

Abstract
Patients with acute myeloid leukemia (AML) and a FLT3 internal tandem duplication (ITD) mutation have a poor prognosis, and FLT3 inhibitors are now under clinical investigation. PIM1, a serine/threonine kinase, is up-regulated in FLT3-ITD AML and may be involved in FLT3-mediated leukemogenesis. We employed a PIM1 inhibitor, AR00459339 (Array Biopharma Inc.), to investigate the effect of PIM1 inhibition in FLT3-mutant AML. Like FLT3 inhibitors, AR00459339 was preferentially cytotoxic to FLT3-ITD cells, as demonstrated in the MV4-11, Molm-14, and TF/ITD cell lines, as well as 12 FLT3-ITD primary samples. Unlike FLT3 inhibitors, AR00459339 did not suppress phosphorylation of FLT3, but did promote the de-phosphorylation of downstream FLT3 targets, STAT5, AKT, and BAD. Combining AR00459339 with a FLT3 inhibitor resulted in additive to mildly synergistic cytotoxic effects. AR00459339 was cytotoxic to FLT3-ITD samples from patients with secondary resistance to FLT3 inhibitors, suggesting a novel benefit to combining these agents. We conclude that PIM1 appears to be closely associated with FLT3 signaling, and that inhibition of PIM1 may hold therapeutic promise, either as monotherapy, or by overcoming resistance to FLT3 inhibitors.
Introduction
Approximately one quarter of patients with acute myeloid leukemia (AML) harbor an internal tandem duplication mutation of the FMS-like tyrosine kinase receptor (FLT3-ITD mutation). These patients often present with leukocytosis, experience a high relapse rate, and have markedly decreased overall survival compared to AML patients lacking FLT3 mutations.[1–6] The exception to this rule may be the coexistence of an NPM1 mutation with the FLT3 mutation, which has been associated with similar relapse-free and overall survival when compared to wild-type AML.[7, 8] In response to the poor prognosis associated with FLT3-ITD mutations, a number of FLT3 tyrosine kinase inhibitors (TKI) are under development, with some being evaluated in clinical trials as single agent therapy while others are being combined with chemotherapy regimens.[9–13]
PIM (proviral integration site for Moloney murine leukemia virus) is a gene that was identified when its expression was noted to be up-regulated by proviral insertion in murine virus-induced T-cell lymphomas.[14] The PIM proteins, the products of a family of proto-oncogenes, are serine-threonine kinases with increased expression in a variety of malignancies. [15–20] PIM kinases have been found to play an important role in enhancing cell survival and suppressing apoptosis in hematopoietic cells.[21, 22] Of the PIM proteins, PIM1 and PIM2 have been most extensively studied in AML. Their expression appears to be up-regulated by STAT5, and they have been found to be over-expressed in primary AML blast samples.[16, 23]
In particular, PIM1 and PIM2 have been associated with FLT3 mediated leukemogenesis in FLT3-ITD AML. PIM1 expression was noted to be 25-fold higher than in FLT3-ITD samples, as compared to wild type FLT3 (WT) AML samples.[18] When FLT3 was inhibited by the TKI lestaurtinib there was also a corresponding decrease in the PIM1 protein product, suggesting PIM1 to be a down-stream target of FLT3, possibly through the latter’s activation of STAT5. In a subsequent study, FLT3 inhibition was shown to lead to a decrease in serine phosphorylation of the anti-apoptotic BAD (Bcl2 antagonist of cell death).[24] It was postulated that PIM1 was involved in FLT3-ITD-mediated leukemogenesis by phosphorylating BAD (at serine 112) and thus promoting blast survival. PIM2 expression has also been studied in FLT3-ITD AML and likewise demonstrated to be up-regulated.[16] Another study suggested that over-expression of PIM2 can transform wild type FLT3 cells, suggesting that PIM2 and FLT3 may act through different, but complementary, pathways to stimulate cell cycling and inhibit apoptosis.[25]
The PIM kinases, therefore, represent potential therapeutic targets in AML, particularly in those cases harboring FLT3-ITD mutations. Indeed, siRNA-mediated down-regulation of PIM proteins has been demonstrated to decrease survival of MV4-11 FLT3-ITD cell lines.[26] We have investigated the effects of a small molecule inhibitor of PIM1, AR00459339, alone and in combination with a FLT3 inhibitor (AR00454200), on AML cell lines and primary samples. We have found that inhibition of PIM1 results in significant cytotoxicity in FLT3-ITD cell lines and patient samples that strikingly parallels the effects of FLT3 inhibition. In addition, we present evidence of downstream effects of PIM1 on proteins in the FLT3-ITD signaling pathway. Our findings support the notion that PIM1 is integral to the process of leukemic transformation in FLT3-ITD AML, and that it may be a valid therapeutic target for this disease.
Materials and Methods
Reagents
AR00459339 and AR00454200 were obtained from Array Biopharma Inc. (Boulder, CO). CEP-701 was obtained from Cephalon Inc (Frazer, PA). AC220 was obtained from Ambit Biosciences Inc (San Diego, CA). Sorafenib was obtained from LC laboratories (Woburn, MA). Inhibitors were dissolved in dimethyl sulfoxide (DMSO), stored at −80°C as a 10 mM stock solution, and diluted as needed. The DMSO concentration within any given experiment was the same for all samples, and in no case was it more than 0.1%. Ficoll-Hypaque was obtained from Amersham (Piscataway, NJ). Anti-FLT3, anti-STAT5, and anti-phosphoserine antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-phosphotyrosine antibody (4G10) was obtained from Upstate Biotechnology (Lake Placid, NY). Anti-PIM1 and anti-PIM2 and anti-phospho-STAT5 antibodies were obtained from Cell Signaling Technology, Inc. (Danvers, MA). Progenitor cell assay medium was obtained from Stem Cell Technologies (Vancouver, Canada). All other reagents were from Sigma (St. Louis, MO).
In vitro PIM Kinase Assays
In vitro kinase assays using the PIM inhibitor were performed in two ways. The first utilized a peptide substrate, and the second utilized purified BAD protein. The human isoforms of PIM1 and PIM2, as well as purified human BAD protein, were obtained from Millipore (Bedford, MA). The rat PIM3 isoform was expressed and purified by Array Biopharma. Each PIM isoform was analyzed using an ATP concentration equal to its Km (35μM, 4μM, 30μM, respectively) and 2.5x Km PIM2tide (7.5μM, 1μM, 3.8μM respectively, PIM2tide provided by Midwest BioTech (Fishers, IN) Cat #MBT2309, sequence H-RSRHSSYPAGT-OH, corresponding to residues 107–117 of murine BAD). Reactions were carried out for one hour at ambient temperature in the presence of 20 mM MOPS pH 7.4, 10 mM MgCl2, 0.005% Tween-20, 1 mM DTT & 1 μCi [γ-33P] ATP. The phosphorylated product was captured and quantitated on a Whatman P81 Unifilter plate (GE Healthcare, Piscataway, NJ). AR00459339 reported IC50 values of 0.8 nM, 52.4 nM & 3.6 nM for PIM1, PIM2 and PIM3 respectively.
For the in vitro kinase assay utilizing purified human BAD protein, reactions were performed in kinase buffer (40 mM Tris pH 7.4, 20 mM MgCl2, 0.01% bovine serum albumin, 0.5 mM ATP) with or without inhibitor. To 50 uL kinase buffer was added 1 nanogram purified PIM1 along with 2 micrograms of purified BAD. The reaction was incubated at 30°C for 30 minutes, then terminated by the addition of electrophoresis loading buffer followed by boiling. Proteins were resolved by western blotting (see below).
To generate the selectivity data, the Array compounds were evaluated with the Kinase Profiler® service from Millipore (Bedford, MA)
Cytotoxicity Assays
Cytotoxicity was assessed using a dimethyl-thiazol diphenyl tetrazolium bromide (MTT) assay. In selected cases, we also used an annexin V binding apoptosis assay to confirm that the cytotoxic effect observed using the MTT method was associated with apoptosis. MTT and annexin V assays were performed as described previously.[27] Briefly, for the MTT assay, 50 μL of 2x medium was aliquoted into triplicate wells of 96-well plates for each drug concentration point. All wells contained the same concentration of DMSO. Fifty μL cell suspension in medium was added to each well (50,000 cells/well for cell lines, 200,000 cells/well for AML blasts). Plates were incubated for 48 hours (cell lines) or 72 hours (AML blasts) at 37°C, 5% CO2, then the MTT assay was performed according to the manufacturer’s instructions (Roche, Indianapolis, IN). For the annexin V apoptosis assay, cells were incubated for 48 hours in medium with increasing AR00459339 concentration. Annexin V binding was then performed and assessed by flow cytometry according to the manufacturer’s instructions (Clontech, Palo Alto, CA).
For synergy studies using combinations of AR00459339 and AR00454200, TF/ITD cells (left) and a FLT3-ITD primary AML sample (right) were incubated with increasing concentrations of the PIM1 inhibitor AR00459339, the FLT3 inhibitor AR00454200, and the combination of the two drugs were fixed at a ratio of 100:1. At 72 hours, metabolic activity was determined using the MTT assay. Combination indices (CI) were generated using a commercially available software program (Calcusyn; Biosoft, Manchester, United Kingdom) after the method of Chou and Talalay.[28]
Cells
Cell lines used for these studies were all cultured at 37°C with 5% CO2 in RPMI medium with 10% fetal bovine serum (FBS) and penicillin/streptomycin (Invitrogen, Carlsbad, CA) and L-glutamine. MV4-11, Molm-14, HL60, and SEMK2 cells were obtained from American Type Culture Collection ([ATCC] Manassas, VA) or the German Collection of Microorganisms and Cell Cultures (Deutsche Sammlung von Mikroorganismen und Zellkulturen [DSMZ], Raunschweig, Germany). The TF/ITD cell line was generated by transfecting TF-1 cells (growth factor dependent) with an expression vector containing the FLT3 coding sequence containing an ITD mutation from an AML patient, as described previously.[10] The resultant TF/ITD cell line is growth factor independent and expresses constitutively phosphorylated FLT3.
Patient Samples
Bone marrow and peripheral blood from AML patients and from normal donors were obtained through a protocol approved by the Johns Hopkins institutional review board. All samples used in this study were from patients who gave informed consent according to the Declaration of Helsinki. A total number of 26 primary AML samples were used to document sensitivity to AR00459339, of which three samples had demonstrated a secondary resistance to the FLT3 inhibitor sorafenib. Mononuclear cells were isolated from these samples using density gradient centrifugation with Ficoll-Hypaque (GE Healthcare, Piscataway, NJ) The specimens were aliquoted and stored in liquid nitrogen in FBS plus 10% DMSO. Before use, the samples were thawed in warm culture medium (RPMI containing 10% FBS with pen/strep and L-glutamine) and incubated for 12 hours at 37°C in 5% CO2. The samples were pelleted and re-suspended in warm medium containing 150 international units/mL DNAse I (GE Healthcare, Piscataway, NJ) for 5 minutes. They were then re-centrifuged over Ficoll-Hypaque to obtain a high fraction of viable cells.
Immunoprecipitation and immunoblotting
Immunoprecipitation and immunoblotting were performed as described previously.[29] In brief, cells were incubated in culture medium with the indicated doses of AR00454200 or AR00459339 for various times at 37°C, then washed with ice-cold phosphate-buffered saline (PBS), and lysed with 1% NP-40 lysis buffer. Clarified lysate (500 μg per sample) was incubated at 4°C overnight with the intended monoclonal antibody for immunoprecipitation, followed by addition of protein A agarose (Millipore, Bedford, MA) for an additional 2 hours. After electrophoresis and transfer to Immobilon membranes (Millipore, Bedford, MA), immunoblotting was performed using the appropriate protein-specific antibodies. Protein bands were visualized using chemiluminescence and scanned with a Biorad GS800 densitometer.
Colony-forming assays
Mononuclear cells from normal donor bone marrow were isolated using Ficoll-Hypaque, then plated in triplicate in 35 mm Petri dishes using MethoCult® medium (H4434) from StemCell Technologies (Vancouver, BC, Canada). Granulocyte macrophage–colony-forming units (CFU-GMs) and erythroid–burst-forming units (BFU-Es) were scored after 14 days of incubation. CFU-GMs were defined as colonies of opaque cells and consisted of at least 50 cells. BFU-Es were defined as colonies of at least 50 hemoglobin-containing cells.
Results
AR00459339 is a selective inhibitor of PIM1
AR00459339 was analyzed using a kinase selectivity panel (Data not shown- Kinase Profiler®, Millipore, Bedford, MA). This analysis revealed that the compound inhibited all 3 PIM kinases, with the greatest inhibitory activity against PIM1. Of the 256 kinases in the panel, only 4 were inhibited to less than 10% of control- PIM1, PIM2, PIM3, and haspin (a histone-associated serine/threonine kinase.[30] We first analyzed AR00459339 using an in vitro kinase assay with a peptide substrate. As shown in figure 1A, this assay revealed potent inhibition of PIM1 (IC50 of 0.8 nM) and PIM3 (IC50 of 3.6 nM). The IC50 of inhibition of the PIM2 kinase was determined to be 52.4 nM, 66-fold higher than the IC50 for PIM1.
Figure 1. AR00459339 inhibits PIM1 kinase.
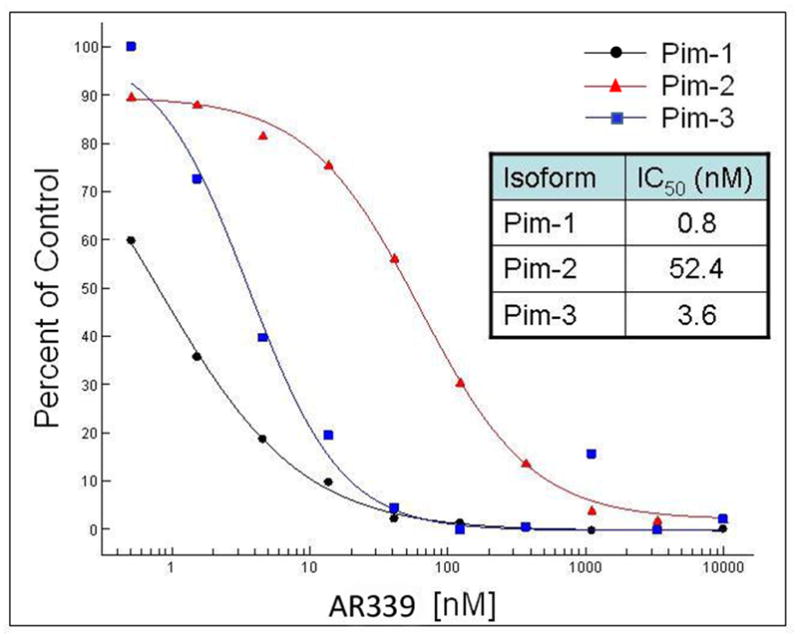
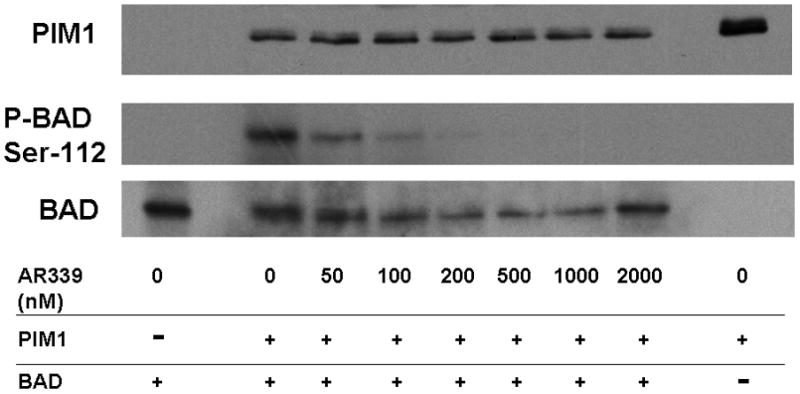
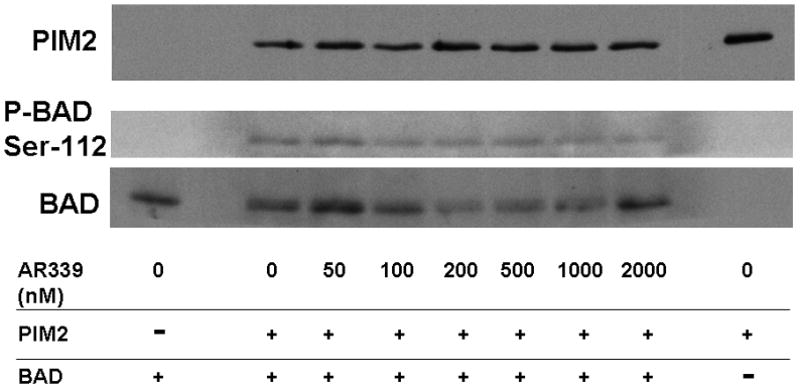
Note: AR00459339 is abbreviated “AR339” in this figure. (A) Using a peptide substrate (PIM2tide; see Methods), in-vitro kinase assays of PIM1, PIM2, and PIM3 were performed in the presence of increasing concentrations of AR00459339. (B) Exogenous PIM1 and (C) PIM2 enzyme, in the presence of BAD protein and increasing concentrations of AR00459339, were incubated in ATP-containing kinase buffer, and then resolved by electrophoresis. Immunoblot analysis using the anti-phospho-BAD antibody at serine 112 was performed, followed by stripping and re-probing with anti-BAD antibody.
We next wished to confirm the activity of AR00459339 with an in vitro kinase assay using one of the established protein substrates for PIM1 and PIM2, namely BAD.[24, 31] As shown in Figure 1B, phosphorylation of BAD by PIM1 at serine 112 is suppressed by AR00459339, where as levels of total BAD and PIM1 persist at levels of up to 2μM. Using this blot for densitometry, we calculated an IC50 for PIM1 inhibition of 39 nM. Consistent with the findings in the peptide substrate assay results described above, AR00459339 was much less effective at inhibiting PIM2 (Figure 1C). Inhibition of phosphorylation of serine 112 of BAD by PIM 2 in this assay was incomplete even at 2000 nM, with an estimated IC50 of 2040 nM.
AR00459339 is preferentially cytotoxic to FLT3-ITD cell lines and primary samples
Having established that AR00459339 could inhibit purified PIM proteins, we wished to examine its effects in AML cells. We first examined inhibition of phosphorylation of BAD at serine 112 in MV4-11 cells by AR00459339 (Figure 2A). In the context of a whole cell assay such as this, the IC50 for inhibiting BAD phosphorylation was 480 nM. This IC50 was considerably higher than that noted above in the in vitro kinase assay. This was not surprising, given the presence of protein binding and protein-protein interactions expected to affect target inhibition in a cell-based assay, in contrast to an in vitro kinase assay, where this is not a complicating factor and in which the proteins are often not in their native configurations. We next tested AR00459339 for cytotoxic effect on multiple AML cell lines, including MV4-11, Molm14, HL60, TF-1, and TF/ITD cells (Figure 2B). AR00459339 was cytotoxic to the FLT3-ITD cell lines MV4-11 (IC50 of 255 nM), Molm14 (IC50 of 484 nM), and TF/ITD cells (IC50 of 593 nM) (Figure 2B), corresponding in range to the IC50 for inhibition of BAD phosphorylation in the experiment shown in Figure 2A. Interestingly, AR00459339 was not significantly cytotoxic to HL60 (IC50 of 5490 nM) or TF-1 cells (IC50 of 4430 nM), which lack expression of mutant FLT3. For comparison, we tested all 5 cell lines against a novel FLT3 inhibitor, AR00454200. As can be seen in Figure 2C, the cytotoxic effects of the FLT3 inhibitor AR00454200 paralleled the pattern of cytotoxicity induced by the PIM inhibitor AR00459339.
Figure 2. AR00459339 inhibits serine phosphorylation of BAD and is preferentially cytotoxic to FLT3/ITD cell lines and primary samples.

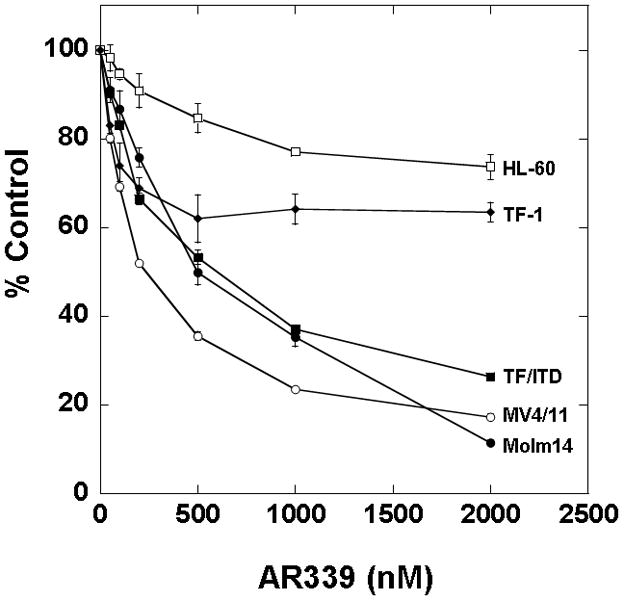
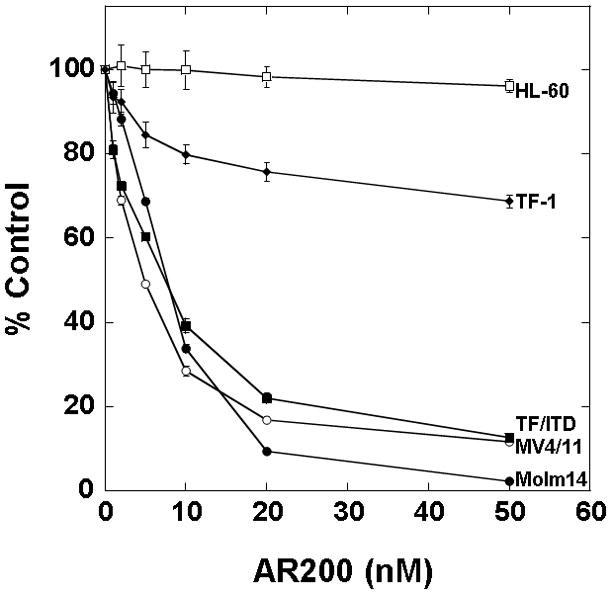
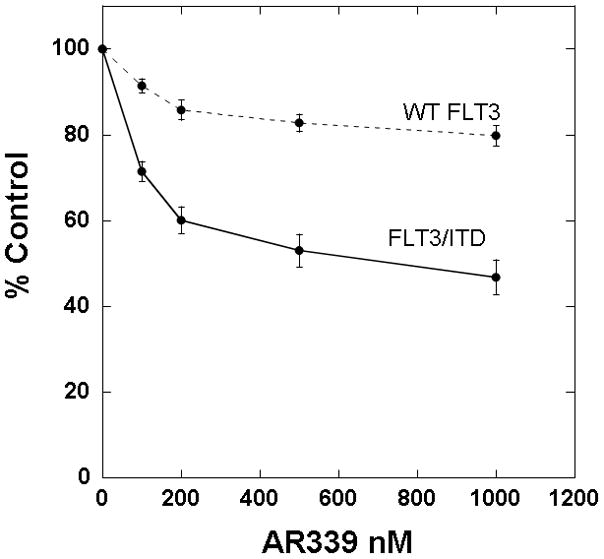
Note: AR00459339 is abbreviated “AR339” and AR00454200 is abbreviated “AR200” in these figures. (A) MV4-11 cells were exposed for 2 hours, in cell culture medium, to increasing concentrations of AR00459339. After resolution by protein electrophoresis, immunoblots were probed with the anti-phospho-BAD antibody at serine 112. The blot was subsequently stripped and re-probed with anti-BAD antibody. (B) Three FLT3/ITD (MV4-11, Molm14, TF/ITD) and two FLT3/WT (HL-60, TF-1) cell lines were incubated in cell culture medium with increasing concentrations of AR00459339. Each drug concentration point was performed in triplicate. At 72 hours, metabolic activity was determined using the MTT assay. Results are plotted for each sample as percentage (optical density [OD]) of untreated control. (C) The same cell lines were similarly incubated with increasing concentrations of the FLT3 inhibitor, AR00454200. (D) Twelve primary myeloblast samples from patients with FLT3/ITD mutations and 12 samples from patients with FLT3/WT AML were incubated with increasing concentrations of AR00459339. At 72 hours, metabolic activity was determined using the MTT assay. For each drug concentration, the percent control for all 12 samples in each group (WT and ITD) was expressed as a mean value, with error bars representing the standard error of the means. The final curves therefore represent the composite response curves for each group of 12 samples.
We then studied the effects of AR00459339 on a series of 24 primary AML samples, consisting of 12 FLT3-ITD and 12 FLT3-WT (FLT3 wild type) blasts (Figure 2D). Although clinical and cytogenetic profiles were incomplete on some of the primary samples tested, we did note that the FLT3-ITD samples consisted of two samples obtained at relapse, whereas the rest were obtained at diagnosis. Of the FLT3-ITD samples, one displayed a complex karyotype, and of the FLT3-WT samples, three displayed a complex karyotype and one displayed isochrome 7 and trisomy 17 alterations. Consistent with what we had observed in the cell lines, AR00459339 was selectively cytotoxic to FLT3-ITD samples. This selective cytotoxicity towards FLT3-ITD primary AML samples is essentially identical to what has been observed by numerous groups using different FLT3 inhibitors.[27, 29, 32, 33] Because the MTT assay is not a direct measurement of cell death, the induction of apoptosis in a FLT3-ITD primary sample by AR00459339 was confirmed using an annexin V assay. After exposure to AR00459339 for 72 hours, there was a dose responsive increase in apoptotic cells, with 76% of cells displaying annexin V positivity (Data not shown).
AR00459339 does not affect FLT3 autophosphorylation, but does lead to inhibition of downstream targets of FLT3
Having established that AR00459339 induces a cytotoxic effect on FLT3-ITD AML cells at concentrations corresponding to PIM1 inhibition in whole cells, we wished to further investigate the basis of AR00459339-induced cytotoxicity We therefore examined the effects of AR00459339 on the phosphorylation status of downstream proteins in the FLT3-ITD signaling pathway. Constitutive activation of FLT3 by ITD mutations is associated with constitutive phosphorylation of STAT5 and AKT. [29, 34–36] As shown in Figure 3A, AR00459339 has no effect on autophosphorylation of ITD-mutated FLT3. It likewise had no effect on wild type or D835Y-mutated FLT3 (data not shown). Somewhat surprisingly, however, exposure to AR00459339 did result in de-phosphorylation of STAT5 and AKT (Figure 3A). The IC50 of inhibition correlated with that of cytotoxicity, but was significantly higher than that noted in the in vitro kinase assay above. We would expect this trend in such a cell-based assay, in which drug-protein binding and protein-protein interaction likely affect target inhibition, in contrast to a kinase assay, in which these are not complicating factors. As expected, FLT3 autophosphorylation, along with AKT and STAT5 phosphorylation, was effectively inhibited by the FLT3 inhibitor AR00454200 (Figure 3B). AR00459339, despite not inhibiting FLT3 autophosphorylation directly, resembles a FLT3 inhibitor both in its cytotoxicity profile as well as its effects on downstream signaling proteins. Thus, the effects of PIM1 inhibition seem to be convergent with those of FLT3 inhibition, at least in FLT3-ITD AML cells.
Figure 3. AR00459339 does not affect phosphorylation of FLT3, but does promote de-phosphorylation of downstream targets of FLT3.

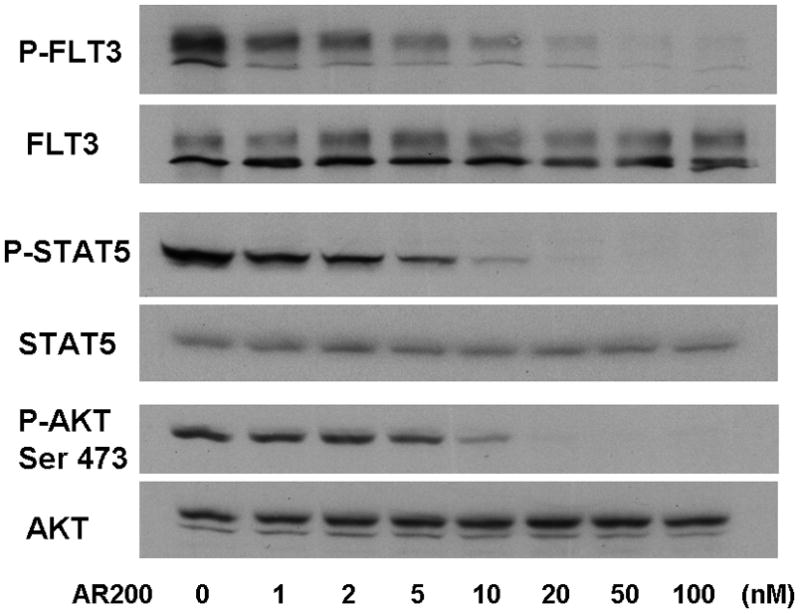
Note: AR00459339 is abbreviated “AR339” and AR00454200 is abbreviated “AR200” in these figures. (A) MV4-11 cells were exposed for 2 hours, in cell culture medium, to increasing concentrations of the PIM1 inhibitor AR00459339. Cells were then lysed, and the lysate was subject to immunoprecipitation with anti-FLT3 antibody, or loaded directly onto gels. After resolution by protein electrophoresis, immunoblots were probed with anti-phosphotyrosine (FLT3 immunoprecipitate, upper row), anti-phospho-STAT5, and anti-AKT at serine 473. The blots was subsequently stripped and reprobed with anti-FLT3, anti-STAT5, and anti-AKT antibodies. In (B), MV4-11 cells were treated with increasing concentrations of AR00454200, followed by similar analysis as in (A).
PIM1 inhibition also appeared to be cytotoxic against FLT3-ITD primary samples from patients who had developed secondary resistance to FLT3 inhbitiors. One of the FLT3-ITD primary samples that we used in the experiment shown in Figure 2C was obtained at diagnosis from a patient who was subsequently treated with the FLT3 inhibitor sorafenib. The patient responded with clearance of peripheral blasts, a phenomenon which has been reported with sorafenib and multiple other FLT3 inhibitors.[9, 10, 37–40] After 10 weeks, the disease progressed with a rapidly rising peripheral blast count. The blasts now had a D835H kinase domain mutation in addition to the ITD mutation (sequencing data not shown). In immunoblot assays, sorafenib had no effect on the phosphorylation status of FLT3, STAT5, and AKT, while other FLT3 inhibitors (CEP-701, AC220, and AR00454200) still showed in vitro efficacy (Figure 4A). AR00459339 had no effect on FLT3 autophosphorylation, but inhibited the phosphorylation of STAT5 and AKT as effectively as the FLT3 inhibitors (Figure 4A). In addition to this first patient, two additional FLT3-ITD patients at our institution were likewise treated successfully with FLT3 inhibition, but subsequently progressed. All patients had de novo AML and did not demonstrate karyotypic abnormalities. Of the three patients with secondary resistance to FLT3 inhibition, blasts obtained prior to the emergence of resistance from two of them were available to us for analysis. Shown in Figure 4B are the results of cytotoxicity assays for these two patients, demonstrating initial in vitro sensitivity to sorafenib (“pre”), then in vitro resistance to sorafenib (“post”). Post-treatment blasts from all three resistant patients were available and showed a cytotoxic response in vitro to AR00459339 (Figure 4C).
Figure 4. AR00459339 is cytotoxic to myeloblasts from patients who have developed clinical resistance to the FLT3 inhibitors.
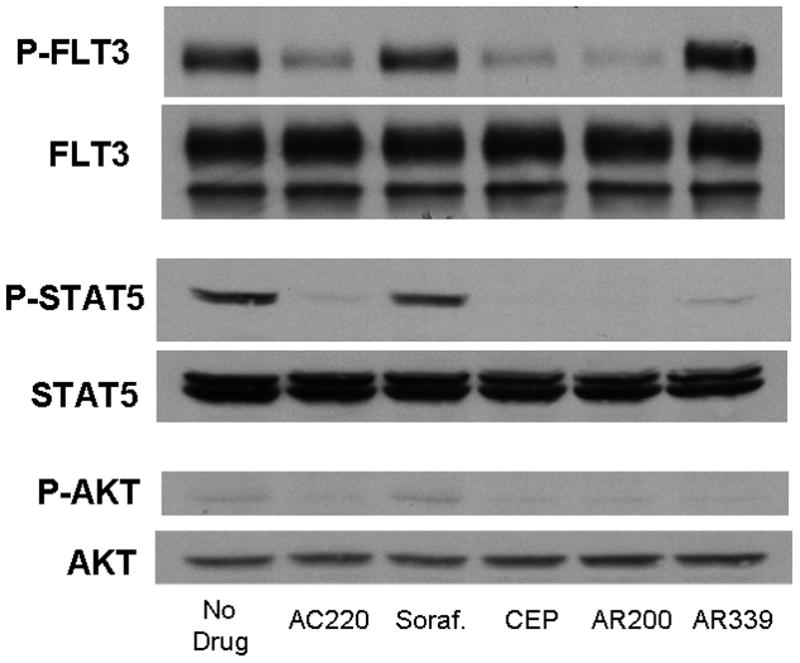
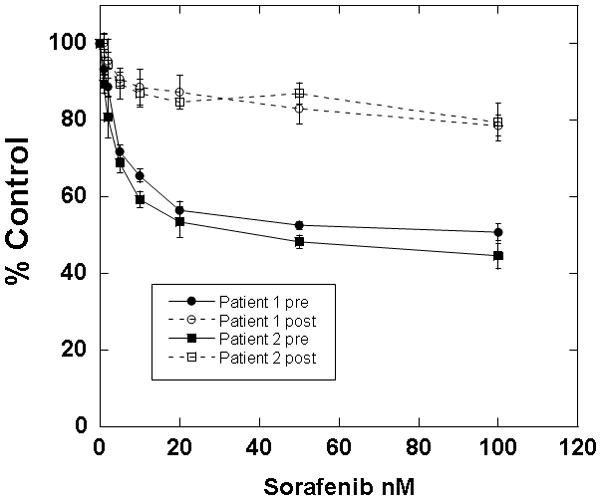
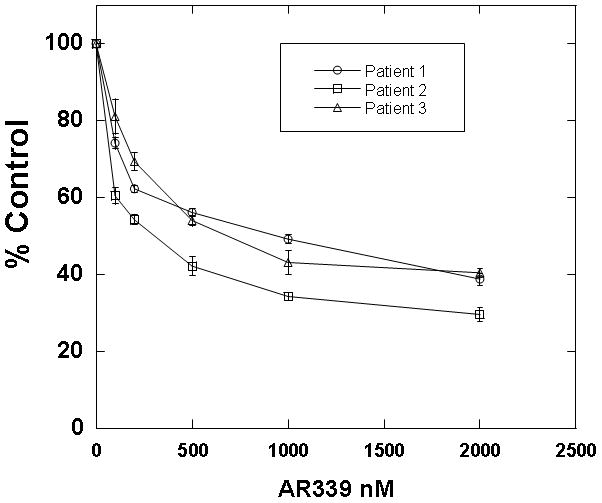
Note: AR00459339 is abbreviated “AR339” and AR00454200 is abbreviated “AR200” in these figures. (A) Myeloblasts from the peripheral blood of a patient with FLT3/ITD AML who progressed on sorafenib therapy were isolated by Ficoll centrifugation and incubated for 1 hour without drug, with 50nM of each of the following FLT3 inhibitors: sorafenib, CEP-701, AR00454200, and AC220, and with 2000 nM of the PIM1 inhibitor AR00459339. Cells were then lysed, and the lysate was subject to immunoprecipitation with anti-FLT3 antibody, or loaded directly onto gels. After resolution by protein electrophoresis, immunoblots were probed with anti-phosphotyrosine (FLT3 immunoprecipitate, upper row), anti-phospho-STAT5, and anti-AKT at serine 473. The blots was subsequently stripped and re-probed with anti-FLT3, anti-STAT5, and anti-AKT antibodies. (B) For two patients with secondary resistance to FLT3 inhibition, blasts were obtained prior to and after the emergence of resistance. These blasts were incubated with increasing concentrations of sorafanib, as well as with AR00459339 (C).
The combination of AR00459339 and AR00454200 leads to additive to weakly synergistic cytotoxicity in FLT3-ITD blasts
Because both AR00459339 (PIM inhibitor) and AR00454200 (FLT3 inhibitor) appear to act on the same downstream effector proteins, we wished to determine if combining them resulted in synergistic cytotoxicity in FLT3-ITD AML cells. However, the combination of AR00459339 and AR00454200 was more additive than synergistic in TF/ITD cells (combination index 0.806–1.129; Figure 5A), and only weakly synergistic (combination index 0.607–.809; Figure 5B) in a primary FLT3-ITD sample.
Figure 5. The combination of the PIM1 inhibitor AR00459339 and the FLT3 inhibitor AR00454200 is additive to mildly synergistic in FLT3/ITD cells.
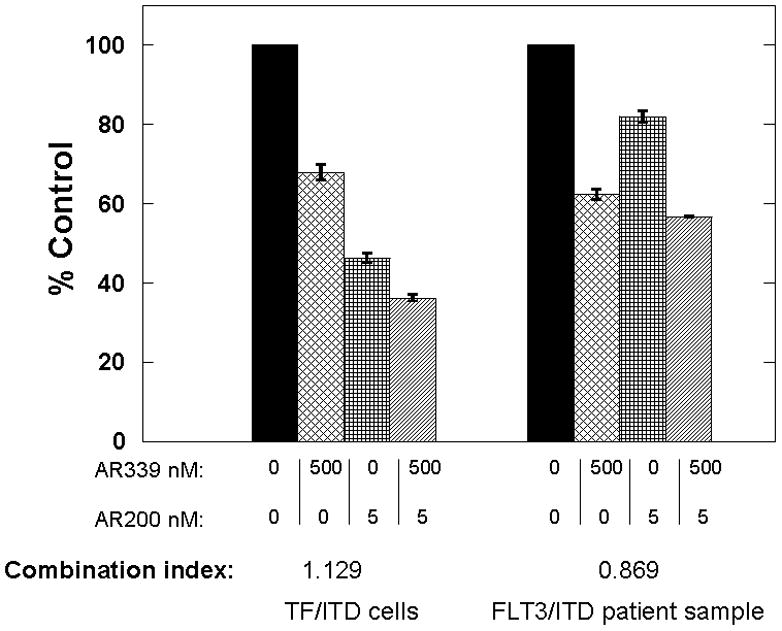
Note: AR00459339 is abbreviated “AR339” and AR00454200 is abbreviated “AR200” in these figures. TF/ITD cells (left) and a FLT3/ITD primary AML sample (right) were incubated with increasing concentrations of the PIM1 inhibitor AR00459339, the FLT3 inhibitor AR00454200, and the combination of the two drugs was fixed at a ratio of 100:1. At 72 hours, metabolic activity was determined using the MTT assay. Results for each sample were obtained as percentage of untreated control. Combination indices (CI) were generated using the Calcusyn software program from Biosoft®. Error bars represent standard deviations.
AR00459339 has a mild inhibitory effect on the normal bone marrow
AR00459339 is preferentially cytotoxic to FLT3-ITD AML cells, suggesting that PIM1 inhibition might be a viable therapeutic modality for this disease. Data from PIM knockout mouse models suggests that PIM proteins are particularly important in mediating response to myeloid growth factors.[41] Therapy with a PIM inhibitor, therefore, might be postulated to cause myelosuppression. We therefore tested the effects of AR00459339 on myeloid progenitor cells in colony forming assays of bone marrow obtained from normal donors. AR00459339 had no significant effect on erythroid colony formation over the dose ranges of PIM1 inhibition (Figure 6). However, AR00459339 induced a modest but reproducible inhibitory effect on CFU-GM numbers in these assays. This suggests that AR00459339 (and potentially other inhibitors of PIM), though possibly clinically useful for the treatment of AML, may also lead to some degree of myelosuppression.
Figure 6. AR00459339 inhibits CFU-GM colonies in a progenitor cell assay.
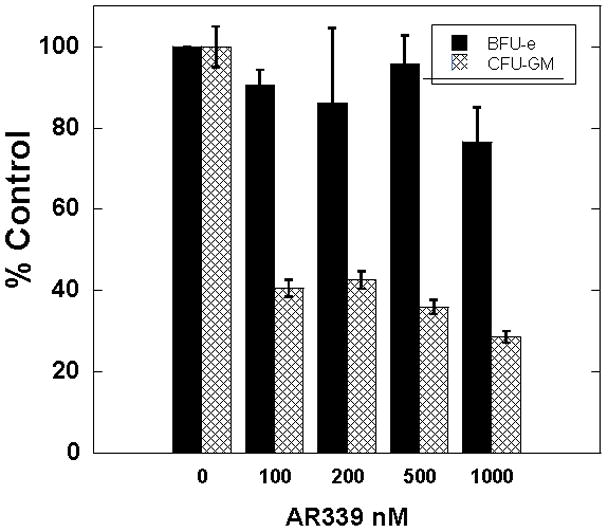
Mononuclear cells from normal donor bone marrow were isolated and plated into methylcellulose medium supplemented with cytokines and containing increasing concentration of AR00459339. Colonies were scored after 10–14 days for the presence of BFU-E and CFU-GM colonies. Error bars represent standard deviation.
Discussion
In this report, we have established that an inhibitor of PIM1, AR00459339, can induce cytotoxicity in FLT3-ITD AML cell lines and patient samples, a pattern of activity that parallels what has been observed with FLT3 inhibitors. The preferential cytotoxicity of PIM1 inhibition on FLT3-ITD cell lines and blasts has been previously reported.[42, 43] In contrast to FLT3 inhibitors, AR00459339 has no effect on FLT3 autophosphorylation but does induce de-phosphorylation of the key downstream proteins, STAT5, AKT, and BAD. Intriguingly, the cytotoxicity of AR00459339 was preserved against AML primary samples from FLT3-ITD patients who had developed resistance to and then progressed on FLT3 inhibitor therapy.
Previous studies have suggested that PIM1 is a down-stream target of FLT3 and a mediator of its activity by phosphorylating pro-apoptotic proteins such as BAD.[24] However, our data suggests that PIM1 may be much more integral to FLT3 signaling and may act more “up-stream” than previously reported. This is supported by our finding that PIM1 inhibition suppresses the phosphorylation of established FLT3 targets like STAT5 and AKT. The lack of pronounced synergy in combining agents that inhibit FLT3 and PIM1 might be explained if these proteins are in a close association more up-stream in a signaling cascade, or perhaps in a complex, which would then theoretically affect the same downstream mediators. This overlapping cytotoxic profile with FLT3 inhibitors is intriguing. However, to date, a definitive clinical benefit has not been demonstrated with FLT3 inhibitors in advanced trials [44, 45], and given a similar spectrum of cytotoxicity, the clinical utility of isolated PIM1 inhibition may be similarly limited.
The effects of AR00459339 do not appear to be mediated through inhibition of PIM2. Like PIM1, PIM2 has been reported to phosphorylate BAD at serine 112.[31] However, the results of our in vitro kinase assays suggest that AR00459339 inhibits PIM2 at much higher concentrations than the drug levels observed to inhibit BAD phosphorylation or to induce cytotoxicity in FLT3-ITD AML cells. PIM2 has been demonstrated by others to be a potentially important component of FLT3-ITD-mediated leukemogenesis, and its expression (like that of PIM1) has clearly been shown to be up-regulated in response to FLT3-ITD signaling.[16, 25, 36, 46] As mentioned above, siRNA-mediated knock-down of PIM1 and PIM2 decreased survival of MV4-11 FLT3-ITD blasts.[26] Our data, however, suggests that PIM1 and PIM2, despite being in the same gene family, may have very different functions. This divergence in function is supported by findings in a murine PIM knockout model, in which bone marrow cells from mice lacking PIM1 displayed significant loss of response to cytokines, while loss of PIM2 by itself had only minimal effects in comparison.[41] Nevertheless, the decreased activity of AR00459339 against PIM2 kinase may still have a limiting impact on its clinical efficacy, given the suspected potential pathogenic role of this serine/threonine kinase in FLT3-ITD AML.
The effects of the loss of PIM1 activity on bone marrow progenitor cells in the mouse knockout model are relevant to the possible use of PIM1 inhibitors for the treatment of AML. We observed a modest but consistent suppression of CFU-GM activity with AR00459339 at doses predicted to inhibit PIM1. We might therefore predict that some degree of myelosuppression will occur when such drugs are used clinically, resulting in a narrow therapeutic index. While this is potentially a considerable limitation of this therapeutic approach, we should note that CFU-GM activity was only partially inhibited and not completely absent.
Treatment of FLT3-ITD AML with tyrosine kinase inhibitors of FLT3 is being actively investigated in clinical trials.[9, 10, 37–40, 47] However, increasing evidence suggests that monotherapy with FLT3 inhibitors can lead to resistance to these agents, and eventual progression of disease on therapy.[48–50] This is a phenomenon that has been well-described with BCR-ABL inhibitors. [51–53] Resistance can manifest in a variety of ways, including the development of point mutations or up-regulation of anti-apoptotic proteins.[48–50] The emergence of resistance will pose a particularly difficult obstacle in the development and long-term efficacy of these agents for patients with AML.
We have observed that AR00459339 is cytotoxic to samples from three patients whose blasts had become resistant to the FLT3 inhibitors. These findings raise the possibility that the emergence of drug resistance to FLT3 inhibition could possibly be overcome by targeting PIM1. Others have indeed reported that cells, transformed by FLT3 mutations resistant to tyrosine kinase inhibitors, maintained their sensitivity to knockdown of PIM1 and PIM2 by RNAi. Although the combination of multiple targeted agents in anti-neoplastic therapy has been used to promote synergistic cytotoxicity, it might also be employed to suppress the emergence of resistant clones and prolong therapeutic efficacy. This is especially applicable in a scenario in which the cancer cell is dependent, or “addicted”, on one predominant pathway such as in FLT3 mutant AML cells.[54] Targeting FLT3 and PIM1, two proteins which closely interact in a common pathway, on which these AML cells are dependent for survival, is therefore a valid therapeutic approach in this regard. This rationale is somewhat analogous to the emergence of highly active anti-retroviral therapy (HAART) for the treatment of HIV-AIDS, by employing combinations of targeted agents which can maintain therapeutic efficacy by limiting tumor escape mechanisms and drug resistance. In summary, the clinical utility of PIM1 inhibition in patients with FLT3-ITD AML, as monotherapy or in combination with FLT3 inhibitors, holds therapeutic promise and warrants further clinical study.
Acknowledgments
None
Role of Funding Source:
This work was supported by grants from the NCI (NCI Leukemia SPORE P50 CA100632-06, R01 CA128864) and the American Society of Clinical Oncology (ML). Mark Levis is a Clinical Scholar of the Leukemia and Lymphoma Society.
Footnotes
Authors’ Contributions: A.T.F. contributed to the study design, performed experiments, and wrote the manuscript; O.A. performed the colony assays; I.S. performed MTT assays; T.S. performed the Annexin V assays; T.R. performed MTT assays and western blots; D.S. contributed to the study design and provided TF/ITD cells; M.M. performed invitro kinase assays; F.M., J.E.R. and S.D.G. contributed to the study design and helped edit the manuscript; S.A. synthesized AR00454200 and N.C.K. synthesized AR00459339 and M.L. designed the study, performed experiments, and edited the manuscript.
Conflicts of Interest: F.M., S.D.G, M.M. and S.A.: Array Biopharma: Employment, Equity Ownership; J.E.R. and N.C.K.: Array BioPharma: Employment, and all other authors have no conflict of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Frohling S, Schlenk RF, Breitruck J, Benner A, Kreitmeier S, Tobis K, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood. 2002;100:4372–80. doi: 10.1182/blood-2002-05-1440. [DOI] [PubMed] [Google Scholar]
- 2.Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98:1752–9. doi: 10.1182/blood.v98.6.1752. [DOI] [PubMed] [Google Scholar]
- 3.Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911–8. [PubMed] [Google Scholar]
- 4.Rombouts WJ, Blokland I, Lowenberg B, Ploemacher RE. Biological characteristics and prognosis of adult acute myeloid leukemia with internal tandem duplications in the Flt3 gene. Leukemia. 2000;14:675–83. doi: 10.1038/sj.leu.2401731. [DOI] [PubMed] [Google Scholar]
- 5.Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood. 2002;100:59–66. doi: 10.1182/blood.v100.1.59. [DOI] [PubMed] [Google Scholar]
- 6.Thiede C, Steudel C, Mohr B, Schaich M, Schakel U, Platzbecker U, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99:4326–35. doi: 10.1182/blood.v99.12.4326. [DOI] [PubMed] [Google Scholar]
- 7.Dohner K, Schlenk RF, Habdank M, Scholl C, Rucker FG, Corbacioglu A, et al. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005;106:3740–6. doi: 10.1182/blood-2005-05-2164. [DOI] [PubMed] [Google Scholar]
- 8.Gale RE, Green C, Allen C, Mead AJ, Burnett AK, Hills RK, et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood. 2008;111:2776–84. doi: 10.1182/blood-2007-08-109090. [DOI] [PubMed] [Google Scholar]
- 9.Pratz KW, Cortes J, Roboz GJ, Rao N, Arowojolu O, Stine A, et al. A pharmacodynamic study of the FLT3 inhibitor KW-2449 yields insight into the basis for clinical response. Blood. 2009;113:3938–46. doi: 10.1182/blood-2008-09-177030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103:3669–76. doi: 10.1182/blood-2003-11-3775. [DOI] [PubMed] [Google Scholar]
- 11.Stone RM, Fischer T, Paquette R, Schiller G, Schiffer CA, Ehninger G, et al. A phase 1b study of midostaurin (PKC412) in combination with daunorubicin and cytarabine induction and high-dose cytarabine consolidation in patients under age 61 with newly diagnosed de novo acute myeloid leukemia: Overall survival of patients whose blasts have FLT3 mutations is similar to those with wild-type FLT3. ASH Annual Meeting Abstracts. 2009;114:Abstract 634. [Google Scholar]
- 12.Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML) Blood. 2009;114:2984–92. doi: 10.1182/blood-2009-05-222034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang W, Konopleva M, Shi YX, McQueen T, Harris D, Ling X, et al. Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. Journal of the National Cancer Institute. 2008;100:184–98. doi: 10.1093/jnci/djm328. [DOI] [PubMed] [Google Scholar]
- 14.Cuypers HT, Selten G, Quint W, Zijlstra M, Maandag ER, Boelens W, et al. Murine leukemia virus-induced T-cell lymphomagenesis: integration of proviruses in a distinct chromosomal region. Cell. 1984;37:141–50. doi: 10.1016/0092-8674(84)90309-x. [DOI] [PubMed] [Google Scholar]
- 15.Amson R, Sigaux F, Przedborski S, Flandrin G, Givol D, Telerman A. The human protooncogene product p33pim is expressed during fetal hematopoiesis and in diverse leukemias. Proc Natl Acad Sci U S A. 1989;86:8857–61. doi: 10.1073/pnas.86.22.8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mizuki M, Schwable J, Steur C, Choudhary C, Agrawal S, Sargin B, et al. Suppression of myeloid transcription factors and induction of STAT response genes by AML-specific Flt3 mutations. Blood. 2003;101:3164–73. doi: 10.1182/blood-2002-06-1677. [DOI] [PubMed] [Google Scholar]
- 17.Cohen AM, Grinblat B, Bessler H, Kristt D, Kremer A, Schwartz A, et al. Increased expression of the hPim-2 gene in human chronic lymphocytic leukemia and non-Hodgkin lymphoma. Leuk Lymphoma. 2004;45:951–5. doi: 10.1080/10428190310001641251. [DOI] [PubMed] [Google Scholar]
- 18.Kim KT, Baird K, Ahn JY, Meltzer P, Lilly M, Levis M, et al. Pim-1 is up-regulated by constitutively activated FLT3 and plays a role in FLT3-mediated cell survival. Blood. 2005;105:1759–67. doi: 10.1182/blood-2004-05-2006. [DOI] [PubMed] [Google Scholar]
- 19.Cibull TL, Jones TD, Li L, Eble JN, Ann Baldridge L, Malott SR, et al. Overexpression of Pim-1 during progression of prostatic adenocarcinoma. J Clin Pathol. 2006;59:285–8. doi: 10.1136/jcp.2005.027672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peltola K, Hollmen M, Maula SM, Rainio E, Ristamaki R, Luukkaa M, et al. Pim-1 kinase expression predicts radiation response in squamocellular carcinoma of head and neck and is under the control of epidermal growth factor receptor. Neoplasia. 2009;11:629–36. doi: 10.1593/neo.81038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lilly M, Kraft A. Enforced expression of the Mr 33,000 Pim-1 kinase enhances factor-independent survival and inhibits apoptosis in murine myeloid cells. Cancer Res. 1997;57:5348–55. [PubMed] [Google Scholar]
- 22.Lilly M, Sandholm J, Cooper JJ, Koskinen PJ, Kraft A. The PIM-1 serine kinase prolongs survival and inhibits apoptosis-related mitochondrial dysfunction in part through a bcl-2-dependent pathway. Oncogene. 1999;18:4022–31. doi: 10.1038/sj.onc.1202741. [DOI] [PubMed] [Google Scholar]
- 23.Miura O, Miura Y, Nakamura N, Quelle FW, Witthuhn BA, Ihle JN, et al. Induction of tyrosine phosphorylation of Vav and expression of Pim-1 correlates with Jak2-mediated growth signaling from the erythropoietin receptor. Blood. 1994;84:4135–41. [PubMed] [Google Scholar]
- 24.Kim KT, Levis M, Small D. Constitutively activated FLT3 phosphorylates BAD partially through pim-1. Br J Haematol. 2006;134:500–9. doi: 10.1111/j.1365-2141.2006.06225.x. [DOI] [PubMed] [Google Scholar]
- 25.Agrawal S, Koschmieder S, Baumer N, Reddy NG, Berdel WE, Muller-Tidow C, et al. Pim2 complements Flt3 wild-type receptor in hematopoietic progenitor cell transformation. Leukemia. 2008;22:78–86. doi: 10.1038/sj.leu.2404988. [DOI] [PubMed] [Google Scholar]
- 26.Adam M, Pogacic V, Bendit M, Chappuis R, Nawijn MC, Duyster J, et al. Targeting PIM kinases impairs survival of hematopoietic cells transformed by kinase inhibitor-sensitive and kinase inhibitor-resistant forms of Fms-like tyrosine kinase 3 and BCR/ABL. Cancer Research. 2006;66:3828–35. doi: 10.1158/0008-5472.CAN-05-2309. [DOI] [PubMed] [Google Scholar]
- 27.Levis M, Tse KF, Smith BD, Garrett E, Small D. A FLT3 tyrosine kinase inhibitor is selectively cytotoxic to acute myeloid leukemia blasts harboring FLT3 internal tandem duplication mutations. Blood. 2001;98:885–7. doi: 10.1182/blood.v98.3.885. [DOI] [PubMed] [Google Scholar]
- 28.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 29.Levis M, Allebach J, Tse KF, Zheng R, Baldwin BR, Smith BD, et al. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood. 2002;99:3885–91. doi: 10.1182/blood.v99.11.3885. [DOI] [PubMed] [Google Scholar]
- 30.Eswaran J, Patnaik D, Filippakopoulos P, Wang F, Stein RL, Murray JW, et al. Structure and functional characterization of the atypical human kinase haspin. Proc Natl Acad Sci U S A. 2009;106:20198–203. doi: 10.1073/pnas.0901989106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan B, Zemskova M, Holder S, Chin V, Kraft A, Koskinen PJ, et al. The PIM-2 kinase phosphorylates BAD on serine 112 and reverses BAD-induced cell death. J Biol Chem. 2003;278:45358–67. doi: 10.1074/jbc.M307933200. [DOI] [PubMed] [Google Scholar]
- 32.Knapper S, Mills KI, Gilkes AF, Austin SJ, Walsh V, Burnett AK. The effects of lestaurtinib (CEP701) and PKC412 on primary AML blasts: the induction of cytotoxicity varies with dependence on FLT3 signaling in both FLT3-mutated and wild-type cases. Blood. 2006;108:3494–503. doi: 10.1182/blood-2006-04-015487. [DOI] [PubMed] [Google Scholar]
- 33.Shiotsu Y, Kiyoi H, Ishikawa Y, Tanizaki R, Shimizu M, Umehara H, et al. KW-2449, a novel multikinase inhibitor, suppresses the growth of leukemia cells with FLT3 mutations or T315I-mutated BCR/ABL translocation. Blood. 2009;114:1607–17. doi: 10.1182/blood-2009-01-199307. [DOI] [PubMed] [Google Scholar]
- 34.Scheijen B, Ngo HT, Kang H, Griffin JD. FLT3 receptors with internal tandem duplications promote cell viability and proliferation by signaling through Foxo proteins. Oncogene. 2004;23:3338–49. doi: 10.1038/sj.onc.1207456. [DOI] [PubMed] [Google Scholar]
- 35.Brandts CH, Sargin B, Rode M, Biermann C, Lindtner B, Schwable J, et al. Constitutive activation of Akt by Flt3 internal tandem duplications is necessary for increased survival, proliferation, and myeloid transformation. Cancer Res. 2005;65:9643–50. doi: 10.1158/0008-5472.CAN-05-0422. [DOI] [PubMed] [Google Scholar]
- 36.Choudhary C, Olsen JV, Brandts C, Cox J, Reddy PN, Bohmer FD, et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol Cell. 2009;36:326–39. doi: 10.1016/j.molcel.2009.09.019. [DOI] [PubMed] [Google Scholar]
- 37.O’Farrell AM, Foran JM, Fiedler W, Serve H, Paquette RL, Cooper MA, et al. An innovative phase I clinical study demonstrates inhibition of FLT3 phosphorylation by SU11248 in acute myeloid leukemia patients. Clin Cancer Res. 2003;9:5465–76. [PubMed] [Google Scholar]
- 38.Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105:54–60. doi: 10.1182/blood-2004-03-0891. [DOI] [PubMed] [Google Scholar]
- 39.DeAngelo DJ, Stone RM, Heaney ML, Nimer SD, Paquette RL, Klisovic RB, et al. Phase 1 clinical results with tandutinib (MLN518), a novel FLT3 antagonist, in patients with acute myelogenous leukemia or high-risk myelodysplastic syndrome: safety, pharmacokinetics, and pharmacodynamics. Blood. 2006;108:3674–81. doi: 10.1182/blood-2006-02-005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quintas-Cardama A, Kantarjian H, Andreef M, Faderl S, Wright JJ, Zhang W, et al. Phase I trial of intermittent administration of sorafenib (BAY 43–9006) for patients (pts) with refractory/relapsed acute myelogenous leukemia (AML) J Clin Oncol. 2007;25:7018. [Google Scholar]
- 41.Mikkers H, Nawijn M, Allen J, Brouwers C, Verhoeven E, Jonkers J, et al. Mice deficient for all PIM kinases display reduced body size and impaired responses to hematopoietic growth factors. Mol Cell Biol. 2004;24:6104–15. doi: 10.1128/MCB.24.13.6104-6115.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pogacic V, Bullock AN, Fedorov O, Filippakopoulos P, Gasser C, Biondi A, et al. Structural analysis identifies imidazo[1,2-b]pyridazines as PIM kinase inhibitors with in vitro antileukemic activity. Cancer Res. 2007;67:6916–24. doi: 10.1158/0008-5472.CAN-07-0320. [DOI] [PubMed] [Google Scholar]
- 43.Lin YW, Beharry ZM, Hill EG, Song JH, Wang W, Xia Z, et al. A small molecule inhibitor of Pim protein kinases blocks the growth of precursor T-cell lymphoblastic leukemia/lymphoma. Blood. 2010;115:824–33. doi: 10.1182/blood-2009-07-233445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levis M, Ravandi F, Wang ES, Baer MR, Perl A, Coutre S, et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood. 2011 Jan 26; doi: 10.1182/blood-2010-08-301796. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Serve H, Wagner R, Sauerland C, Brunnberg U, Krug U, Schaich M, et al. Sorafenib in combination with standard induction and consolidation therapy In elderly AML patients: Results from a randomized, placebo-controlled phase II trial. Blood (ASH Annual Meeting Abstracts) 2010;116:Abstract 333. [Google Scholar]
- 46.Choudhary C, Brandts C, Schwable J, Tickenbrock L, Sargin B, Ueker A, et al. Activation mechanisms of STAT5 by oncogenic Flt3-ITD. Blood. 2007;110:370–4. doi: 10.1182/blood-2006-05-024018. [DOI] [PubMed] [Google Scholar]
- 47.Ravandi F, Cortes JE, Jones D, Faderl S, Garcia-Manero G, Konopleva MY, et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol. 2010;28:1856–62. doi: 10.1200/JCO.2009.25.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heidel F, Solem FK, Breitenbuecher F, Lipka DB, Kasper S, Thiede MH, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107:293–300. doi: 10.1182/blood-2005-06-2469. [DOI] [PubMed] [Google Scholar]
- 49.von Bubnoff N, Engh RA, Aberg E, Sanger J, Peschel C, Duyster J. FMS-like tyrosine kinase 3-internal tandem duplication tyrosine kinase inhibitors display a nonoverlapping profile of resistance mutations in vitro. Cancer Res. 2009;69:3032–41. doi: 10.1158/0008-5472.CAN-08-2923. [DOI] [PubMed] [Google Scholar]
- 50.Weisberg E, Barrett R, Liu Q, Stone R, Gray N, Griffin JD. FLT3 inhibition and mechanisms of drug resistance in mutant FLT3-positive AML. Drug Resist Updat. 2009;12:81–9. doi: 10.1016/j.drup.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hochhaus A, Kreil S, Corbin AS, La Rosee P, Muller MC, Lahaye T, et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. 2002;16:2190–6. doi: 10.1038/sj.leu.2402741. [DOI] [PubMed] [Google Scholar]
- 52.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–25. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 53.Apperley JF. Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007;8:1018–29. doi: 10.1016/S1470-2045(07)70342-X. [DOI] [PubMed] [Google Scholar]
- 54.Pratz KW, Sato T, Murphy KM, Stine A, Rajkhowa T, Levis M. FLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood. 2010;115:1425–32. doi: 10.1182/blood-2009-09-242859. [DOI] [PMC free article] [PubMed] [Google Scholar]