

Abstract
The genomic and non-genomic signaling pathways are well-known estrogen signaling pathways. The 66-kDa estrogen receptor-α (ER-α66) is a typical ligand-inducible transcription factor that mainly mediates genomic estrogen signaling. Recently, we identified and cloned a 36-kDa variant of ER-α66, known as ER-α36. This variant lacks intrinsic transcription activity and predominantly mediates non-genomic estrogen signaling. Thus, the expression of ER-α66 and ER-α36 should be dynamically regulated and carefully coordinated to maintain a balance between genomic and non-genomic estrogen signaling. However, the molecular mechanisms underlying this correlation remain poorly understood. The Wilms’ tumor suppressor gene, wt1, encodes a zinc-finger protein WT1 that functions as a dual transcription regulator to activate or suppress gene transcription. High levels of WT1 expression are associated with breast cancer malignancy. In the present study, high-passage ER-positive breast cancer MCF7 cells were found to express ER-α66 and WT1 at higher levels and ER-α36 at a very low level. Using the small hairpin RNA method, stable MCF7 cells were established that expressed knocked-down levels of WT1. The cells expressed a reduced level of ER-α66 but an increased level of ER-α36, suggesting that WT1 regulates the expression of ER-α66 and ER-α36 oppositely. Further co-transfection assays showed that all isoforms of WT1 directly activated the promoter activity of the ER-α66 gene while suppressing ER-α36 promoter activity. Our results therefore indicate that WT1 is a dual transcription factor that regulates the promoter activity of ER-α66 and ER-α36 oppositely, implicating WT1 as one of the coordinators that orchestrate genomic and non-genomic estrogen signaling.
Keywords: WT1, estrogen receptor-α, estrogen receptor-α36, breast cancer
Introduction
Long-term exposure to estrogen is a well-known risk factor for the development of breast cancer (1). Estrogen signaling pathways, in particular the mitogenic pathway, mediated by the estrogen receptor-α (ER-α) is crucial in the development of breast cancer stimulated by estrogen (2,3). ER-α is a ligand-activated transcription factor comprising three independent but interacting functional domains: the N-terminal A/B domain, the C or DNA-binding domain, and the D/E/F or ligand-binding domain. The N-terminal domain of ER-α encodes a ligand-independent activation function (AF-1). The DNA-binding or C domain contains a two zinc-finger structure that plays an important role in receptor dimerization and binds to specific DNA sequences. The C-terminal E/F domain is a ligand-binding domain that mediates ligand binding, receptor dimerization, nuclear translocation, and a ligand-dependent transactivation function (AF-2) (2,3). Stimulation of the target gene expression by ER-α in response to 17β-estradiol is predominantly thought to be responsible for cell proliferation (2).
ER-α was shown to act as a transcription factor. However, not all of the physiological effects mediated by estrogens are achieved through a direct effect on gene transcription. On the other hand, a ‘non-classic’, ‘non-genomic’ or ‘membrane signaling’ pathway exists that involves cytoplasmic proteins, growth factors and other membrane-initiated signaling pathways (4–6).
Previously, we identified and cloned a 36-kDa variant of ER-α, i.e., ER-α36, which is mainly expressed on the plasma membrane and mediates non-genomic estrogenic signaling (Fig. 1A) (7,8). ER-α36 lacks transcription activation domains AF-1 and AF-2 of the 66-kDa full-length ER-α (ER-α66), and possesses an altered ligand-binding domain and an intact DNA-binding domain, consistent with the fact that ER-α36 possesses no intrinsic transcriptional activity (8). ER-α36 is predominantly expressed on the plasma membrane and in the cytoplasm, and mediates non-genomic estrogen signaling (8,9). ER-α36 is generated from a promoter located in the first intron of the ER-α66 gene (10), indicating that ER-α36 expression is regulated differently from ER-α66, consistent with the findings that ER-α36 is expressed in specimens from ER-negative breast cancer patients and established ER-negative breast cancer cells that lack ER-α66 expression (8,11,12). Thus, the nuclear ER-α66 is mainly involved in genomic estrogen signaling whereas extra-nuclear ER-α36 is involved in non-genomic estrogen signaling.
Figure 1.
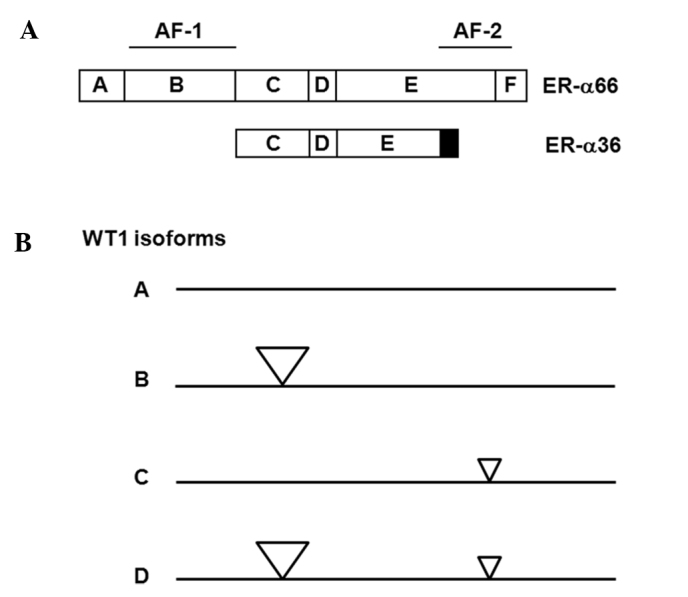
Schematic protein structures of ER-α66, ER-α36 and WT1 isoforms. (A) Schematic protein structures of ER-α66 and ER-α36. (B) Schematic protein structures of WT1 isoforms A, B, C and D. The large triangle is the 17-amino-acid insert and the small triangle is the 3-amino-acid (KTS) insert.
Previously, the extra-nuclear ER-α36 was found to act as a dominant-negative inhibitor of genomic estrogen signaling through impeding the transcription activities mediated by the AF-1 and AF-2 domains of ER-α66 (8). Recently, ER-α66 was found to suppress the promoter activity of ER-α36 via a half estrogen response element (ERE) site located in the 5′-flanking sequence of the ER-α36 gene (10). These findings suggest that the genomic and non-genomic estrogen signaling pathways mediated by ER-α66 and ER-α36 are dynamically and strictly regulated at different levels. Dysregulated genomic and/or non-genomic estrogen signaling may lead to various diseases including cancer. Thus, the expression levels of ER-α66 and ER-α36 in a particular cell context require strict coordination. However, the underlying mechanisms of this coordination remain to be elucidated.
The Wilms’ tumor susceptibility gene, wt1, at chromosome locus 11p13 (13–15) encodes a C2-H2-type zinc-finger protein, WT1. Alternative splicing results in four protein isoforms of WT1 that differ due to the presence of one 17-amino acid insert between the transcription regulatory and DNA-binding domains, and one 3-amino-acid (KTS) insert between the third and fourth zinc fingers (16,17). The different isoforms are referred to as A, B, C and D, whereby the A isoform lacks both 17-amino-acid and KTS inserts, the B isoform contains the 17-amino-acid insert but lacks the KTS insert, the C isoform lacks the 17-amino-acid insert but contains the KTS insert, and the D isoform contains both inserts (Fig. 1B). Mutations of wt1 were found to be correlated with subsets of Wilms’ tumor (16,17), mesothelioma and ovarian tumors (18), consistent with the role of WT1 as a tumor suppressor. However, high levels of the wild-type WT1 mRNA and protein have been found in leukemia (19), lung cancer (20) and breast cancer (21–23). Breast cancer patients with tumors that highly express WT1 usually have a lower 5-year disease-free survival rate than patients with tumors of low WT1 expression (23), indicating that WT1 expression is associated with aggressive phenotype of breast cancer. However, the biological function and underlying mechanisms of WT1 in the development of aggressive breast tumors have yet to be investigated.
In the present study, the Wilms’ tumor suppressor WT1 activated promoter activity of the ER-α66 gene and suppressed ER-α36 promoter activity, suggesting that WT1 acts as a ‘coordinator’ of the genomic and non-genomic estrogen signaling pathways through the opposite regulation of the expression of ER-α36 and ER-α66.
Materials and methods
Cell culture and establishment of stable cell lines
Human embryonic kidney (HEK293) cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in DMEM and 10% fetal calf serum at 37°C in a 5% CO2 incubator. Relatively high-passage MCF7 cells were initially obtained from Dr Thomas F. Deuel’s laboratory at the Scripps Research Institute. The subline of MCF7 cells used in this study had been cultured for >75 passages and were maintained at 37°C in a 5% CO2 atmosphere in Improved Modified Eagle’s Medium (IMEM) supplemented with 5% fetal calf serum. To establish stable cells that express knocked-down levels of the Wilms’ tumor suppressor, WT1, MCF7 cells were plated at a density of 1×105 cells per 60-mm dish and transfected 24 h later with a mixture of four WT1 small hairpin (sh) RNA expressing constructs purchased from Origene (TR300442, Rockville, MD, USA) using the FuGene 6 transfection reagent (Roche Applied Sciences, Indianapolis, IN, USA). The control expression vector was also transfected into MCF7 cells to serve as a control. Following transfection (48 h), the cells were replated and selected with 5 μg/ml of puromycin (Invitrogen Corporation, Carlsbad, CA, USA) for two weeks. The medium was changed every three days until colonies appeared. A number of clonal cell lines were established that express the knocked-down levels of WT1. Two of these cell lines are described in this study, i.e., MCF7/sh-WTl-1 and -2. More than 20 individual clones from cells transfected with the empty expression vector were pooled and used as control MCF7/V cells.
Western blot analysis
Cells were washed three times with cold phosphate-buffered saline (PBS) and lysed with lysis buffer [50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.25 mM EDTA pH 8.0, 0.1% sodium dodecyl sulfate (SDS), 1% Triton® X-100, 50 mM NaF and the protease inhibitor cocktail from Sigma (St. Louis, MO, USA)]. Following adjustment to the same total protein content, cell lysates were analyzed by Western blot analysis. Cell lysates (25 μg) were boiled for 5 min in SDS gel-loading buffer and separated on a 10% SDS-PAGE gel. Following electrophoresis, the proteins were transferred to a PVDF membrane (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were probed with different primary antibodies, incubated with appropriate HRP-conjugated secondary antibodies and visualized with enhanced chemiluminescence (ECL) detection reagents (Amersham Pharmacia Biotech., Piscataway, NJ, USA). The same membranes were stripped and reprobed with an antibody against β-actin (I-19) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) to confirm equal loading.
Polyclonal anti-ER-α36 antibody was generated and characterized as previously described (8). Anti-ER-α66 antibody (Ab-15) was obtained from Lab Vision Products (Fremont, CA, USA). Polyclonal anti-WT1 antibody was from Invitrogen Corporation.
Luciferase assay
HEK293 cells were transfected using FuGene 6 transfection reagent with the reporter plasmids encoding the firefly luciferase gene driven by the 5′-flanking sequence of ER-α66 or ER-α36 gene, ER-α66 promoter-Luc and ER-α36 promoter-Luc, respectively. The ER-α66 promoter-Luc reporter plasmid was purchased from Switchgear Genomics (Menlo Park, CA, USA). The plasmid contains the DNA sequence from −748 to +324 (relative to the major transcription initiation site) of the ER-α66 promoter region. The ER-α36 promoter-Luc containing the 715-bp 5′-flanking sequence of the ER-α36 gene was generated and characterized as previously described (10). Expression vectors containing the WT1 isoforms A, B, C and D were previously described (24). A cytomegalovirus-driven Renilla luciferase plasmid, pRL-CMV (Promega, San Luis Obispo, CA, USA), was also included in the transfection to establish transfection efficacy. Following transfection (48 h), cell extracts were prepared, and the luciferase activity was determined and normalized using the Dual-Luciferase Assay System (Promega) and a TD 20/20 Luminometer (Turner BioSystems, Inc. Sunnyvale, CA, USA) according to the manufacturer’s instructions.
Statistical analysis
Data were summarized as the mean ± standard error (SE) using an GraphPad InStat software program. The Tukey-Kramer multiple comparisons test was also used. P<0.05 was considered to be statistically significant.
Results
Knockdown of WT1 expression in MCF7 cells alters the expression of ER-α66 and ER-α36
Recently, we found that high-passage MCF7 cells express increased levels of the Wilms’ tumor suppressor WT1 compared to low-passage MCF7 cells (25). To determine the role played by WT1 in MCF7 cells, expression levels of WT1 were knocked down in high-passage MCF7 cells, using the shRNA method. The clonal cell lines MCF7/sh-WTl-1 and -2 were transfected with the WT1 shRNA expression vectors. A cell line was generated from a mixture of >20 clones transfected with the empty expression vector (MCF7/V).
Western blot analysis using the antibody against WT1 confirmed that the WT1 protein (~52 kDa) was significantly down-regulated in the MCF7/sh-WTl-1 and -2 cell lines compared to the control MCF7 cells transfected with the empty vector (MCF7/V) (Fig. 2). The expression levels of ER-α66 were markedly decreased in the WT1 shRNA-transfected MCF7 cells, MCF7/sh-WTl-1 and -2, compared to the control (MCF7/V) cells (Fig. 2). We also noted that ER-α36 expression was increased in the MCF7/sh-WTl-1 and -2 cells (Fig. 2). These results suggest that as a dual transcription regulator, WT1 modulates the promoter activities of ER-α66 and ER-α36 oppositely.
Figure 2.
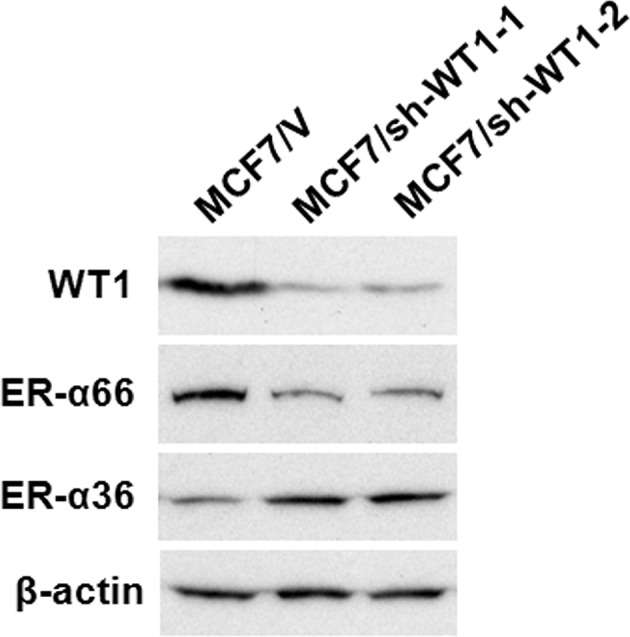
Expression levels of ER-α66 and ER-α36 are modified in MCF7 cells with knocked-down levels of WT1. Western blot analysis of WT1, ER-α66 and ER-α36 expression in a number of MCF7 cell variants: control cells (MCF7/V, transfected with the empty expression vector) and ER-α36 expression knocked-down cells (MCF7/sh-WT-1 and -2).
WT1 activates the promoter activity of ER-α66
To determine whether WT1 regulates ER-α66 promoter activity, we performed transient co-transfection assays in HEK293 cells that express undetectable levels of WT1, ER-α66 and ER-α36. HEK293 cells were co-transfected with a luciferase reporter gene driven by the 5′-flanking sequence of the ER-α66 gene (−748 to +324, relative to the major transcription initiation site) with expression vectors encoding four different isoforms of WT1 separately to evaluate the effects of different isoforms of WT1 on ER-α66 promoter activity. Findings showed that all four isoforms of WT1 activated the promoter activity of ER-α66; the WT1-B isoform exhibited the strongest activity whereas the WT1-D isoform exbitited the weakest activity (Fig. 3B). Computer analysis of the 5′-flanking sequence of ER-α66 revealed the existence of a perfect WT1 binding site located downstream of the TATA box (Fig. 3A). Our data thus indicated that WT1 positively regulates ER-α66 promoter activity presumably via the WT1 binding site located at −98 to −88 (relative to the major transcription initiation site).
Figure 3.
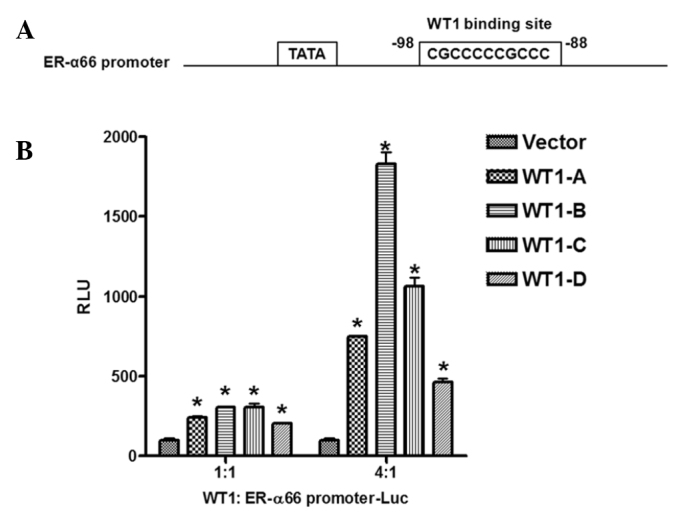
WT1 isoforms regulate ER-α66 promoter activity. (A) Schematic DNA structure of the ER-α66 promoter region. The WT1 binding site is shown. (B) HEK293 cells were transiently co-transfected with the expression vectors encoding the four WT1 isoforms, A, B, C, or D, and the ER-α66 promoter luciferase reporter. The ratio of WT1:ER-α66 promoter-Luc was 1:1 or 4:1. The luciferase activity of HEK293 cells co-transfected with empty vector and ER-α66 promoter-Luc was arbitrarily set to 100 RLU as a control. *P<0.05, the remaining groups were compared to the control. All experiments were repeated three times, and three parallel samples were used for each group.
WT1 suppresses the promoter activity of ER-α36
Recently, we cloned and characterized the 5′-flanking sequence of ER-α36 that is located in the first intron of the ER-α66 gene (10). A computer analysis of the promoter region of ER-α36 revealed two WT1 binding sites located both upstream and downstream of the TATA box (Fig. 4A). We then examined whether WT1 regulates the promoter activity of ER-α36. HEK293 cells were co-transfected with a luciferase reporter driven by the 5′-flanking sequence of the ER-α36 gene (−736 to +16, relative to the transcription initiation site) with expression vectors encoding four isoforms of WT1 to examine the effects of these WT1 isoforms on ER-α36 promoter activity. The four isoforms of WT1 inhibited the promoter activity of ER-α36 with different efficiency in that the WT1-D isoform exhibited the strongest activity whereas the WT1-A isoform exhibited the weakest activity (Fig. 4B).
Figure 4.
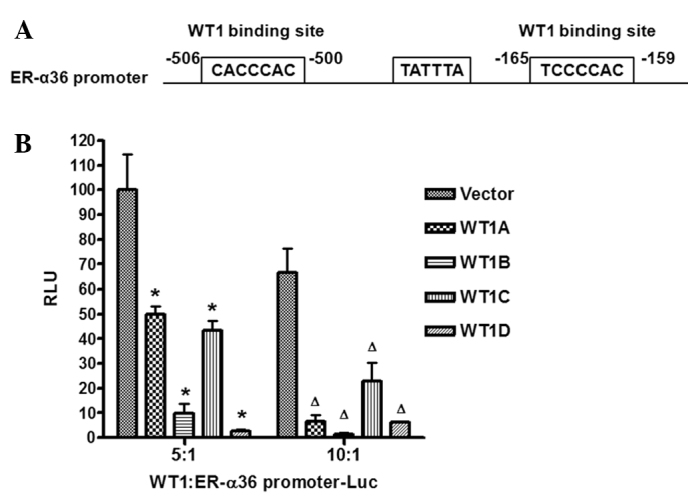
WT1 isoforms suppress ER-α36 promoter activity. (A) Schematic DNA structure of the ER-α36 promoter region. The WT1 binding sites are shown. (B) HEK293 cells were transiently co-transfected with the WT1 isoforms, A, B, C, or D, and the ER-α36 promoter luciferase reporter. The ratio of WT1:ER-α36 promoter-Luc was 5:1 or 10:1. The luciferase activity of HEK293 cells co-transfected with 5:1 empty vector and ER-α36 promoter-Luc was arbitrarily set to 100 RLU as a control. *P<0.05, the remaining groups were compared to the 5:1 control; ΔP<0.05, the remaining groups were compared to the 10:1 control. All experiments were repeated three times, and three parallel samples were used for each group.
Discussion
The diverse functions of estrogens are mediated by the estrogen receptors, ER-α and ER-β, both of which play a role as ligand-dependent transcription factors. The liganded ERs readily form homodimers or heterodimers that interact with the palindromic ERE in the promoter regions of estrogen responsive genes and stimulate gene transcription (2,3). Alternatively, ER-α may act indirectly by tethering to other transcription factors, such as Sp1 and AP1, to modulate activities of these transcription factors, thereby regulating downstream gene expression (2,3).
Previously, accumulating evidence suggested a rapid (within seconds or minutes) estrogen action that cannot be explained by the genomic signaling pathway which usually requires a long period of time to reach maximal gene activation (4–6,26). This non-genomic estrogen signaling pathway cross-talks with various signaling pathways, such as the adenylate cyclase, cAMP-dependent signaling and the MAPK pathways (4–6,26). Thus, the genomic and non-genomic pathways of estrogen action may integrate with one another to achieve a complete cellular response to estrogens.
It is well known that ER-α66 predominantly mediates genomic estrogen signaling by regulating target gene expression, although a previous study showed that ER-α66 is also involved in non-genomic estrogen signaling (27). ER-α36, on the other hand, lacks intrinsic transcription activity and mainly mediates non-genomic estrogen signaling (8). Thus, the expression levels of ER-α66 and ER-α36 should be dynamically and strictly regulated in order to maintain a balance between the genomic and non-genomic estrogen signaling pathways.
In the present study, ER-positive breast cancer MCF7 cells expressed high levels of WT1 and ER-α66, whereas MCF7 cells with a knocked-down level of WT1 expressed a decreased level of ER-α66, suggesting that WT1 up-regulates ER-α66 expression. We also found that the same WT1 knocked-down MCF7 cells expressed an increased level of ER-α36, suggesting that WT1 plays a role as a negative regulator of ER-α36 expression. Further co-transfection assays showed that WT directly activated the promoter activity of the ER-α66 gene and suppressed ER-α36 promoter activity. Thus, this study showed that WT1 plays a role as a dual transcription factor in the regulation of the promoter activities of ER-α66 and ER-α36 oppositely.
Current evidence indicates a potentially oncogenic role of WT1 in breast cancer (28). WT1 expression was found in primary breast tumors (21–23), and high levels of WT1 expression were shown to predict a poor prognosis in breast cancer patients (23), consistent with a putative oncogenic role of WT1. WT1 is a dual transcription regulator and that plays a role in the activation or suppression of gene transcription depending on the cell and promoter context (24,29–32). Previously, we demonstrated that WT1 acts as a transcription suppressor on promoters harboring WT1 binding sites both upstream and downstream of the transcription initiation site. WT1 also promotes transcription activity with WT1 binding sites located either upstream or downstream of the transcription site (24). Our computer analysis revealed the existence of two putative WT1 binding sites in the promoter region of ER-α36 located both upstream and downstream of the TATA box. By contrast, the ER-α66 promoter contained one perfect WT1 binding site downstream of the TATA box. Consequently, WT1 functions to oppositely regulate the promoter activities of ER-α66 and ER-α36.
Han et al have reported that the forced expression of WT1-B and -D isoforms in MCF7 cells down-regulated ER-α66 expression. Additionally, the co-transfection of WT1-B and -D isoforms moderately suppressed ER-α66 promoter activity (33). However, in the present study, it was noted that both WT1-B and -D isoforms up-regulated the promoter activity of ER-α66, and knockdown of all WT1 isoforms with shRNA down-regulated ER-α66 expression. The exact mechanisms underlying this discrepancy have yet to be elucidated. One possibility is that various ER-α66 promoter reporter constructs were used that contained a different length of the 5′-flanking sequence of the ER-α66 gene with different transcription factor binding sites. In a recent study, the forced expression of only WT1-B and -D isoforms was used (33). It was reported that various isoforms of WT1 clearly affected mammary epithelial cells differently (34). Another possibility is that changes noted in the ratios among various isoforms of WT1 following the forced expression of specific isoforms of WT1 may provide different outcomes. Our results suggest that the ratios of different WT1 isoforms expressed in mammary epithelial cells are involved in the coordination of the genomic and non-genomic signaling pathways by regulation of ER-α66 and ER-α36 expression oppositely.
Acknowledgements
This study was funded by the Nebraska Tobacco Settlement Biomedical Research Program Award (LB-595) to (Z.-Y. Wang) and NIH grant DK070016 (Z.-Y. Wang).
References
- 1.Vorherr H. Breast cancer: epidemiology, endocrinology, biochemistry, and pathobiology. Urban and Schwarzenberg; Baltimore: 1980. [Google Scholar]
- 2.Nilsson S, Makela S, Treuter E, et al. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 3.Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001;29:2905–2919. doi: 10.1093/nar/29.14.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Segars JH, Driggers PH. Estrogen action and cytoplasmic signaling cascades. Part I: membrane-associated signaling complexes. Trends Endocrinol Metab. 2002;13:349–354. doi: 10.1016/s1043-2760(02)00633-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Driggers PH, Segars JH. Estrogen action and cytoplasmic signaling pathways. Part II: the role of growth factors and phosphorylation in estrogen signaling. Trends Endocrinol Metab. 2002;13:422–427. doi: 10.1016/s1043-2760(02)00634-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelly MJ, Levin ER. Rapid actions of plasma membrane estrogen receptors. Trends Endocrinol Metab. 2001;12:152–156. doi: 10.1016/s1043-2760(01)00377-0. [DOI] [PubMed] [Google Scholar]
- 7.Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. Identification, cloning, and expression of human estrogen receptor-α36, a novel variant of human estrogen receptor-α66. Biochem Biophys Res Commun. 2005;336:1023–1027. doi: 10.1016/j.bbrc.2005.08.226. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. A variant of estrogen receptor-{α}, hER-{α}36: transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc Natl Acad Sci USA. 2006;103:9063–9068. doi: 10.1073/pnas.0603339103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin SL, Yan LY, Liang XW, et al. A novel variant of ER-α, ER-α36, mediates testosterone-stimulated ERK and Akt activation in endometrial cancer Hec1A cells. Reprod Biol Endocrinol. 2009;7:102. doi: 10.1186/1477-7827-7-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zou Y, Ding L, Coleman M, Wang Z. Estrogen receptor-α (ER-α) suppresses expression of its variant ER-α 36. FEBS Lett. 2009;583:1368–1374. doi: 10.1016/j.febslet.2009.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee LM, Cao J, Deng H, Chen P, Gatalica Z, Wang ZY. ER-α36, a novel variant of ER-α, is expressed in ER-positive and -negative human breast carcinomas. Anticancer Res. 2008;28:479–483. [PMC free article] [PubMed] [Google Scholar]
- 12.Shi L, Dong B, Li Z, et al. Expression of ER-(α)36, a novel variant of estrogen receptor-(α), and resistance to tamoxifen treatment in breast cancer. J Clin Oncol. 2009;27:3423–3429. doi: 10.1200/JCO.2008.17.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonetta L, Kuehn SE, Huang A, et al. Wilms tumor locus on 11p13 defined by multiple CpG island-associated transcripts. Science. 1990;250:994–997. doi: 10.1126/science.2173146. [DOI] [PubMed] [Google Scholar]
- 14.Call KM, Glaser T, Ito CY, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell. 1990;60:509–520. doi: 10.1016/0092-8674(90)90601-a. [DOI] [PubMed] [Google Scholar]
- 15.Gessler M, Poustka A, Cavenee W, Neve RL, Orkin SH, Bruns GA. Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature. 1990;343:774–778. doi: 10.1038/343774a0. [DOI] [PubMed] [Google Scholar]
- 16.Coppes MJ, Campbell CE, Williams BR. The role of WT1 in Wilms tumorigenesis. FASEB J. 1993;7:886–895. doi: 10.1096/fasebj.7.10.8393819. [DOI] [PubMed] [Google Scholar]
- 17.Lee SB, Haber DA. Wilms tumor and the WT1 gene. Exp Cell Res. 2001;264:74–99. doi: 10.1006/excr.2000.5131. [DOI] [PubMed] [Google Scholar]
- 18.Little M, Wells C. A clinical overview of WT1 gene mutations. Hum Mutat. 1997;9:209–225. doi: 10.1002/(SICI)1098-1004(1997)9:3<209::AID-HUMU2>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 19.Inoue K, Ogawa H, Sonoda Y, et al. Aberrant overexpression of the Wilms tumor gene (WT1) in human leukemia. Blood. 1997;89:1405–1412. [PubMed] [Google Scholar]
- 20.Oji Y, Miyoshi S, Maeda H, et al. Overexpression of the Wilms’ tumor gene WT1 in de novo lung cancers. Int J Cancer. 2002;100:297–303. doi: 10.1002/ijc.10476. [DOI] [PubMed] [Google Scholar]
- 21.Silberstein GB, Van Horn K, Strickland P, Roberts CT, Jr, Daniel CW. Altered expression of the WT1 wilms tumor suppressor gene in human breast cancer. Proc Natl Acad Sci USA. 1997;94:8132–8137. doi: 10.1073/pnas.94.15.8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loeb DM, Evron E, Patel CB, et al. Wilms’ tumor suppressor gene (WT1) is expressed in primary breast tumors despite tumor-specific promoter methylation. Cancer Res. 2001;61:921–925. [PubMed] [Google Scholar]
- 23.Miyoshi Y, Ando A, Egawa C, et al. High expression of Wilms’ tumor suppressor gene predicts poor prognosis in breast cancer patients. Clin Cancer Res. 2002;8:1167–1171. [PubMed] [Google Scholar]
- 24.Wang ZY, Qiu QQ, Deuel TF. The Wilms’ tumor gene product WT1 activates or suppresses transcription through separate functional domains. J Biol Chem. 1993;268:9172–9175. [PubMed] [Google Scholar]
- 25.Wang L, Wang ZY. The Wilms’ tumor suppressor WT1 induces estrogen-independent growth and anti-estrogen insensitivity in ER-positive breast cancer MCF7 cells. Oncol Rep. 2010;23:1109–1117. doi: 10.3892/or_00000739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levin ER. Integration of the extranuclear and nuclear actions of estrogen. Mol Endocrinol. 2005;19:1951–1959. doi: 10.1210/me.2004-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pedram A, Razandi M, Levin ER. Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol. 2006;20:1996–2009. doi: 10.1210/me.2005-0525. [DOI] [PubMed] [Google Scholar]
- 28.Yang L, Han Y, Suarez Saiz F, Minden MD. A tumor suppressor and oncogene: the WT1 story. Leukemia. 2007;21:868–876. doi: 10.1038/sj.leu.2404624. [DOI] [PubMed] [Google Scholar]
- 29.Liu XW, Gong LJ, Guo LY, et al. The Wilms’ tumor gene product WT1 mediates the down-regulation of the rat epidermal growth factor receptor by nerve growth factor in PC12 cells. J Biol Chem. 2001;276:5068–5073. doi: 10.1074/jbc.M008776200. [DOI] [PubMed] [Google Scholar]
- 30.Han Y, San-Marina S, Liu J, Minden MD. Transcriptional activation of c-myc proto-oncogene by WT1 protein. Oncogene. 2004;23:6933–6941. doi: 10.1038/sj.onc.1207609. [DOI] [PubMed] [Google Scholar]
- 31.Hewitt SM, Hamada S, McDonnell TJ, Rauscher FJ, III, Saunders GF. Regulation of the proto-oncogenes bcl-2 and c-myc by the Wilms’ tumor suppressor gene WT1. Cancer Res. 1995;55:5386–5389. [PubMed] [Google Scholar]
- 32.Englert C, Hou X, Maheswaran S, et al. WT1 suppresses synthesis of the epidermal growth factor receptor and induces apoptosis. EMBO J. 1995;14:4662–4675. doi: 10.1002/j.1460-2075.1995.tb00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han Y, Yang L, Suarez-Saiz F, San-Marina S, Cui J, Minden MD. Wilms’ tumor 1 suppressor gene mediates antiestrogen resistance via down-regulation of estrogen receptor-α expression in breast cancer cells. Mol Cancer Res. 2008;6:1347–1355. doi: 10.1158/1541-7786.MCR-07-2179. [DOI] [PubMed] [Google Scholar]
- 34.Burwell EA, McCarty GP, Simpson LA, Thompson KA, Loeb DM. Isoforms of Wilms’ tumor suppressor gene (WT1) have distinct effects on mammary epithelial cells. Oncogene. 2007;26:3423–3430. doi: 10.1038/sj.onc.1210127. [DOI] [PubMed] [Google Scholar]