCAAGGCGTACTTTTG-3’ and 5’-TTGAAA CTAAAAATCCTTTGCAGGAC-3’, at 65 °C), PDGFRA exon 12 (5’-CTCTGGTGCACTGGGACTTT-3’ and 5’-GCAAGGGAAAAGGGAGTCT T-3’, at 60 °C), and PDGFRA exon 18 (5’-ATGGCTTGATCCTGAGTCATT-3’ and 5’-GTGTGGGAAGTGTGGACG-3’, at 60 °C). Gene mutations were analysed through the direct sequencing of uncloned polymerase chain reaction (PCR) fragments. Samples that appeared to contain mutations were further examined for the presence of the wild-type c-kit gene by subcloning the purified PCR products using a TA cloning vector system (Stratagene, CA). Five independent subclones from each PCR were sequenced.
Cell isolation and short-term primary cell culture
GIST primary cells were isolated and cultured as described in the literature with minor modifications[23]. The nonnecrotic tissue was separated from fresh GIST samples and finely minced with curved scissors. The minced tissue was homogenised by being passed through a 15-gauge needle with a syringe five times and subsequently being passed through a stainless steel mesh (200 wires/inch). The cells were counted with a haemocytometer, and cell viability was determined by propidium iodide staining.
After centrifugation in phosphate buffered solution (PBS) at 4 °C, cell pellets were resuspended in 1640 RPMI medium supplemented with 20% heat-inactivated foetal bovine serum (FBS), 1.0 mmol/L nonessential amino acids, 1.0 mmol/L sodium pyruvate, and 0.5 mmol/L 3-isobutyl-1- methylxanthine to inhibit fibroblast growth. Medium changes were performed at 24 h after seeding and every two or three days before cell analysis.
Enzyme-linked immunosorbent assay for stem cell factor production
For the measurement of SCF production, the concentration of GIST primary cells was adjusted to 5 × 106 cells/mL, and the supernatants were collected 24 h later. Samples were stored at -80 °C for further analysis. SCF concentration was determined by enzyme-linked immunosorbent assay (ELISA) using 96-well plates coated with catching antibody (diluted to 5 μg/mL in PBS, 100 μL/well) at 4 °C overnight according to the manufacturer’s instructions.
Western blotting
Protein extracts were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and incubated with a specific antibody to demonstrate protein loading. Anti-phospho-KIT (pY703) antibody was purchased from Abcam (United Kingdom). Glyceraldehyde-3-phosphate dehydrogenase mouse monoclonal antibody (Santa Cruz Biotechnology) was used as an internal control. To examine the effects of exogenous SCF on KIT phosphorylation, GIST primary cells were incubated overnight with medium containing 0.5% FBS. Cells were subsequently stimulated with various concentrations of SCF for 15 min, and phospho-KIT was examined as described above.
In vitro proliferation assay
Cell proliferation was determined using a methyl thiazolyl tetrazolium (MTT) assay (Roche, United States) according to the manufacturer’s instructions. In brief, GIST primary cells were seeded at a density of 8 × 103 cells/100 μL into 96-well plates and allowed to adhere. After 24 h, the original media were replaced with fresh media containing various concentrations of SCF (PeproTech, United States) or KIT immunoblocking antibody (Sigma, United States) with 0.5% FBS. After 48 h of incubation, MTT was added to each well. The quantity of the formazan product measured at 572 nm was directly proportional to the number of live cells in the culture.
Flow cytometric analysis of apoptosis
GIST primary cells were subjected to serum withdrawal for 12 h in the presence or absence of exogenous SCF or KIT immunoblocking antibody to induce apoptosis. Subsequently, apoptosis was detected using an annexin V-fluorescein isothiocyanate staining kit (R and D Systems). The apoptotic index was reported as the percentage of annexin V-positive cells in the early and late stages of apoptosis.
Statistical analysis
Data are expressed as the mean ± SD or the median and 25th and 75th percentiles [median (Q1, Q3)] for continuous variables and as percentages for categorical variables. Continuous variables were compared using Student’s t test or the Wilcoxon rank sum test for nonnormally distributed data. Correlations between categorical and continuous variables were assessed using the χ2 or Fisher’s exact test and t test, respectively. P values of less than 0.05 were considered to be significant. All analysis were performed with SPSS version 17.0 (SPSS, Chicago, IL).
RESULTS
Tumor genotypes
All 51 cryopreserved specimens were screened for mutations in the c-kit gene. Overall, 32 of 51 (62.7%) tumors harboured c-kit mutations. Among the GISTs with c-kit mutations, 27 (52.9%) had mutations in exon 11 of c-kit, five had mutations in exon 9 (Figure 1), and none had mutations in exon 13, 14 or 17. The amino acid changes observed in the 27 tumors with exon 11 mutations were as follows: deletion in 16 (59.3%) tumors, substitution in 7 (25.9%), both deletion and substitution in 2 (7.4%), insertion in 1 (3.7%), and duplication in 1 (3.7%). Five cases harboured exon 9 c-kit mutations, including in-frame insertions (3 tumors; 60.0%) and missense mutations (2 tumors; 40%). GISTs without c-kit mutations were further examined for PDGFRA mutations; only four (7.8%) tumors harboured substitution mutations in PDGFRA exon 18. All c-kit mutations were heterozygous, as indicated by the presence of a wild-type c-kit allele in the nucleotide sequence (Figure 1A).
Figure 1.

Heterozygous c-kit mutations in primary gastrointestinal stromal tumors. The mutant nucleotide sequence is indicated in the upper diagram, and the wild-type nucleotide sequence is indicated in the lower diagram. Nucleotide changes are shown in red capital letters. A: Cryopreserved specimens; B: Fresh specimens.
In the three fresh specimens analysed in this experiment, sequencing showed that GIST1 and GIST3 each harboured heterozygous c-kit exon 11 mutations, while GIST2 had no mutations in either c-kit or PDGFRA (Figure 1B).
Expression of stem cell factor in primary GISTS
The immunohistochemistry experiments shown in Figure 2 demonstrated that SCF expression was observed in both GIST cells and fibroblasts, while KIT and SCF were co-expressed in tumor cells. Given that GISTs contains only a small number of fibroblasts, a specimen was considered “positive” for SCF expression if only the tumor cells showed distinct cytoplasmic or membrane staining; otherwise, the specimen was considered “negative” for SCF expression. SCF protein was present in 39 (76.5%) of the 51 GIST samples. A morphometric study revealed more pHH3 and Ki-67 positive cells and lower apoptotic cells in SCF-positive GIST cases compared with SCF-negative GIST cases (Figure 2). These results suggest that SCF may participate in an autocrine-paracrine stimulatory loop within primary GISTs.
Figure 2.

The expression of stem cell factor in primary gastrointestinal stromal tumors. Sections of gastrointestinal stromal tumors (GISTs) (n = 51) were immunostained with stem cell factor (SCF), KIT (CD117), pHH3 and Ki-67 antibodies (magnification, ×200). Apoptosis was assessed in situ by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) staining (magnification, ×200). Representative images are shown. Bar = 50 μm.
Correlations between SCF and clinicopathological variables
As shown in Table 1, the co-expression of wild-type KIT receptor and SCF was associated with known prognostic variables, including larger tumor size (P = 0.0118), higher mitotic count (P = 0.0058), higher proliferative index (P = 0.0012), higher mitotic index (P = 0.0282), lower apoptosis index (P = 0.0484), and an increased NIH risk level (P = 0.0012). These results suggest that the SCF-mediated activation of wild-type KIT may play an important role in tumor cell growth by directly promoting cell proliferation and inhibiting apoptosis. The co-expression of the wild-type KIT receptor and SCF was not associated with any other variable tested, including patient’s age, sex, histological phenotype, cellularity, tumor location and c-kit gene mutation.
KIT activation in primary GISTS
In line with other published findings[24,25], KIT was phosphorylated at tyrosine 703 in 74.5% (38/51) of GISTs (Figure 3A), although the levels of phosphorylated KIT varied substantially from tumor to tumor, even between those with identical c-kit gene mutations. Furthermore, KIT phosphorylation was not significantly associated with the presence of c-kit mutations (P = 0.6625). Thirteen tumors without detectable c-kit mutations showed strong KIT phosphorylation, which was consistent with a nonmutational mechanism of KIT activation.
Figure 3.
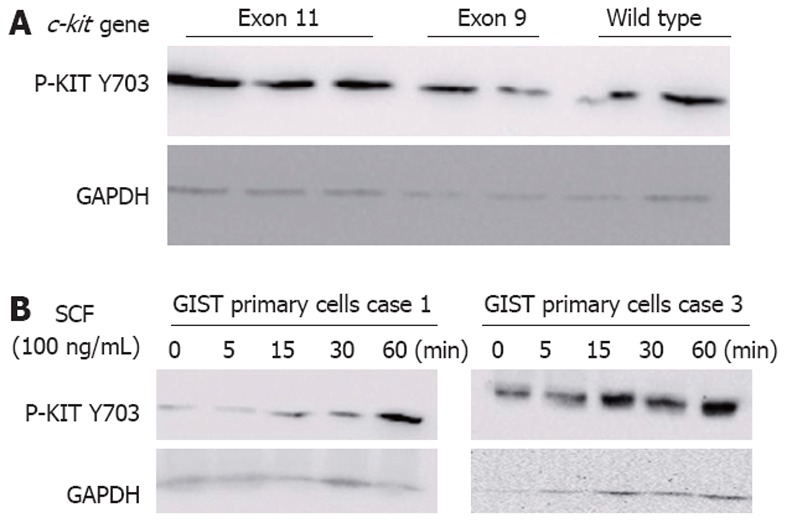
Western blotting analysis of phosphorylated KIT. A: KIT phosphorylation (p-KIT Y703) was analysed in gastrointestinal stromal tumor (GIST) samples (n = 51). A representative western blotting of p-KIT Y703 (top) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (bottom) in patients with exon 11 or 9 mutations or wild-type c-kit-bearing tumors; B: GIST primary cells were incubated with 100 ng/mL stem cell factor (SCF) for the indicated times. KIT phosphorylation (p-KIT Y703) was analysed by western blotting.
To determine whether the KIT receptor was activated due to the presence of the wild-type KIT receptor in c-kit-mutant GISTs, we cultivated GIST primary cells in the presence of various concentrations of exogenous SCF. KIT was rapidly phosphorylated in response to exogenous SCF in a time-dependent manner in GIST primary cells (Figure 3B). These findings suggest that the hyperphosphorylation of the wild-type KIT receptor was induced by exogenous SCF, confirming that SCF mediated the activation of the wild-type KIT receptor in c-kit-mutant GISTs.
Effects of wild-type KIT receptor activation on proliferation in GIST primary cells
To assess whether SCF-mediated wild-type KIT activation participated in the proliferation of GIST tumor cells, we analysed the proliferation rate of GIST primary cells exposed to exogenous SCF. Consistent with the finding that SCF induced KIT phosphorylation, exogenous SCF significantly increased cell proliferation in a dose-dependent manner after 72 h of treatment (Figure 4A).
Figure 4.

Stem cell factor stimulates gastrointestinal stromal tumors cell proliferation in vitro. Gastrointestinal stromal tumor (GIST) primary cells were incubated for 72 h with exogenous stem cell factor (SCF) (0-100 ng/mL, A) or KIT immunoblocking antibody (100 ng/mL, B), cell proliferation was analysed by methyl thiazolyl tetrazolium assay. The data are presented as the mean ± SD (n = 4 for each). aP < 0.05, bP < 0.01 vs untreated cells.
The ELISA results showed that all three primary cell lines produced significant amounts of SCF (3.5 ± 2.33 pg/mL per 106 cells). Therefore, we examined the effect of SCF-mediated wild-type KIT activation on cell proliferation by inhibiting the interactions between the KIT receptor and endogenous SCF. As shown in Figure 4B, cell proliferation was significantly inhibited (by > 50%) after 72 h of treatment with a KIT immunoblocking antibody, suggesting that SCF-mediated wild-type KIT receptor activation plays a key role in controlling GIST cell proliferation through the SCF/KIT autocrine-paracrine loop.
Effects of wild-type KIT receptor activation on apoptosis in GIST primary cells
The capacity to regulate survival is an important feature of tumor cells. Therefore, we tested whether wild-type KIT receptor activation could rescue GIST primary cells from serum deprivation-induced death. As shown in Figure 5, the addition of exogenous SCF (100 ng/mL, 12 h) significantly reduced the percentage of apoptotic cells, from 32.05% ± 2.65% to 18.55% ± 1.83% (P < 0.01) in GIST primary cells. In contrast, treatment with KIT immunoblocking antibody significantly increased the percentage of apoptotic cells from 32.05% ± 2.65% to 43.58% ± 2.94% (P < 0.01).
Figure 5.
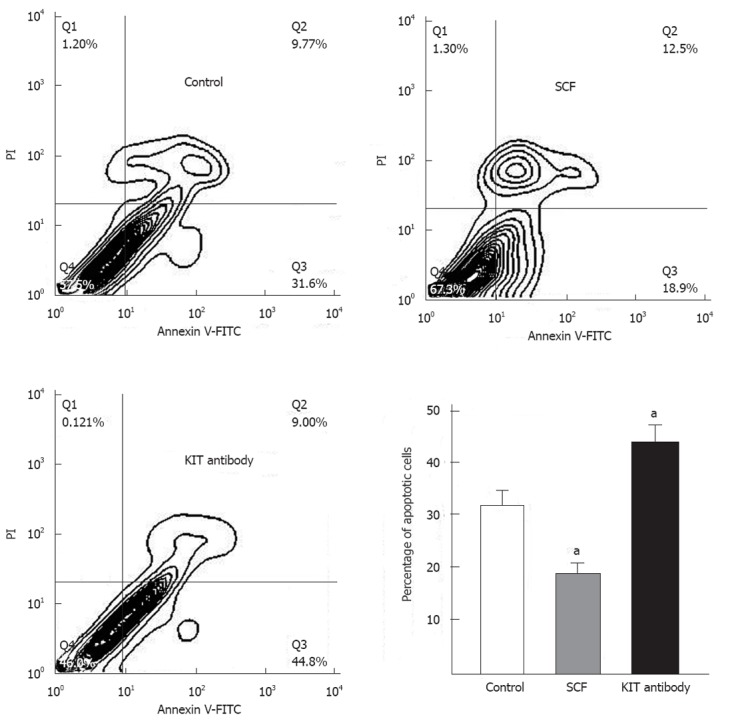
Stem cell factor inhibits gastrointestinal stromal tumor cell apoptosis in vitro. Representative flow cytometric contour of annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) dual-colour flow cytometry after 12 h of treatment with exogenous stem cell factor (SCF) or KIT immunoblocking antibody. The lower right quadrant represents early apoptotic cells. The data are presented as the mean ± SD (n = 4 for each). aP < 0.01 vs untreated cells.
DISCUSSION
The present study showed that c-kit gene mutations occurred in 32 (62.7%) of the 51 GIST clinical samples, which is consistent with previous results[14-16]. All of these mutations were heterozygous, i.e., at least one wild-type c-kit allele was present in all tumors, which is consistent with a previous report that most GIST mutations were heterozygous[26]. The heterozygous nature of the receptor status in GISTs suggests that KIT receptor activation is not induced by c-kit gene mutations alone but rather by other mechanisms, such as the activation of ligand-dependent signal transduction pathways. Indeed, we noted that KIT activation, as manifested by receptor tyrosine phosphorylation, is a general phenomenon in GISTs, even those without c-kit mutations.
Constitutive receptor tyrosine kinase activation is believed to be important for tumor proliferation and progression, and in GISTs, KIT activation can serve as an initiating event in oncogenesis[27-29]. However, the results obtained in our study showed that SCF expression was positively correlated with mitotic activity. KIT was rapidly phosphorylated upon stimulation with exogenous SCF, suggesting that ligand-dependent hyperactivation is also a strong mitogen in GIST cells. This observation is consistent with the data reported by Hirano et al[18], who found that SCF-positive GIST cases had a significantly higher average MIB-1 labelling index and a larger average tumor size than did the SCF-negative cases. Similarly, Théou-Anton et al[17] detected SCF in up to 93% of the GISTs studied and speculated that KIT activation in GISTs may be caused partly by the presence of SCF within the tumors. However, no in vitro measurements were conducted in either of these two studies, and the possible role of SCF-mediated wild-type KIT receptor activation in GIST proliferation should be explored in vitro to assess its independent contribution to cell growth. Accordingly, we further examined the mitogenic activity of this molecule on GIST primary cells harbouring a heterozygous c-kit mutation. It was found that exogenous SCF markedly stimulated cell proliferation and KIT phosphorylation in all GIST primary cells, while the inhibition of the SCF/KIT interaction reduced cell proliferation, confirming that exogenous or endogenous SCF-mediated activation of wild-type KIT provided an important signal for GIST cell proliferation. Further, GIST882, which has a homozygous activating c-kit mutation[30], did not exhibit increased proliferation in response to supplemental SCF, and the exogenous SCF stimulation of GIST544 cells, which express a heterozygous c-kit mutation, induces stronger KIT phosphorylation and cell growth[31]; these observations further validate our results.
Apoptosis is an active process that plays a key role in the development and maintenance of tissue homeostasis. The tumor growth rate partly depends on an excess of proliferation over apoptosis[32]. Indeed, the relationship between SCF-mediated wild-type KIT receptor activation and apoptosis has been explored extensively. For example, c-kit activation was found to suppress the apoptosis of normal murine melanocyte precursors[33], soft tissue sarcomas of neuroectodermal origin[34], neuroblastomas[35], and normal and malignant human haematopoietic cells[36]. Our results also support a role for SCF mediated wild-type KIT receptor activation in the survival of GIST primary cells because exogenous SCF rescues GIST primary cells from serum deprivation-induced death and may consequently prolong cell survival, while blocking the interactions between KIT receptor and endogenous SCF markedly reduces the viability of these cells.
In summary, our results demonstrated that the SCF-dependent activation of the wild-type KIT receptor is specifically involved in promoting the cell growth of GISTs via autocrine-paracrine loop activation. Therefore, drugs targeted against GISTs should switch off the activation of both mutant and wild-type receptors to achieve an effective response.
COMMENTS
Background
Gastrointestinal stromal tumor (GIST) is the most common sarcoma of the intestinal tract. Imatinib has shown remarkable efficacy in the treatment of GISTs, which are notoriously refractory to conventional chemotherapy or radiation. However, a considerable proportion of GIST patients show primary or acquired resistance to Imatinib.
Innovations and breakthroughs
In this study, the authors showed that the co-expression of the wild-type KIT receptor and stem cell factor (SCF) was associated with known prognostic variables in a series of 51 patients. Authors further demonstrated that SCF-mediated wild-type KIT receptor activation participated in GIST growth by stimulating the proliferation and inhibiting the apoptosis of GIST primary cells. These results suggest that SCF-mediated wild-type KIT receptor activation represents a new and potentially promising target for GIST therapy. Therefore, to achieve an effective response, drugs targeted against GISTs should switch off the activation of both mutant and wild-type receptors.
Applications
The study demonstrated that the SCF-dependent activation of the wild-type KIT receptor is specifically involved in promoting the cell growth of GISTs via autocrine-paracrine loop activation. Therefore, drugs that target wild-type receptor activation may be viable candidates for the treatment of GISTs and should be explored in future studies.
Peer review
This is a well written manuscript. The use of confocal microscopy would have been ideal for studying both KIT and SCF expression.
Footnotes
Supported by The National Natural Science Foundation of China, No.30700809 and No.30972876
Peer reviewer: Dina G Tiniakos, Laboratory of Histology and Embryology, Medical School, University of Athens, 75, M. Asias str, Goudi, 11527 Athens, Greece
S- Editor Gou SX L- Editor A E- Editor Li JY
References
- 1.Yarden Y, Kuang WJ, Yang-Feng T, Coussens L, Munemitsu S, Dull TJ, Chen E, Schlessinger J, Francke U, Ullrich A. Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987;6:3341–3351. doi: 10.1002/j.1460-2075.1987.tb02655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson DM, Lyman SD, Baird A, Wignall JM, Eisenman J, Rauch C, March CJ, Boswell HS, Gimpel SD, Cosman D. Molecular cloning of mast cell growth factor, a hematopoietin that is active in both membrane bound and soluble forms. Cell. 1990;63:235–243. doi: 10.1016/0092-8674(90)90304-w. [DOI] [PubMed] [Google Scholar]
- 3.Flanagan JG, Leder P. The kit ligand: a cell surface molecule altered in steel mutant fibroblasts. Cell. 1990;63:185–194. doi: 10.1016/0092-8674(90)90299-t. [DOI] [PubMed] [Google Scholar]
- 4.Martin FH, Suggs SV, Langley KE, Lu HS, Ting J, Okino KH, Morris CF, McNiece IK, Jacobsen FW, Mendiaz EA. Primary structure and functional expression of rat and human stem cell factor DNAs. Cell. 1990;63:203–211. doi: 10.1016/0092-8674(90)90301-t. [DOI] [PubMed] [Google Scholar]
- 5.Zsebo KM, Williams DA, Geissler EN, Broudy VC, Martin FH, Atkins HL, Hsu RY, Birkett NC, Okino KH, Murdock DC. Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell. 1990;63:213–224. doi: 10.1016/0092-8674(90)90302-u. [DOI] [PubMed] [Google Scholar]
- 6.Blechman JM, Lev S, Givol D, Yarden Y. Structure-function analyses of the kit receptor for the steel factor. Stem Cells. 1993;11 Suppl 2:12–21. doi: 10.1002/stem.5530110804. [DOI] [PubMed] [Google Scholar]
- 7.Fleischman RA. From white spots to stem cells: the role of the Kit receptor in mammalian development. Trends Genet. 1993;9:285–290. doi: 10.1016/0168-9525(93)90015-a. [DOI] [PubMed] [Google Scholar]
- 8.Huizinga JD, Thuneberg L, Klüppel M, Malysz J, Mikkelsen HB, Bernstein A. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature. 1995;373:347–349. doi: 10.1038/373347a0. [DOI] [PubMed] [Google Scholar]
- 9.Boissan M, Feger F, Guillosson JJ, Arock M. c-Kit and c-kit mutations in mastocytosis and other hematological diseases. J Leukoc Biol. 2000;67:135–148. doi: 10.1002/jlb.67.2.135. [DOI] [PubMed] [Google Scholar]
- 10.Testa U. Membrane Tyrosine Kinase Receptors are an Important Target for the Therapy of Acute Myeloid Leukemia. Current Cancer Therapy Reviews. 2008;4:31–49. [Google Scholar]
- 11.Sircar K, Hewlett BR, Huizinga JD, Chorneyko K, Berezin I, Riddell RH. Interstitial cells of Cajal as precursors of gastrointestinal stromal tumors. Am J Surg Pathol. 1999;23:377–389. doi: 10.1097/00000478-199904000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. 2006;130:1466–1478. doi: 10.5858/2006-130-1466-GSTROM. [DOI] [PubMed] [Google Scholar]
- 13.Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol. 1998;152:1259–1269. [PMC free article] [PubMed] [Google Scholar]
- 14.Hirota S, Ohashi A, Nishida T, Isozaki K, Kinoshita K, Shinomura Y, Kitamura Y. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology. 2003;125:660–667. doi: 10.1016/s0016-5085(03)01046-1. [DOI] [PubMed] [Google Scholar]
- 15.Antonescu CR, Sommer G, Sarran L, Tschernyavsky SJ, Riedel E, Woodruff JM, Robson M, Maki R, Brennan MF, Ladanyi M, et al. Association of KIT exon 9 mutations with nongastric primary site and aggressive behavior: KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res. 2003;9:3329–3337. [PubMed] [Google Scholar]
- 16.Rubin BP, Heinrich MC, Corless CL. Gastrointestinal stromal tumour. Lancet. 2007;369:1731–1741. doi: 10.1016/S0140-6736(07)60780-6. [DOI] [PubMed] [Google Scholar]
- 17.Théou-Anton N, Tabone S, Brouty-Boyé D, Saffroy R, Ronnstrand L, Lemoine A, Emile JF. Co expression of SCF and KIT in gastrointestinal stromal tumours (GISTs) suggests an autocrine/paracrine mechanism. Br J Cancer. 2006;94:1180–1185. doi: 10.1038/sj.bjc.6603063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirano K, Shishido-Hara Y, Kitazawa A, Kojima K, Sumiishi A, Umino M, Kikuchi F, Sakamoto A, Fujioka Y, Kamma H. Expression of stem cell factor (SCF), a KIT ligand, in gastrointestinal stromal tumors (GISTs): a potential marker for tumor proliferation. Pathol Res Pract. 2008;204:799–807. doi: 10.1016/j.prp.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 19.Tamborini E, Bonadiman L, Negri T, Greco A, Staurengo S, Bidoli P, Pastorino U, Pierotti MA, Pilotti S. Detection of overexpressed and phosphorylated wild-type kit receptor in surgical specimens of small cell lung cancer. Clin Cancer Res. 2004;10:8214–8219. doi: 10.1158/1078-0432.CCR-04-1013. [DOI] [PubMed] [Google Scholar]
- 20.West RB, Corless CL, Chen X, Rubin BP, Subramanian S, Montgomery K, Zhu S, Ball CA, Nielsen TO, Patel R, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol. 2004;165:107–113. doi: 10.1016/S0002-9440(10)63279-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rubin BP. Gastrointestinal stromal tumours: an update. Histopathology. 2006;48:83–96. doi: 10.1111/j.1365-2559.2005.02291.x. [DOI] [PubMed] [Google Scholar]
- 22.Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, Miettinen M, O’Leary TJ, Remotti H, Rubin BP, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol. 2002;33:459–465. doi: 10.1053/hupa.2002.123545. [DOI] [PubMed] [Google Scholar]
- 23.Prenen H, Cools J, Mentens N, Folens C, Sciot R, Schöffski P, Van Oosterom A, Marynen P, Debiec-Rychter M. Efficacy of the kinase inhibitor SU11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate. Clin Cancer Res. 2006;12:2622–2627. doi: 10.1158/1078-0432.CCR-05-2275. [DOI] [PubMed] [Google Scholar]
- 24.Janeway KA, Liegl B, Harlow A, Le C, Perez-Atayde A, Kozakewich H, Corless CL, Heinrich MC, Fletcher JA. Pediatric KIT wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res. 2007;67:9084–9088. doi: 10.1158/0008-5472.CAN-07-1938. [DOI] [PubMed] [Google Scholar]
- 25.Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, Hibbard MK, Chen CJ, Xiao S, Tuveson DA, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001;61:8118–8121. [PubMed] [Google Scholar]
- 26.Théou N, Tabone S, Saffroy R, Le Cesne A, Julié C, Cortez A, Lavergne-Slove A, Debuire B, Lemoine A, Emile JF. High expression of both mutant and wild-type alleles of c-kit in gastrointestinal stromal tumors. Biochim Biophys Acta. 2004;1688:250–256. doi: 10.1016/j.bbadis.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 27.Nishida T, Hirota S, Taniguchi M, Hashimoto K, Isozaki K, Nakamura H, Kanakura Y, Tanaka T, Takabayashi A, Matsuda H, et al. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat Genet. 1998;19:323–324. doi: 10.1038/1209. [DOI] [PubMed] [Google Scholar]
- 28.Maeyama H, Hidaka E, Ota H, Minami S, Kajiyama M, Kuraishi A, Mori H, Matsuda Y, Wada S, Sodeyama H, et al. Familial gastrointestinal stromal tumor with hyperpigmentation: association with a germline mutation of the c-kit gene. Gastroenterology. 2001;120:210–215. doi: 10.1053/gast.2001.20880. [DOI] [PubMed] [Google Scholar]
- 29.Corless CL, McGreevey L, Haley A, Town A, Heinrich MC. KIT mutations are common in incidental gastrointestinal stromal tumors one centimeter or less in size. Am J Pathol. 2002;160:1567–1572. doi: 10.1016/S0002-9440(10)61103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lux ML, Rubin BP, Biase TL, Chen CJ, Maclure T, Demetri G, Xiao S, Singer S, Fletcher CD, Fletcher JA. KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol. 2000;156:791–795. doi: 10.1016/S0002-9440(10)64946-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duensing A, Medeiros F, McConarty B, Joseph NE, Panigrahy D, Singer S, Fletcher CD, Demetri GD, Fletcher JA. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs) Oncogene. 2004;23:3999–4006. doi: 10.1038/sj.onc.1207525. [DOI] [PubMed] [Google Scholar]
- 32.Henson PM, Hume DA. Apoptotic cell removal in development and tissue homeostasis. Trends Immunol. 2006;27:244–250. doi: 10.1016/j.it.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 33.Ito M, Kawa Y, Ono H, Okura M, Baba T, Kubota Y, Nishikawa SI, Mizoguchi M. Removal of stem cell factor or addition of monoclonal anti-c-KIT antibody induces apoptosis in murine melanocyte precursors. J Invest Dermatol. 1999;112:796–801. doi: 10.1046/j.1523-1747.1999.00552.x. [DOI] [PubMed] [Google Scholar]
- 34.Ricotti E, Fagioli F, Garelli E, Linari C, Crescenzio N, Horenstein AL, Pistamiglio P, Vai S, Berger M, di Montezemolo LC, et al. c-kit is expressed in soft tissue sarcoma of neuroectodermic origin and its ligand prevents apoptosis of neoplastic cells. Blood. 1998;91:2397–2405. [PubMed] [Google Scholar]
- 35.Timeus F, Crescenzio N, Valle P, Pistamiglio P, Piglione M, Garelli E, Ricotti E, Rocchi P, Strippoli P, Cordero di Montezemolo L, et al. Stem cell factor suppresses apoptosis in neuroblastoma cell lines. Exp Hematol. 1997;25:1253–1260. [PubMed] [Google Scholar]
- 36.Hassan HT, Zander A. Stem cell factor as a survival and growth factor in human normal and malignant hematopoiesis. Acta Haematol. 1996;95:257–262. doi: 10.1159/000203893. [DOI] [PubMed] [Google Scholar]