

Abstract
Basal-like breast cancer is an aggressive disease with limited therapeutic options because these tumors frequently express the ‘triple-negative’ phenotype. We have recently reported that inducible nitric oxide synthase (NOS2) is a strong predictor of survival in patients with estrogen receptor negative [ER(−)] breast cancer, and that NOS2 expression is correlated with a basal-like phenotype. Recent reports also describe the pro-tumor effects of NO in breast and many other types of cancer. NO promotes cancer progression by activating several oncogenic signaling pathways such as extracellular signal-regulated kinases (ERK)-1/2, phosphoinositide 3-kinases (PI3K)/Akt, and c-Myc. Protein phosphatase 2A (PP2A) is a tumor suppressor that negatively regulates the same cancer-related signaling pathways that are activated by NO. PP2A activity is suppressed in tumor cells, but potential pharmacological agents have recently been described to increase PP2A activity in ER(−) breast cancer cells. We examine here the various functions of NO and PP2A in breast cancer and propose a novel mechanism by which activation of PP2A antagonizes NO signaling that promotes ER(−) breast cancer.
NO signaling in breast cancer
The role of NO in cancer was one of the earliest biological effects described for this endogenously synthesized, unique signaling molecule [1]. Interestingly, NO has both tumor suppressing and tumor promoting effects [2]. For example, NO is essential in the macrophage-mediated eradication of leukemia cells and activates p53-mediated apoptosis [1,3]. p53 is a tumor suppressor protein that is activated by multiple cellular stresses and possesses multiple anti-cancer functions such as inducing cell apoptosis, cell-cycle arrest and activating DNA repair enzymes [4]. By contrast, NO increases genotoxicity and inhibits DNA repair enzymes [1,3]. Endogenous production of NO is catalyzed by a family of NOS enzymes, and the inducible isoform (NOS2) is associated with immune cell function and inflammation [5]. NOS2 overexpression in tumor cells is generally associated with increased proliferation and migration. For example, NOS2 expression promotes colon and breast cancer cell growth and invasiveness [3].
The relationship between NOS2 and p53 provides insight into the divergent properties of NO in cancer [6]. Wild-type p53 antagonizes NOS2 via two mechanisms. p53 binds to the TATA-binding element in the promoter region of the NOS2 gene to inhibit expression [7]. Furthermore, p53 binds directly to the NOS2 protein, which attenuates NOS activity [8]. By contrast, NO stabilizes p53, leading to apoptosis [9,10]. NO-mediated apoptosis of leukemia cells requires wild-type p53, because NO does not induce apoptosis in p53 null cells [11]. In addition, p53 mutant carcinoma cells expressing NOS2 have accelerated tumor growth and increased vascular endothelial growth factor (VEGF) production compared to wild-type p53 tumors [12], suggesting that p53 status plays a significant role in determining the pro- or anti-tumorigenic role of NO. These studies revealed an important relationship between p53 status, NO, and seemingly disparate cancer outcomes.
NOS2 has been shown to have a role in a wide variety of cancers. High NOS2 expression is associated with favorable prognoses in ovarian [13] and lung cancers [14]. By contrast, many clinical studies have correlated elevated NOS2 expression with poor patient survival in various cancers, including breast, colon, gastric, esophageal, prostate, cervical, squamous cell carcinoma, hepatocellular carcinoma, melanoma, ovarian and leukemia [3]. Moreover, these findings are often linked with other inflammatory biomarkers. For example, NOS2 and COX-2 were associated with increased microvessel density in non-small-cell lung cancer [15]. Therefore, NOS2 is emerging as a biomarker of poor cancer patient prognosis (i.e. decreased patient survival).
In this review we first discuss the clinical relevance of NOS2 expression and the pro-tumor signaling effects of NO. Because NOS2 expression is strongly associated with poor patient survival for many cancer types, it is a potential therapeutic target. We discuss how NO activates multiple signaling pathways associated with tumor progression and metastasis. Then we briefly review the tumor suppressor protein phosphatase 2A (PP2A), which negatively regulates the same signaling pathways that are activated by NO. Evidence is growing for the potential of PP2A agonists as a therapeutic approach that could counteract the pro-tumor effects of NO signaling (Figure 1).
Figure 1.
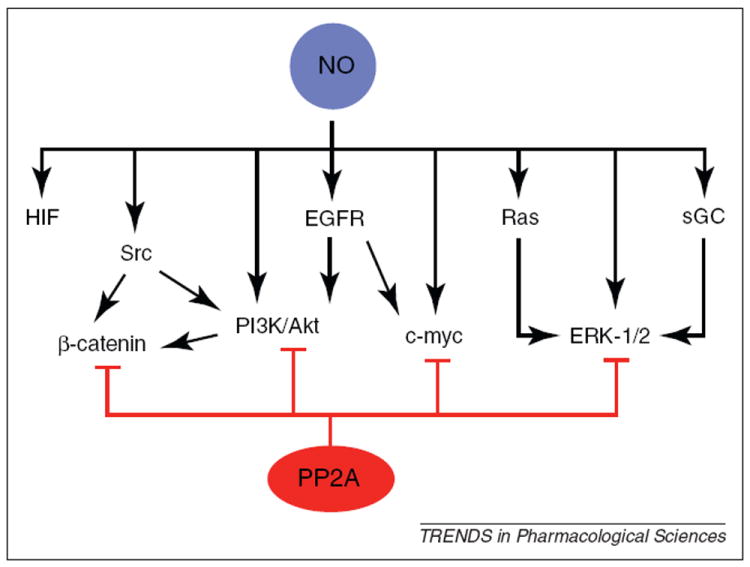
Oncogenic signaling pathways regulated by NO and PP2A. NO activates numerous signaling pathways associated with cancer cell proliferation, survival, chemoresistance and migration. Some of the molecular and pathway targets of NO signaling include sGC, HIF-1α, Src, PI3K/Akt, EGFR, Ras, c-Myc and ERK-1/2. Furthermore, NO activates oncogenic pathways by redundant mechanisms; for example, ERK-1/2 is activated from either Ras signaling or sGC activation and PI3K/Akt signaling is activated by either EGFR or Src kinases. These redundant mechanisms indicate that single agent therapies could fail to limit oncogenic NO signaling. The tumor suppressor PP2A is the endogenous negative regulator of many NO-activated signaling pathways. We propose that pharmacological activation of PP2A could represent an effective strategy to limit the myriad of pro-tumor NO effects.
Clinical markers of NOS2 in breast cancer
NO signaling is emerging as a key factor that enhances breast cancer aggressiveness. An early report showed that NOS2 expression is significantly associated with a high tumor grade in invasive ductal carcinoma patients [16]. More recent studies revealed that NOS2 is expressed in about 70% of all breast tumors, and that NOS2 expression correlates with markers of poor prognosis such nuclear epidermal growth factor receptor (EGFR) and STAT3 (signal transducer and activator of transcription 3), phospho-Akt and phospho-STAT3 [17-19]. Increased NOS2 expression is significantly associated with phospho-Akt in human breast tumors regardless of breast cancer subtype, indicating that NO is clinically associated with increased Akt activation, consistent with cell-culture-based data [17]. This suggests that oncogenic pathways associated with Akt signaling are probably activated by NOS2.
Recently NOS2 was shown to be a novel prognostic marker in ER(−) breast cancer patients [19] (Box 1). The association between high NOS2 expression and decreased ER(−) patient survival is remarkably strong (hazard ratio of 6.19 for survival after five years), suggesting that NOS2 expression is an important biomarker for ER(−) diseases [19]. Furthermore, high NOS2 expression is correlated with decreased survival among patients with basal-like breast cancer, another form of aggressive disease (Box 1) [19]. The NOS2 association with prognosis is independent of other predictive factors, such as tumor grade, lymph node involvement, and degree of metastasis, suggesting that early induction of metastasis is perhaps favored by proinflammatory NOS2 overexpression [19]. In addition, a recent report indicates that cyclooxygenase 2 (COX-2) is also a predictor of poor survival among ER(−) patients [20]. It is possible that the activities of NOS2 and COX-2 synergize to form a tumor-induced inflammatory response that results in an aggressive tumor phenotype in ER(−) breast cancer patients.
Box 1. Breast cancer subtypes.
Breast cancers are not homogeneous. Breast tumors are stratified according to their expression of estrogen receptor-alpha (ER-α). The therapeutic options and clinical outcomes are drastically different for ER-negative [ER(−)] compared to ER-positive [ER(+)] tumors. ER(+) tumors require estrogen for proliferation and therefore pharmacological agents that inhibit ER function or the production of estrogen have positive effects on patient survival, whereas patients with ER(−) tumors do not respond to these types of treatments. Furthermore, ER(−) tumors are usually more aggressive (e.g. increased incidence of metastasis, increased rates of disease relapse and lower survival rates) than ER(+) tumors [112,113].
With the advent of the genomic era, the sub-classification of breast tumors has advanced beyond ER-α expression. Tumor subtypes are defined by unique genetic profiles; this understanding has changed the way researchers and clinicians view breast cancer by revealing the underlying differences between tumors and clinical outcomes among breast cancer patients [114,115]. ER(+) tumors are now further stratified into luminal A and luminal B subtypes and ER(−) tumors are classified as normal-like, Her2/neu (ErbB2)+ or basal-like [115]. Basal-like breast cancer is a very aggressive subtype and these tumors express a phenotype that is similar in genetic profile and morphology to basal breast cells [116]. Furthermore, the basal-like subtype usually exhibits the ‘triple-negative’ phenotype that is defined as tumor cells lacking the expression of progesterone receptor, estrogen receptor and ErbB2 (Her2/neu) [117]. Both the basal-like and triple-negative phenotypes are associated with limited therapeutic options and high mortality rates compared with other breast cancers [116,118].
A signature set of 44 genes is associated with NOS2 overexpression, and 15 of these upregulated genes are well-established markers of basal-like breast cancer, such as P-cadherin, cytokeratins, Wnt5A, and KLF5 [19]. Furthermore, these basal-like markers are individually associated with aggressiveness and poor survival in patients. For example, Wnt5A has recently been shown to be associated with brain metastasis [21]. High KLF5 expression predicts shorter disease-free survival and overall survival than breast cancer patients with lower KLF5 expression [22]. KLF5 expression is higher in younger patients, consistent with triple-negative (Box 1) and/or basal-like breast cancer [22]. Other genes associated with NOS2 expression, such as CD44 and inflammatory molecules such as IL-6, IL-8 and S100A8 are associated with metastasis and poor outcome [19]. Therefore, the function of NOS2 as a predictor of ER(−) patient survival is perhaps mediated by NO-dependent induction of these markers in ER(−) breast tumors.
Although NOS2 expression is correlated with poor clinical outcomes in ER(−) breast cancers, it is possible that NO is not the NOS2 end-product that elicits the malignant phenotype. All three isoforms of NOS have been reported to produce superoxide under conditions of substrate (argi-nine) or cofactor (tetrahydrobiopterin) depletion [23]; therefore it is possible that superoxide generation can contribute to the pro-tumor environment provided by NOS2 expression. Although superoxide has cancer-promoting signaling effects [24], it does not account for the wide array of signaling effects seen with both NOS2 activity and exogenous NO-donor compounds described below. However, this alternative role of NOS2 could increase the tumorigenic effects of NOS2.
NO activates multiple pro-tumor pathways
NO signaling in breast cancer results in altered expression of many genes implicated in breast cancer proliferation, cellular invasion, tumor angiogenesis and metastasis. Thus, NO signaling affects multiple signaling pathways. For example, NO signaling in breast cancer cells is known to activate many cancer-related pathways, and NO increases hypoxia inducible factor (HIF), ERK-1/2 and phosphatidylinositol 3-kinase (PI3K/Akt) [25]. These signaling pathways have strong links to cancer proliferation, angiogenesis and tumor metastasis [3].
Recent reports show that discrete concentrations of NO over different time frames induce specific signal transduction pathways (Figure 2) [25]. NOS2 is capable of generating concentrations in a range from nanomolar to micromolar NO, and this range includes both cell regulatory functions and immunotoxic properties [5,25]. Here we briefly review some of the cancer-related signaling pathways associated with increasing levels of NO.
Figure 2.
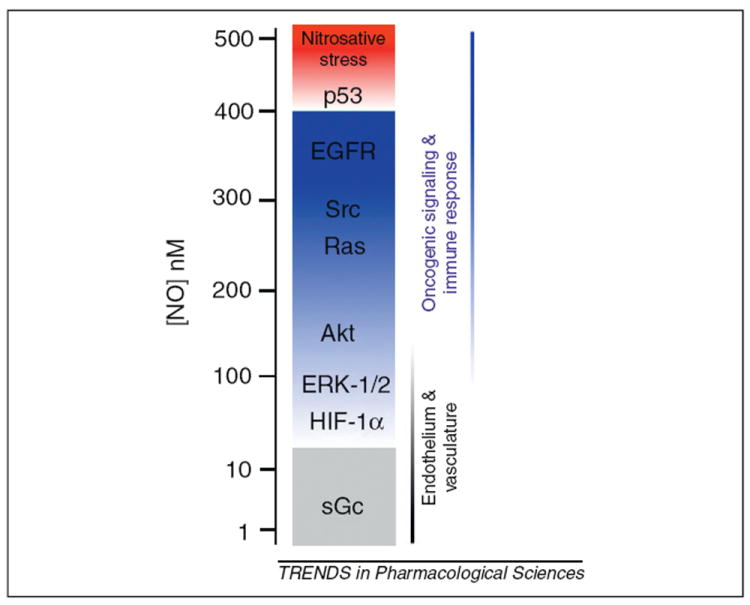
NO activates multiple signaling pathways at different concentrations. The biological effects of NO depend on the concentration of this unique signaling molecule. Physiological maintenance of endothelial cells and vascular tone is achieved at very low NO concentration (from below 1 nM to 30 nM). At this level, NO signals through sGC and HIF-1α. An increase in NO levels, as a result of an immune response or in tumor microenvironments, leads to activation of oncogenic signaling pathways including ERK-1/2, Akt, Src, Ras and EGFR. When NO levels reach ~500 nM the p53 tumor suppressor is activated (provided the cell expresses wild-type p53) and anti-tumor effects of NO are observed. Beyond this level of NO signaling, nitrosative stress accumulates and cells undergo necrosis, apoptosis or senescence. The NO concentrations eliciting oncogenic signaling and immune responses are predominantly produced by NOS2 (inducible isoform) catalysis, whereas lower levels are due to either NOS1 (neuronal) or NOS3 (endothelial) activity. This model of NO signaling explains the pro- and anti-tumor effects observed by NO.
Soluble guanylate cyclase
Levels of NO that activate soluble guanylate cyclase (sGC) are below nanomolar concentrations for endothelial cells and neurons, and 20–30 nM for vascular smooth muscle cell relaxation. NO levels that increase angiogenesis in endothelial cells are extremely low – in the picomolar to nanomolar range. These lower levels of NO are usually associated with NOS1 (neuronal) or NOS3 (endothelial) catalysis [5]. NO activation of sGC and the increased production of cGMP has both positive and negative effects on cancer cell proliferation [26] and this level of NO signaling in cancer needs further elucidation.
HIF-1α
NO concentrations ≥100 nM stabilize HIF-1α protein by inhibiting iron-containing prolyl hydroxylases that regulate HIF-1α turnover [27,28]. NO stabilization of HIF-1α also leads to increases in HIF transcriptional activity [25]. HIF controls many cancer-related genes such as MMP2, uPA and VEGF [27,29]. This model of NO signaling shows that direct reaction of NO with various metalloproteins can increase cancer cell proliferation and angiogenesis.
ERK-1/2
NO at nanomolar concentrations has been reported to activate ERK-1/2 by both sGC-dependent and independent mechanisms [28,30]. ERK-1/2 is a mitogen-activated protein (MAP) kinase that induces cell proliferation [31] and migration [32] by phosphorylating transcription factors associated with cancer such as c-Myc [33] and Elk1 [34].
Phosphatidylinositol 3-kinases and Akt
As discussed above, NOS2 is clinically associated with Akt phosphorylation in breast cancer patients, and NO results in PI3K and Akt activation and downstream signaling, at levels similar to those required for ERK-1/2 activation [17,30,35]. The PI3K/Akt signaling axis is a major pathway in cancer cells that promotes proliferation, survival and migration [36]. As such, the PI3K/Akt pathway is a candidate target for therapeutic intervention [37]. PI3K/Akt signaling is induced by multiple upstream kinases such as receptor tyrosine kinases, integrin-linked kinases and focal adhesion kinases [38].
EGFR
We recently reported that NO signaling results in EGFR tyrosine phosphorylation that is consistent with kinase activation [19]. The levels of NO required to phosphorylate EGFR appear to be in the 200–400 nM range (D.W., unpublished observations). At this level of NO the autoxidation reactions become increasingly dominant and oxidized NO products are formed (such as N2O3). The formation of N2O3 is physiologically relevant because nitrosylating species react with protein thiols to produce protein S-nitrosothiols (SNO). Protein-SNO is emerging as an important post-translational signaling moiety [39], however the mechanism by which NO leads to EGFR activation is unclear. EGFR activation is associated with many cancers and its kinase function activates multiple oncogenic signaling pathways such as PI3K/Akt, Ras, FAK, c-Abl, c-Myc and STAT3 [40].
S-nitrosylation of Src and Ras
S-nitrosylation of cancer-related protein kinases appears to be a physiologically relevant post-translational modification because NO signaling results in S-nitrosylation of a specific cysteine residue on the proto-oncogene Src [41,42]. S-nitrosylation results in Src kinase activation and increases breast cancer cell migration [42]. Src signaling results in cancer cell proliferation, survival, migration and invasiveness [43].
Similar to Src, the proto-oncogene Ras is also S-nitro-sylated by NO, leading to activation of this signaling molecule [44]. Ras activation leads to Raf–MEK–ERK-1/2 signal transduction and cell proliferation [45]. Furthermore, Ras is commonly mutated in many human cancers [46]. These two examples of NO signaling at relatively high concentrations indicate that S-nitrosylation of proto-oncogenes is a plausible contributor to human cancers. We predict that this unique signaling mechanism of NO will be discovered for other oncogenic proteins and could represent a mechanistic link between NOS2 expression, patient survival and oncogene activation.
β-Catenin
β-Catenin signaling is central to many human cancers and is a predictor of survival in patients with basal-like breast cancer [47,48]. β-Catenin transcriptional activity is induced upon multiple stimuli, such as Wnt, EGFR, Src and PI3K/Akt signaling. A few reports indicate that NO signaling results in the activation of β-catenin transcriptional regulation in colitis [49], osteocytes [50] and colorectal cancer cells [51]. Given the evidence that NOS2 and NO signaling are involved in the basal-like phenotype [19], and that β-catenin activation is predictive of basal-like breast cancer mortality [47], we predict that NOS2 signaling contributes to the basal-like phenotype via β-catenin activation. This further strengthens the need to limit NOS2 signaling in ER(−) breast cancer.
An immediate implication of oncogenic NO signaling described here is the potential use of NOS inhibitors as a novel therapeutic approach to limiting ER(−) breast cancer. NOS inhibitors such as aminoguanidine and L-NG-mono-methyl arginine (L-NMMA) have been tested in clinical trials for ailments such as asthma, chronic obstructive pulmonary disease (COPD) and cardiogenic shock with varying outcomes [52-54]. Although short-term administration could have beneficial effects, chronic NOS2 inhibition could lead to dysregulation of constitutive NOS2 functions (i.e. in the kidney, lung and immune response) and unwelcome side effects that could limit the use of these inhibitors. Similarly, nonspecific NOS inhibitors that target all three isoforms would affect the numerous physiological roles of NO, and this would probably have adverse results. Furthermore, general NOS inhibition would abate the anti-tumor effects observed with extremely low levels of NO [55]. This suggests that alternative means to inhibit these NO-activated pathways are needed. One such strategy is to activate endogenous phosphatases with tumor suppressor function that target the same pathways which are activated by NO signaling. A potential candidate is the tumor suppressor PP2A that negatively regulates the pathways shown in Figure 1.
PP2A tumor suppressor functions in cancer
PP2A tumor suppressor and NO signaling pathways
NO signaling activates multiple oncogenic kinases in breast cancer cells. The role of kinase activation in tumorigenesis and metastasis has been thoroughly investigated in human cancers [56]. Equally important to cancer initiation and proliferation, and perhaps insufficiently investigated compared with kinase research, is the down-regulation of tumor-suppressing protein phosphatases (PP) [57,58]. The PP2A family of proteins comprises a major class of cellular serine/threonine phosphatases with tumor suppressor activities [57,59,60]. PP2A activity negatively regulates many proliferative signaling pathways associated with cancer progression by depho-sphorylating crucial proteins in these pathways [57,61]. Furthermore, PP2A suppression is required for cell transformation, signifying that PP2A regulation is crucial for tumorigenesis [57,62]. As detailed below, PP2A activity is an endogenous negative regulator of oncogenic NO signaling (Figure 1).
PP2A structure and substrates
The substrate specificity, subcellular localization, and phosphatase activity of PP2A are controlled by its subunit composition. PP2A has wide substrate specificity due to the vast array of regulatory (B) and scaffolding (A) subunits that form a tripeptide holoenzyme with a catalytic subunit (C) [60,61,63]. The C and A subunits have two isoforms (α and β) of similar sequence and activity [60]. Mutations of the A subunit are found in multiple cancers and are sufficient to induce tumorigenesis [60,61,64]. The B subunit has great diversity because it consists of B, B′, B″ and B′″ classes of proteins [60,61,65]. The B subunit can direct PP2A C subunit – and therefore phosphatase activity – to distinct protein complexes and subcellular compartments [65]. Thus PP2A can catalyze a wide array of protein substrates by varying subunit composition.
PP2A regulates NO-activated pathways
Activation of receptor tyrosine kinases (RTK) such as EGFR initiates multiple signaling pathways involved in proliferation and survival. The MAPK/ERK1/2 is a major signaling pathway in cancer proliferation activated by RTK. PP2A activation blocks ERK-1/2 pathway activation [66]. Similarly, RTK activation results in increased PI3K/Akt signaling activity. As described above, NO also activates PI3K/Akt signaling, which is central to cell proliferation and survival. PP2A limits Akt kinase activity by dephosphorylating both Ser473 and Thr308 residues [67,68]. PP2A B56 subunit interacts with β-catenin and phosphatase activity causes β-catenin protein degradation [69]. c-Myc is also regulated by PP2A because the B56γ subunit is required for phosphatase activity [70]. PP2A dephosphorylation of c-Myc results in proteasomal degradation of the oncoprotein [70]. The B56γ subunit is also required for PP2A to regulate p53-induced apoptosis in response to DNA damage by dephosphorylating Thr55 [71]. In addition to these NO-activated pathways, PP2A also negatively regulates other cancer-related signaling pathways such as FOXO [72], IKK-β/NF-κB [73] and Shh [74]. Therefore, PP2A activity is central to multiple signaling pathways that are commonly dysregulated in human cancers and activated by NO signaling (Figure 1).
Regulation of PP2A in cancer
Cancer cells must reduce PP2A activity to allow for uncontrolled proliferation mediated by kinase signaling cascades [57,60]. Furthermore, the inhibition of PP2A is necessary in cancer initiation [75,76]. Because PP2A is essential for many biological processes and has a diverse set of substrates, its activity is regulated on many levels. As described, the subunit composition controls the specific activity and localization of PP2A [61]. However mutations or viral infection can alter PP2A subunit activity [57,75]. The polyomavirus simian virus 40 (SV40) produces a protein called small t antigen that contributes to carcinogenesis by binding to the A subunit [62,75]. Small t antigen blocks the B subunit from forming an AB heterodimer, and thus inhibits proper PP2A functions. Similarly, somatic mutation in either A or B subunits allows for unregulated oncogenic signaling, because functional PP2A holoenzymes that regulate signaling are absent [77].
The catalytic subunit of PP2A is further regulated by phosphorylation and methylation. Methylation of a conserved lysine residue in the C terminus is required for holoenzyme assembly [63,78] and phosphorylation of PP2A C subunit at Tyr307 by Src, EGFR and other kinases inhibits phosphatase activity [79]. Furthermore, the catalytic subunit forms complexes with inhibitory proteins such as CIP2A, I2PP2A (alternatively referred to as protein SET or template-activating factor I) and I1PP2A [80-82]. The catalytic site of PP2A is a low pKa cysteine that is susceptible to oxidative stress [83]. Reactive oxygen species (i.e. O2− and H2O2) produced from tumor cell nicotinamide adenine dinucleotide phosphate (NADPH) oxidases after growth factor receptor activation can oxidize the active site thiol [84,85]. This reversible modification results in phosphatase inhibition and is another mechanism by which PP2A activity is suppressed.
Pharmacological PP2A activation
Small molecule agents have been found that increase PP2A phosphatase activity in cancer cells [86-88]. Agents that increase PP2A tumor suppressor activity should counteract oncogenic NO signaling. Although pharmacological activation of PP2A activity might not overcome genomic mutations of PP2A subunits, small molecules can potentially alter PP2A phosphorylation, chemical and protein inhibitors. Here we briefly review agents that increase PP2A activity.
Endostatin is an anti-angiogenic peptide that increases PP2A activity, which dephosphorylates eNOS and inhibits ERK-1/2 signaling [66,89]. The mechanism of PP2A activation by endostatin is unknown, and clinical trials in cancer patients have reported mixed efficacy. Endostatin did not provide significant tumor regression in advanced neuroendocrine tumors [90]. In non-small-cell lung cancer patients, endostatin in combination with cisplatin and gemcitabine resulted in increased survival over therapy alone [91].
Forskolin and other activators of cellular cAMP levels cause PP2A activity to increase [88]. The role of cAMP on PP2A activity has not been elucidated; however the activation of adenylate cyclase is not likely to be a specific mechanism of action for a potential anticancer drug. In view of the indiscriminate manner in which these compounds increase intracellular cAMP, forskolin and similar drugs do not have a promising clinical application.
Ceramides are structural components of the cell membrane and also have potent signaling properties that result in cell apoptosis, senescence, or cell-cycle arrest [92]. One effect of ceramides is the increase of PP2A phosphatase activity [93]. Ceramides are known to bind to I2PP2A, and it is proposed that ceramide activation of PP2A is mediated through SET inhibition [82]. The potential use of ceramides directly as pharmacological agents appears to be unlikely because these compounds are rapidly metabolized and have a wide range of biological effects. However pharmacological agents that increase ceramide biosynthesis, such as vitamin D3, daunorubicin and mitoxantrone, could indicate a successful approach to activating PP2A in vivo [94-96]. Recently a novel approach of delivering nanoliposomal ceramides reduced hepatocellular carcinoma tumor xenograft growth [97]. As promising as this method of ceramide administration appears, further studies will be needed to validate PP2A activation and clinical efficacies.
Dithiolethiones are a class of compounds found in dietary vegetables and that have chemopreventive effects in humans [98]. Dithiolethiones increase the antioxidant response element (ARE) by interacting with sensitive thiols of the KEAP1/Nrf2 complex, causing Nrf2 to translocate to the nucleus to bind to ARE sites in target gene promoter regions [99]. Target genes involved in xenobiotic metabolizing and cellular defense systems contribute to the chemopreventive effects of these compounds [100] Oltipraz, a prototypical dithiolethione, was protective against aflatoxin-induced hepatocellular carcinoma in a clinical trial [98]. Similarly, anethole dithiolethione (ADT) limited bronchial dysplasia in smokers [101]. Recently we reported that dithiolethiones increase PP2A phosphatase activity in cancer cells, which inhibit EGF-induced Akt and c-Myc signaling [87]. Dithiolethiones also inhibited cell proliferation without inducing apoptosis [87]. Furthermore, we found that PP2A activation events preceded Nrf2 activation, suggesting that PP2A activation is a novel biochemical effect of dithiolethiones [87]. Given that these compounds are well tolerated in humans, are clinically available and have unique anti-cancer properties in vitro, we propose that dithiolethiones could have beneficial clinical effects in cancer chemotherapy.
Apolipoprotein E (ApoE) is a major protein involved in lipid, vitamin and cholesterol transport. In addition to its metabolic roles, ApoE plays a part in disorders such as Alzheimer’s and cardiovascular diseases [102]. ApoE is also an immune modulator because it suppresses T cell proliferation and generally has anti-inflammatory effects [103]. However, immune suppression by ApoE could counteract its potential anti-tumor effects; this is an issue that needs to be resolved for this class of PP2A agonists. The anti-inflammatory effects of ApoE are also observed with ApoE-based peptides [104]. Recently an ApoE-based peptide, COG112, was described to interact with the I2PP2A oncoprotein, and this interaction resulted in increased PP2A activity [105]. Furthermore, this effect was recently observed in human cancer cell lines because the ApoE peptide causes a dose-dependent release of PP2A C subunit from I2PP2A, resulting in increased PP2A activity [86]. COG112 also suppressed EGF-induced Akt and c-Myc signaling in an okadaic acid-sensitive manner and inhibited cancer cell proliferation [86]. These new reports indicate that targeting I2PP2A is a novel approach to increasing tumor suppressor PP2A activity [86].
Targeting I2PP2A has multiple potential benefits. In addition to inhibiting PP2A, I2PP2A also inhibits nm23-H1, a metastatic suppressor protein [106]. nm23-H1 is sequestered in the cytosol by binding to I2PP2A. Upon T cell activation, granzyme A cleaves the I2PP2A protein, which releases nm23-H1 to enter the nucleus and act as an exonuclease to cleave DNA [106]. In addition, I2PP2A regulates Rac1-mediated cell migration [107]. As part of the Rac1 complex, I2PP2A presumably acts to inhibit PP2A to allow for Rac1 phosphorylation [107]. We recently demonstrated that the ApoE-based peptide COG112 inhibited multiple I2PP2A oncoprotein functions – including PP2A and nm23-H1 inactivation – and decreased Rac1-mediated cell migration [86]. Migration, metastasis and tumor suppressor inhibition are manifested by NO signaling in breast cancer cells. Therefore, targeting the many I2PP2A oncoprotein functions alters multiple pathways activated by NO.
Concluding remarks
The processes of cancer initiation, proliferation and metastasis are mediated by several oncogenic signaling pathways that work in concert to advance the disease state. Many recent reports have confirmed the role of NO in promoting various cancers, including ER(−) and basal-like breast cancer [19,108,109]. NOS2 expression is a strong predictor of poor survival in ER(−) and basal-like patients and is associated with the expression of basal-like signature genes [19]. Some of these genes are independently poor prognostic markers in breast cancer, such as CD44, IL-8, Wnt5A and KLF5 [19]. NO signaling activates multiple cancer-related signaling pathways to promote the basal-like phenotype and cancer cell aggressiveness [110]. For example, ERK-1/2, PI3K/Akt, c-Myc, and HIF are some of the signaling pathways activated by NO in breast cancer cells [19,25,110]. NO does not activate all of these pathways in a uniform manner. Rather, the activation of signaling pathways by NO is achieved at threshold concentrations, and this accounts for the disparate effects of NO signaling in cancer (Figure 2) [25,35]. Therapeutic intervention against NO signaling in breast cancer can either target NOS2 activity itself or the multiple pathways activated by NO signaling. The tumor suppressor PP2A is the endogenous negative regulator of many cancer-related signaling pathways [57,61], including those activated by NO. PP2A activity is inhibited in cancer cells by multiple mechanisms [57]; however, PP2A phosphatase activity can be pharmacologically increased [86-88]. We propose that NO signaling is a major driving force in ER(−) and basal-like breast cancer and that PP2A activation could be a novel therapeutic mechanism to limit the diverse effects of NO signaling in cancer (Figure 1).
To further advance the pharmacology of PP2A agonists, high-throughput assays need to be developed and chemical libraries should be screened for PP2A activation. In addition, the chemical biology of PP2A activation needs to be further elucidated to advance pharmacophore discovery. Furthermore, pharmacological agents that target inhibitors of PP2A, such as Src and EGFR, could represent another approach to blocking PP2A inhibition by cancer cells.
Oncogenic kinases have a clear role in promoting and proliferating cancers; however, the inhibition of tumor suppressors is equally culpable in cancer biology [111]. Another area of research that needs attention is the role of NO signaling in inhibiting crucial tumor suppressors such as PP2A, PTEN and BRCA1. We predict that NO chemical biology will have a profound effect on a number of tumor suppressor genes and that this represents a novel area of basic and translational research.
Given the vast array of NO signaling effects on cancer-related oncogenic pathways and the clinical relevance of NOS2 in relation to patient survival, we suggest that NOS2 is a proto-oncogene in aggressive ER(−) breast tumors. NOS2 signaling and its putative role as a proto-oncogene warrants further examination in other cancers such as prostate, hepatocellular carcinoma, colorectal and non-small-cell lung tumors.
References
- 1.Wink DA, et al. The reemergence of nitric oxide and cancer. Nitric Oxide. 2008;19:65–67. doi: 10.1016/j.niox.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mocellin S, et al. Nitric oxide, a double edged sword in cancer biology: Searching for therapeutic opportunities. Med Res Rev. 2007;27:317–352. doi: 10.1002/med.20092. [DOI] [PubMed] [Google Scholar]
- 3.Cheng R, et al. Nitric oxide and cancer: an overview. In: Bonavida B, editor. Nitric Oxide (NO) and Cancer. Springer; 2010. pp. 3–20. [Google Scholar]
- 4.Hofseth LJ, et al. p53: 25 years after its discovery. Trends Pharmacol Sci. 2004;25:177–181. doi: 10.1016/j.tips.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 5.Wink DA, et al. Nitric oxide and redox mechanisms in the immune response. J Leukoc Biol. 2011;89:873–891. doi: 10.1189/jlb.1010550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ambs S, et al. Cancer-prone oxyradical overload disease. IARC Sci Publ. 1999:295–302. [PubMed] [Google Scholar]
- 7.Forrester K, et al. Nitric oxide-induced p53 accumulation and regulation of inducible nitric oxide synthase expression by wild-type p53. Proc Natl Acad Sci U S A. 1996;93:2442–2447. doi: 10.1073/pnas.93.6.2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ambs S, et al. Up-regulation of inducible nitric oxide synthase expression in cancer-prone p53 knockout mice. Proc Natl Acad Sci U S A. 1998;95:8823–8828. doi: 10.1073/pnas.95.15.8823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hofseth LJ, et al. Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proc Natl Acad Sci U S A. 2003;100:143–148. doi: 10.1073/pnas.0237083100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brune B, Schneiderhan N. Nitric oxide evoked p53-accumulation and apoptosis. Toxicol Lett. 2003;139:119–123. doi: 10.1016/s0378-4274(02)00426-5. [DOI] [PubMed] [Google Scholar]
- 11.Wang C, et al. Thresholds of nitric oxide-mediated toxicity in human lymphoblastoid cells. Chem Res Toxicol. 2003;16:1004–1013. doi: 10.1021/tx0340448. [DOI] [PubMed] [Google Scholar]
- 12.Ambs S, et al. p53 and vascular endothelial growth factor regulate tumor growth of NOS2-expressing human carcinoma cells. Nat Med. 1998;4:1371–1376. doi: 10.1038/3957. [DOI] [PubMed] [Google Scholar]
- 13.Anttila MA, et al. Prognostic significance of iNOS in epithelial ovarian cancer. Gynecol Oncol. 2007;105:97–103. doi: 10.1016/j.ygyno.2006.10.049. [DOI] [PubMed] [Google Scholar]
- 14.Puhakka A, et al. High expression of nitric oxide synthases is a favorable prognostic sign in non-small cell lung carcinoma. APMIS. 2003;111:1137–1146. doi: 10.1111/j.1600-0463.2003.apm1111210.x. [DOI] [PubMed] [Google Scholar]
- 15.Marrogi AJ, et al. Nitric oxide synthase, cyclooxygenase 2, and vascular endothelial growth factor in the angiogenesis of non-small cell lung carcinoma. Clin Cancer Res. 2000;6:4739–4744. [PubMed] [Google Scholar]
- 16.Miles D, et al. Association between biosynthesis of nitric oxide and changes in immunological and vascular parameters in patients treated with interleukin-2. Eur J Clin Invest. 1994;24:287–290. doi: 10.1111/j.1365-2362.1994.tb01087.x. [DOI] [PubMed] [Google Scholar]
- 17.Prueitt RL, et al. Inflammation and IGF-I activate the Akt pathway in breast cancer. Int J Cancer. 2007;120:796–805. doi: 10.1002/ijc.22336. [DOI] [PubMed] [Google Scholar]
- 18.Lo H-W, et al. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005;7:575–589. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 19.Glynn SA, et al. Increased NOS2 predicts poor survival in estrogen receptor-negative breast cancer patients. J Clin Invest. 2010;120:3843–3854. doi: 10.1172/JCI42059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glynn S, et al. COX-2 activation is associated with Akt phosphorylation and poor survival in ER-negative, HER2-positive breast cancer. BMC Cancer. 2010;10:626. doi: 10.1186/1471-2407-10-626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klemm F, et al. β-Catenin-independent WNT signaling in basal-like breast cancer and brain metastasis. Carcinogenesis. 2011;32:434–442. doi: 10.1093/carcin/bgq269. [DOI] [PubMed] [Google Scholar]
- 22.Tong D, et al. Expression of KLF5 is a prognostic factor for disease-free survival and overall survival in patients with breast cancer. Clin Cancer Res. 2006;12:2442–2448. doi: 10.1158/1078-0432.CCR-05-0964. [DOI] [PubMed] [Google Scholar]
- 23.Stuehr D, et al. Oxygen reduction by nitric-oxide synthases. J Biol Chem. 2001;276:14533–14536. doi: 10.1074/jbc.R100011200. [DOI] [PubMed] [Google Scholar]
- 24.Finkel T. Redox-dependent signal transduction. FEBS Lett. 2000;476:52–54. doi: 10.1016/s0014-5793(00)01669-0. [DOI] [PubMed] [Google Scholar]
- 25.Thomas DD, et al. The chemical biology of nitric oxide: implications in cellular signaling. Free Radic Biol Med. 2008;45:18–31. doi: 10.1016/j.freeradbiomed.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mujoo K, et al. Role of soluble guanylyl cyclase–cyclic GMP signaling in tumor cell proliferation. Nitric Oxide. 2010;22:43–50. doi: 10.1016/j.niox.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liao D, Johnson RS. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007;26:281–290. doi: 10.1007/s10555-007-9066-y. [DOI] [PubMed] [Google Scholar]
- 28.Thomas DD, et al. Hypoxic inducible factor 1α, extracellular signal-regulated kinase, and p53 are regulated by distinct threshold concentrations of nitric oxide. Proc Natl Acad Sci U S A. 2004;101:8894–8899. doi: 10.1073/pnas.0400453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benita Y, et al. An integrative genomics approach identifies Hypoxia Inducible Factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res. 2009;37:4587–4602. doi: 10.1093/nar/gkp425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pervin S, et al. Nitric oxide in physiologic concentrations targets the translational machinery to increase the proliferation of human breast cancer cells: involvement of mammalian target of rapamycin/eIF4E pathway. Cancer Res. 2007;67:289–299. doi: 10.1158/0008-5472.CAN-05-4623. [DOI] [PubMed] [Google Scholar]
- 31.Meloche S, Pouyssegur J. The ERK1//2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26:3227–3239. doi: 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- 32.Chen H, et al. Extracellular signal-regulated kinase signaling pathway regulates breast cancer cell migration by maintaining slug expression. Cancer Res. 2009;69:9228–9235. doi: 10.1158/0008-5472.CAN-09-1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gupta S, Davis RJ. MAP kinase binds to the NH2-terminal activation domain of c-Myc. FEBS Lett. 1994;353:281–285. doi: 10.1016/0014-5793(94)01052-8. [DOI] [PubMed] [Google Scholar]
- 34.Marais R, et al. The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell. 1993;73:381–393. doi: 10.1016/0092-8674(93)90237-k. [DOI] [PubMed] [Google Scholar]
- 35.Ridnour LA, et al. Molecular mechanisms for discrete nitric oxide levels in cancer. Nitric Oxide. 2008;19:73–76. doi: 10.1016/j.niox.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim D, et al. AKT/PKB signaling mechanisms in cancer and chemoresistance. Front Biosci. 2005;10:975–987. doi: 10.2741/1592. [DOI] [PubMed] [Google Scholar]
- 37.Carnero A, et al. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets. 2008;8:187–198. doi: 10.2174/156800908784293659. [DOI] [PubMed] [Google Scholar]
- 38.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 39.Foster MW, et al. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med. 2009;15:391–404. doi: 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oda K, et al. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol Syst Biol. 2005;1 doi: 10.1038/msb4100014. 2005.0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akhand AA, et al. Nitric oxide controls src kinase activity through a sulfhydryl group modification-mediated Tyr-527-independent and Tyr-416-linked mechanism. J Biol Chem. 1999;274:25821–25826. doi: 10.1074/jbc.274.36.25821. [DOI] [PubMed] [Google Scholar]
- 42.Rahman MA, et al. S-nitrosylation at cysteine 498 of c-Src tyrosine kinase regulates nitric oxide-mediated cell invasion. J Biol Chem. 2010;285:3806–3814. doi: 10.1074/jbc.M109.059782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guarino M. Src signaling in cancer invasion. J Cell Physiol. 2010;223:14–26. doi: 10.1002/jcp.22011. [DOI] [PubMed] [Google Scholar]
- 44.Lander HM, et al. A molecular redox switch on p21(ras). Structural basis for the nitric oxide-p21(ras) interaction. J Biol Chem. 1997;272:4323–4326. doi: 10.1074/jbc.272.7.4323. [DOI] [PubMed] [Google Scholar]
- 45.McCubrey JA, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 47.Khramtsov AI et al. Wnt/β-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am J Pathol. 2010;176:2911–2920. doi: 10.2353/ajpath.2010.091125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Geyer FC, et al. β-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mod Pathol. 2011;24:209–231. doi: 10.1038/modpathol.2010.205. [DOI] [PubMed] [Google Scholar]
- 49.Wang H, et al. Nitric oxide increases Wnt-induced secreted protein-1 (WISP-1/CCN4) expression and function in colitis. J Mol Med. 2009;87:435–445. doi: 10.1007/s00109-009-0445-4. [DOI] [PubMed] [Google Scholar]
- 50.Santos A, et al. Early activation of the β-catenin pathway in osteocytes is mediated by nitric oxide, phosphatidyl inositol-3 kinase/Akt, and focal adhesion kinase. Biochem Biophys Res Commun. 2010;391:364–369. doi: 10.1016/j.bbrc.2009.11.064. [DOI] [PubMed] [Google Scholar]
- 51.Liu Y, et al. Polyoma enhancer activator 3, an ets transcription factor, mediates the induction of cyclooxygenase-2 by nitric oxide in colorectal cancer cells. J Biol Chem. 2004;279:18694–18700. doi: 10.1074/jbc.M308136200. [DOI] [PubMed] [Google Scholar]
- 52.Bailey A, et al. The tragedy of TRIUMPH for nitric oxide synthesis inhibition in cardiogenic shock: where do we go from here? Am J Cardiovasc Drugs. 2007;7:337–345. doi: 10.2165/00129784-200707050-00003. [DOI] [PubMed] [Google Scholar]
- 53.Brindicci C, et al. Effects of aminoguanidine, an inhibitor of inducible nitric oxide synthase, on nitric oxide production and its metabolites in healthy control subjects, healthy smokers, and COPD patients. Chest. 2009;135:353–367. doi: 10.1378/chest.08-0964. [DOI] [PubMed] [Google Scholar]
- 54.Hesslinger C, et al. Inhibition of inducible nitric oxide synthase in respiratory diseases. Biochem Soc Trans. 2009;37:886–891. doi: 10.1042/BST0370886. [DOI] [PubMed] [Google Scholar]
- 55.Frederiksen LJ, et al. Chemosensitization of cancer in vitro and in vivo by nitric oxide signaling. Clin Cancer Res. 2007;13:2199–2206. doi: 10.1158/1078-0432.CCR-06-1807. [DOI] [PubMed] [Google Scholar]
- 56.Zhang J, et al. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 57.Westermarck J, Hahn WC. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol Med. 2008;14:152–160. doi: 10.1016/j.molmed.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 58.Ostman A, et al. Protein-tyrosine phosphatases and cancer. Nat Rev Cancer. 2006;6:307–320. doi: 10.1038/nrc1837. [DOI] [PubMed] [Google Scholar]
- 59.Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sablina AA, Hahn WC. The role of PP2A A subunits in tumor suppression. Cell Adhes Migr. 2007;1:140–141. doi: 10.4161/cam.1.3.4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eichhorn PJA, et al. Protein phosphatase 2A regulatory subunits and cancer. Biochim Biophys Acta: Rev Cancer. 2009;1795:1–15. doi: 10.1016/j.bbcan.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 62.Arroyo JD, Hahn WC. Involvement of PP2A in viral and cellular transformation. Oncogene. 2005;24:7746–7755. doi: 10.1038/sj.onc.1209038. [DOI] [PubMed] [Google Scholar]
- 63.Xu Y, et al. Structure of the protein phosphatase 2A holoenzyme. Cell. 2006;127:1239–1251. doi: 10.1016/j.cell.2006.11.033. [DOI] [PubMed] [Google Scholar]
- 64.Ruediger R, et al. Disruption of protein phosphatase 2A subunit interaction in human cancers with mutations in the A alpha subunit gene. Oncogene. 2001;20:10–15. doi: 10.1038/sj.onc.1204059. [DOI] [PubMed] [Google Scholar]
- 65.Virshup DM, Shenolikar S. From promiscuity to precision: protein phosphatases get a makeover. Mol Cell. 2009;33:537–545. doi: 10.1016/j.molcel.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 66.Schmidt A, et al. Endostatin influences endothelial morphology via the activated ERK1/2-kinase endothelial morphology and signal transduction. Microvasc Res. 2006;71:152–162. doi: 10.1016/j.mvr.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 67.Ugi S, et al. Protein phosphatase 2A negatively regulates insulin’s metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol Cell Biol. 2004;24:8778–8789. doi: 10.1128/MCB.24.19.8778-8789.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Resjö S, et al. Protein phosphatase 2A is the main phosphatase involved in the regulation of protein kinase B in rat adipocytes. Cell Signal. 2002;14:231–238. doi: 10.1016/s0898-6568(01)00238-8. [DOI] [PubMed] [Google Scholar]
- 69.Seeling JM, et al. Regulation of β-catenin signaling by the B56 subunit of protein phosphatase 2A. Science. 1999;283:2089–2091. doi: 10.1126/science.283.5410.2089. [DOI] [PubMed] [Google Scholar]
- 70.Arnold HK, Sears RC. Protein phosphatase 2A regulatory subunit B56{alpha} associates with c-Myc and negatively regulates c-Myc accumulation. Mol Cell Biol. 2006;26:2832–2844. doi: 10.1128/MCB.26.7.2832-2844.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li H-H, et al. A specific PP2A regulatory subunit, B56g, mediates DNA damage-induced dephosphorylation of p53 at Thr55. EMBO J. 2007;26:402–411. doi: 10.1038/sj.emboj.7601519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yan L, et al. PP2A regulates the pro-apoptotic activity of FOXO1. J Biol Chem. 2008;283:7411–7420. doi: 10.1074/jbc.M708083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Witt J, et al. Mechanism of PP2A-mediated IKKbeta dephosphorylation: a systems biological approach. BMC Syst Biol. 2009;3:71. doi: 10.1186/1752-0509-3-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Krauss S, et al. Protein phosphatase 2A and rapamycin regulate the nuclear localization and activity of the transcription factor GLI3. Cancer Res. 2008;68:4658–4665. doi: 10.1158/0008-5472.CAN-07-6174. [DOI] [PubMed] [Google Scholar]
- 75.Sablina AA, Hahn WC. SV40 small T antigen and PP2A phosphatase in cell transformation. Cancer Metastasis Rev. 2008;27:137–146. doi: 10.1007/s10555-008-9116-0. [DOI] [PubMed] [Google Scholar]
- 76.Chen W, et al. Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell. 2004;5:127–136. doi: 10.1016/s1535-6108(04)00026-1. [DOI] [PubMed] [Google Scholar]
- 77.Chen W, et al. Cancer-associated PP2A Aα subunits induce functional haploinsufficiency and tumorigenicity. Cancer Res. 2005;65:8183–8192. doi: 10.1158/0008-5472.CAN-05-1103. [DOI] [PubMed] [Google Scholar]
- 78.Xing Y, et al. Structural mechanism of demethylation and inactivation of protein phosphatase 2A. Cell. 2008;133:154–163. doi: 10.1016/j.cell.2008.02.041. [DOI] [PubMed] [Google Scholar]
- 79.Chen J, et al. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science. 1992;257:1261–1264. doi: 10.1126/science.1325671. [DOI] [PubMed] [Google Scholar]
- 80.Junttila MR, et al. CIP2A inhibits PP2A in human malignancies. Cell. 2007;130:51–62. doi: 10.1016/j.cell.2007.04.044. [DOI] [PubMed] [Google Scholar]
- 81.Dobson S, et al. Characterization of a unique aspartate-rich protein of the SET/TAF-family in the human malaria parasite, Plasmodium falciparum, which inhibits protein phosphatase 2A. Mol Biochem Parasitol. 2003;126:239–250. doi: 10.1016/s0166-6851(02)00293-1. [DOI] [PubMed] [Google Scholar]
- 82.Mukhopadhyay A, et al. Direct interaction between the inhibitor 2 and ceramide via sphingolipid-protein binding is involved in the regulation of protein phosphatase 2A activity and signaling. FASEB J. 2009;23:751–763. doi: 10.1096/fj.08-120550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Foley TD, et al. Oxidative inhibition of protein phosphatase 2A activity: role of catalytic subunit disulfides. Neurochem Res. 2007;32:1957–1964. doi: 10.1007/s11064-007-9394-x. [DOI] [PubMed] [Google Scholar]
- 84.Kamata T. Roles of Nox1 and other Nox isoforms in cancer development. Cancer Sci. 2009;100:1382–1388. doi: 10.1111/j.1349-7006.2009.01207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen L, et al. Hydrogen peroxide-induced neuronal apoptosis is associated with inhibition of protein phosphatase 2A and 5, leading to activation of MAPK pathway. Int J Biochem Cell Biol. 2009;41:1284–1295. doi: 10.1016/j.biocel.2008.10.029. [DOI] [PubMed] [Google Scholar]
- 86.Switzer CH, et al. Targeting SET/I2PP2A oncoprotein functions as a multi-pathway strategy for cancer therapy. Oncogene. 2011;30:2504–2513. doi: 10.1038/onc.2010.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Switzer CH, et al. Dithiolethione compounds inhibit Akt signaling in human breast and lung cancer cells by increasing PP2A activity. Oncogene. 2009;28:3837–3846. doi: 10.1038/onc.2009.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Perrotti D, Neviani P. Protein phosphatase 2A (PP2A), a drugable tumor suppressor in Ph1+ leukemias. Cancer Metastasis Rev. 2008;27:159–168. doi: 10.1007/s10555-008-9119-x. [DOI] [PubMed] [Google Scholar]
- 89.URBICH C, et al. Dephosphorylation of endothelial nitric oxide synthase contributes to the anti-angiogenic effects of endostatin. FASEB J. 2002;16:706–708. doi: 10.1096/fj.01-0637fje. [DOI] [PubMed] [Google Scholar]
- 90.Kulke MH, et al. Phase II study of recombinant human endostatin in patients with advanced neuroendocrine tumors. J Clin Oncol. 2006;24:3555–3561. doi: 10.1200/JCO.2006.05.6762. [DOI] [PubMed] [Google Scholar]
- 91.Zhao X, et al. A randomized phase II study of recombinant human endostatin plus gemcitabine/cisplatin compared with gemcitabine/cisplatin alone as first-line therapy in advanced non-small-cell lung cancer. Invest New Drugs. 2011 doi: 10.1007/s10637-011-9631-7. [DOI] [PubMed] [Google Scholar]
- 92.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 93.Cornell TT, et al. Ceramide-dependent PP2A regulation of TNFα-induced IL-8 production in respiratory epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2009;296:L849–L856. doi: 10.1152/ajplung.90516.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Magrassi L, et al. Vitamin D metabolites activate the sphingomyelin pathway and induce death of glioblastoma cells. Acta Neurochir (Wien) 1998;140:707–713. doi: 10.1007/s007010050166. [DOI] [PubMed] [Google Scholar]
- 95.Canals D, et al. Drug targeting of sphingolipid metabolism: sphingomyelinases and ceramidases. Br J Pharmacol. 2011;163:694–712. doi: 10.1111/j.1476-5381.2011.01279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bettaïeb A, et al. Daunorubicin- and mitoxantrone-triggered phosphatidylcholine hydrolysis: implication in drug-induced ceramide generation and apoptosis. Mol Pharmacol. 1999;55:118–125. doi: 10.1124/mol.55.1.118. [DOI] [PubMed] [Google Scholar]
- 97.Tagaram HRS, et al. Nanoliposomal ceramide prevents in vivo growth of hepatocellular carcinoma. Gut. 2011;60:695–701. doi: 10.1136/gut.2010.216671. [DOI] [PubMed] [Google Scholar]
- 98.Zhang Y, Munday R. Dithiolethiones for cancer chemoprevention: where do we stand? Mol. Cancer Ther. 2008;7:3470–3479. doi: 10.1158/1535-7163.MCT-08-0625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee J-S, Surh Y-J. Nrf2 as a novel molecular target for chemoprevention. Cancer Lett. 2005;224:171–184. doi: 10.1016/j.canlet.2004.09.042. [DOI] [PubMed] [Google Scholar]
- 100.Bass SE, et al. Novel dithiolethione-modified nonsteroidal anti-inflammatory drugs in human hepatoma HepG2 and colon LS180 Cells. Clin Cancer Res. 2009;15:1964–1972. doi: 10.1158/1078-0432.CCR-08-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lam S, et al. A randomized phase IIb trial of anethole dithiolethione in smokers with bronchial dysplasia. J Natl Cancer Inst. 2002;94:1001–1009. doi: 10.1093/jnci/94.13.1001. [DOI] [PubMed] [Google Scholar]
- 102.Huang Y. Mechanisms linking apolipoprotein E isoforms with cardiovascular and neurological diseases. Curr Opin Lipidol. 2010;21:337–345. doi: 10.1097/MOL.0b013e32833af368. [DOI] [PubMed] [Google Scholar]
- 103.Jofre-Monseny L, et al. Impact of apoE genotype on oxidative stress, inflammation and disease risk. Mol Nutr Food Res. 2008;52:131–145. doi: 10.1002/mnfr.200700322. [DOI] [PubMed] [Google Scholar]
- 104.Singh K, et al. The apolipoprotein E-mimetic peptide COG112 inhibits the inflammatory response to Citrobacter rodentium in colonic epithelial cells by preventing NF-kappaB Activation. J Biol Chem. 2008;283:16752–16761. doi: 10.1074/jbc.M710530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Christensen D, et al. Apolipoprotein-E and peptide mimetics modulate inflammation by binding the SET Protein and activating protein phosphatase 2A. J Immunol. 2011;186:2535–2542. doi: 10.4049/jimmunol.1002847. [DOI] [PubMed] [Google Scholar]
- 106.Fan Z, et al. Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell. 2003;112:659–672. doi: 10.1016/s0092-8674(03)00150-8. [DOI] [PubMed] [Google Scholar]
- 107.ten Klooster JP, et al. Rac1-induced cell migration requires membrane recruitment of the nuclear oncogene SET. EMBO J. 2007;26:336–345. doi: 10.1038/sj.emboj.7601518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fukumura D, et al. The role of nitric oxide in tumour progression. Nat Rev Cancer. 2006;6:521–534. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 109.Yang GY, et al. Induced nitric oxide synthase as a major player in the oncogenic transformation of inflamed tissue. Methods Mol Biol. 2009;512:119–156. doi: 10.1007/978-1-60327-530-9_8. [DOI] [PubMed] [Google Scholar]
- 110.Ambs S, Glynn SA. Candidate pathways linking inducible nitric oxide synthase to a basal-like transcription pattern and tumor progression in human breast cancer. Cell Cycle. 2011;10:619–624. doi: 10.4161/cc.10.4.14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 112.Platet N, et al. Estrogens and their receptors in breast cancer progression: a dual role in cancer proliferation and invasion. Crit Rev Oncol Hematol. 2004;51:55–67. doi: 10.1016/j.critrevonc.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 113.Parl FF, et al. Prognostic significance of estrogen receptor status in breast cancer in relation to tumor stage, axillary node metastasis, and histopathologic grading. Cancer. 1984;54:2237–2242. doi: 10.1002/1097-0142(19841115)54:10<2237::aid-cncr2820541029>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 114.Sørlie T, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sorlie T, et al. Distinct molecular mechanisms underlying clinically relevant subtypes of breast cancer: gene expression analyses across three different platforms. BMC Genomics. 2006;7:127. doi: 10.1186/1471-2164-7-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rakha EA, et al. Basal-like breast cancer: a critical review. J Clin Oncol. 2008;26:2568–2581. doi: 10.1200/JCO.2007.13.1748. [DOI] [PubMed] [Google Scholar]
- 117.Reis-Filho JS, Tutt ANJ. Triple negative tumours: a critical review. Histopathology. 2008;52:108–118. doi: 10.1111/j.1365-2559.2007.02889.x. [DOI] [PubMed] [Google Scholar]
- 118.Cleator S, et al. Triple-negative breast cancer: therapeutic options. Lancet Oncol. 2007;8:235–244. doi: 10.1016/S1470-2045(07)70074-8. [DOI] [PubMed] [Google Scholar]