

Abstract
Object
Ischemic injury is a potential complication in a variety of surgical procedures and is a particular impediment to the success of surgeries involving highly vulnerable neural tissue. One approach to limiting this form of injury is to enhance metabolic supply to the affected tissue. Trans-sodium crocetinate (TSC) is a carotenoid compound that has been shown to increase tissue oxygenation by facilitating the diffusivity of small molecules, such as oxygen and glucose. The present study examined the ability of TSC to modify oxygenation in ischemic neural tissue and tested the potential neuroprotective effects of TSC in permanent and temporary models of focal cerebral ischemia.
Methods
Adult male rats (330–370 g) were subjected to either permanent or temporary focal ischemia by simultaneous occlusion of both common carotid arteries and the left middle cerebral artery (3-vessel occlusion [3-VO]). Using the permanent ischemia paradigm, TSC was administered intravenously beginning 10 minutes after the onset of ischemia at 1 of 8 dosages, ranging from 0.023 to 4.580 mg/kg. Cerebral infarct volume was measured 24 hours after the onset of ischemia. The effect of TSC on infarct volume was also tested after temporary (2-hour) ischemia using a dosage of 0.092 mg/kg. In other animals undergoing temporary ischemia, tissue oxygenation was monitored in the ischemic penumbra using a Licox probe.
Results
Administration of TSC reduced infarct volume in a dose-dependent manner in the permanent ischemia model, achieving statistical significance at dosages ranging from 0.046 to 0.229 mg/kg. The most effective dosage of TSC in the permanent ischemia experiment (0.092 mg/kg) was further tested using a temporary (2-hour) ischemia paradigm. Infarct volume was reduced significantly by TSC in this ischemia-reperfusion model as well. Recordings of oxygen levels in the ischemic penumbra of the temporary ischemia model showed that TSC increased tissue oxygenation during vascular occlusion, but reduced the oxygen overshoot (hyperoxygenation) that occurs upon reperfusion.
Conclusions
The novel carotenoid compound TSC exerts a neuroprotective influence against permanent and temporary ischemic injury when administered soon after the onset of ischemia. The protective mechanism of TSC remains to be confirmed; however, the permissive effect of TSC on the diffusivity of small molecules is a plausible mechanism based on the observed increase in tissue oxygenation in the ischemic penumbra. This represents a form of protection based on “metabolic reflow” that can occur under conditions of partial vascular perfusion. It is particularly noteworthy that TSC could conceivably limit the progression of a wide variety of cellular injury mechanisms by blunting the ischemic challenge to the brain.
Keywords: trans-sodium crocetinate, focal ischemia, oxygen delivery, neuroprotection, metabolic reflow
Focal ischemia is an anticipated consequence of certain neurosurgical procedures and a common complication of others. Many attempts to limit the progression of intraoperative ischemic brain injury have focused on the suppression of metabolic demand or the inhibition of cellular injury cascades. These strategies share a common goal of limiting the cellular/molecular consequences that result from a reduction in the supply of metabolic substrates. An alternative approach to limiting ischemic tissue damage is to enhance metabolic supply in areas that retain partial perfusion. One such strategy, which has been tested in a variety of models of brain injury, is therapeutic hyperoxia. Hyperbaric hyperoxia has shown beneficial effects in experimental models of intraoperative ischemia, stroke, and traumatic brain injury.4,6,7,9,14,22,25,28,32,33,41,42,44,47,57,61,63,64,66–68 Clinical studies of hyperbaric hyperoxia have been less conclusive, but such studies have been hampered by small sample sizes and relatively long delays prior to initiating treatment.1–3,5,10–12,18–20,36–38,46,50,69 More recent studies indicate that normobaric hyperoxia can also improve outcomes in experimental models of cerebral ischemia and injury.16,21,29,31,49,52,53,62 Moreover, early-stage human studies using normobaric hyperoxia have shown promising results in treating cerebral injury.34,35,39,43,51,58,60 However, significant controversy as to the efficacy of normobaric hyperoxia remains, as highlighted in recent commentaries on the progress in this field.8,13,15,59 Despite this controversy, the potential for such therapies to ameliorate multiple forms of CNS injury for which there are no or inadequate medical countermeasures continues to warrant careful experimental and clinical assessment.
When considering the challenges of ischemic conditions, it is important to keep in mind that the delivery of metabolic substrates from the blood to tissue involves a series of resistances. A limiting resistance in this series is diffusion through the plasma boundary layer.26,27,65 As described by Fick’s law, oxygen delivery is directly proportional to both the oxygen concentration gradient and diffusivity, the latter being a measure of the ease with which a compound diffuses through a medium. It is thus possible to enhance substrate delivery to tissue not only through hyperoxia, but also by enhancing the diffusion of substrates through the plasma boundary layer. One compound that has been shown to increase the diffusion rate of oxygen through plasma is trans-sodium crocetinate (TSC).30,55 Trans-sodium crocetinate is a carotenoid compound (vitamin A analog) whose ability to enhance diffusivity in plasma is attributable to an alteration in the intermolecular spacing in aqueous solutions.30 It is also noteworthy that TSC can facilitate the diffusivity of other small molecules, including glucose.55 Although the baseline diffusivities for oxygen and glucose through aqueous media are different, they both increase by a similar percentage in the presence of TSC. Thus, TSC does not affect oxygen or glucose directly. Rather, TSC can increase hydrogen bonding of neighboring water molecules, a process known as “structure-building,” which reduces the resistance to the diffusion of small molecules such as oxygen and glucose.
The present study examined the ability of TSC to modify tissue oxygenation in the ischemic brain. In addition, the potential impact of TSC on tissue injury was studied in both permanent and temporary models of focal ischemia.
Methods
General Procedures
All procedures were approved by the University of Virginia Animal Care and Use Committee. Adult male Sprague-Dawley rats (330–370 g) were initially anesthetized in a plastic chamber with 4% halothane. Once they were anesthetized, orotracheal intubation was performed with the tube connected to a ventilator (Rodent Ventilator model 638, Harvard Apparatus) and FiO2 (fraction of inspired oxygen) was standardized at 50% for all experiments. The right femoral artery was cannulated for BP monitoring and repeated blood gas analysis (348 Blood Gas Analyzer, Bayer HealthCare). Rectal temperature was monitored continuously and maintained at 37°C with a heating lamp. Inspired halothane was controlled around 1.5% to maintain anesthesia and keep the MABP at approximately 110 mm Hg.
Both CCAs were exposed and a 5–0 monofilament polypropylene suture was passed through a polyethylene tube to create snares, which were loosely placed around each CCA for subsequent occlusion. The MCA was exposed according to the method of Hiramatsu et al.23 Briefly, animals were placed in a right decubitus position and a 1.5-cm incision was made between the left margin of the orbit and the tragus. The exposed temporal muscle was dissected from the cranium to reveal the infratemporal bone. After removal of the zygomatic arch, a craniectomy was made slightly anterior to the foramen ovale by using an electric drill under a surgical microscope. The dura mater was opened carefully and reflected using a 30-gauge needle, thus exposing the MCA bifurcation.
Focal ischemia was established by clipping (Sundt AVM microclip No. 1, Codman & Shurtleff, Inc.) the MCA at a point distal to the origin of the lenticulostriate arteries. The polypropylene suture loops around the CCAs were closed at the same time to complete the 3-vessel occlusion (3-VO). Loss of blood flow was confirmed visually with the surgical microscope, and occlusion of the MCA and both CCAs was maintained for 24 hours in the permanent ischemia protocol. In the transient ischemia protocol, the microclip blocking the MCA and polypropylene snares around both CCAs were removed 2 hours after onset of ischemia and reflow was confirmed visually by observation through the surgical microscope. Animals were anesthetized during the surgical procedure but were permitted to recover from anesthesia postoperatively. The animals were monitored carefully for 1 hour after the surgery and then returned to the vivarium.
Measurement of Infarct Volume
Twenty-four hours after the onset of ischemia, animals were anesthetized with an overdose of pentobarbital and killed by decapitation. The brains were removed rapidly and coronal sections (2-mm thick) were cut with a McIlwain tissue chopper. The sections were stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC) in phosphate-buffered saline for 5 minutes at 37°C and then placed in 4% paraformaldehyde solution.
Infarct volume was calculated by summing the infarct areas in individual sections, as measured using image analysis software (Scion Image Beta 4.02, Scion Corp.). In addition, the areas of the hemispheres ipsilateral and contralateral to the occluded MCA were measured, and the total volume of each hemisphere was calculated in a similar manner. The actual infarct volume, adjusted for swelling, was calculated using the following formula: actual infarct volume = total infarct volume × (contralateral hemisphere volume/ipsilateral hemisphere volume). The values presented below are the actual infarct volumes.
Statistical comparisons for 3 groups or more used a 1-way ANOVA followed by the LSD post hoc test. The Student t-test was used for comparisons between 2 groups. A probability value of < 0.05 was considered to be statistically significant.
Experiment 1: Effect of TSC on Cerebral Infarction in a Model of Permanent Focal Ischemia
Ten minutes after permanent 3-VO was established, animals were treated with vehicle or 1 of 8 different dosages of TSC (0.023, 0.046, 0.092, 0.229, 0.458, 0.916, 2.29, or 4.58 mg/kg). Seven animals were included in each group. Vehicle or TSC (in equivalent volumes) was administered via the femoral vein using a “bolus-infusion-bolus” treatment paradigm. Ten minutes after the onset of ischemia, a bolus injection of 0.1 ml was administered, followed by continuous infusion at 0.01 ml per minute for 60 minutes. Thirty minutes after the cessation of infusion, another 0.1 ml was injected. This treatment protocol was based on previous evidence demonstrating TSC-induced elevation of tissue oxygenation in the brain.40 All of the dosages reported herein reflect the total dosage of TSC administered during the 2 bolus injections and the infusion period. Animals were killed 24 hours after the onset of ischemia and infarct volume was calculated as described above.
Experiment 2: Effect of TSC on Cerebral Infarction in a Model of Transient Focal Ischemia
The most effective dosage of TSC, as determined from the dose-response curve generated in Experiment 1, was used to test the effect of TSC in a model of transient focal ischemia. Trans-sodium crocetinate (0.092 mg/kg) or vehicle (6 animals per group) was administered using the same bolus-infusion-bolus treatment protocol beginning 10 minutes after the onset of ischemia, as in Experiment 1. Reflow was established after 2 hours of 3-VO and animals were killed 24 hours after the onset of ischemia—that is, 22 hours after reflow. Cerebral infarct volume was determined as described above.
Experiment 3: Effect of TSC on Brain Tissue Oxygenation in a Model of Transient Focal Ischemia
This experiment assessed the effect of TSC on brain tissue oxygenation during ischemia. The preparative stages of the 3-VO surgery were performed as described earlier until the point of clipping the arteries. The skull of the rat was then secured to a stereotactic frame. A mid-line scalp incision was made and a small craniotomy was performed over the region of the ischemic penumbra (0.5 mm posterior to bregma and 2.5 mm lateral to the mid-line). These coordinates were used based on our previous studies in which distinct core and penumbra regions were defined with the 3-VO model.54 An oxygen-sensing probe (Licox, Integra Neuroscience) was lowered stereotactically into the neocortex (2.5 mm deep) to measure brain PO2. An equilibration period of at least 60 minutes was allowed for the Licox probe, after which baseline values were recorded for at least 15 minutes. Once a stable baseline was established, the vessels for the 3-VO were blocked. Either TSC (0.092 mg/kg; 5 animals) or vehicle (7 animals) was administered starting 10 minutes after the onset of ischemia using the standard bolus-infusion-bolus treatment protocol. Reflow was established after 2 hours of 3-VO and tissue oxygen recordings were terminated approximately 15 minutes thereafter. For the quantitative analyses, the recorded tissue oxygen levels were normalized for each animal to their preischemic baseline to facilitate comparisons among animals. Animals were killed at the end of this recording period. The animals in these groups were not used for the assessment of infarct volume because of the additionally invasive nature of the oxygen recording procedure in the ischemic tissue.
Results
Experiment 1: Effect of TSC on Cerebral Infarction in a Model of Permanent Focal Ischemia
Treatment with TSC beginning 10 minutes after the onset of ischemia produced a dose-dependent neuroproective effect, reducing the volume of cerebral infarction in the model of permanent focal cerebral ischemia (Fig. 1). A biphasic (U-shaped) reduction in infarct volume was observed, with intermediate dosages of TSC providing the greatest protective effect. The reductions in infarct volume achieved statistical significance at dosages ranging from 0.023 to 0.229 mg/kg. A maximal protective effect, representing a reduction in infarct volume of approximately 60%, was achieved at a dosage of 0.092 mg/kg (Fig. 1). Examples of the cerebral infarcts observed in the Vehicle Group and the group treated with 0.092 mg/kg TSC are shown in Fig. 2. Comparisons between matching section levels show a substantial reduction in infarct size in the TSC-treated animal. Physiological parameters were monitored 1) before ischemia, 2) during ischemia and TSC infusion, and 3) during ischemia after the cessation of TSC treatment (Table 1). No significant differences among groups were observed for the physiological parameters, which included MABP, heart rate, blood pH, blood PCO2, and blood PO2.
Fig. 1.
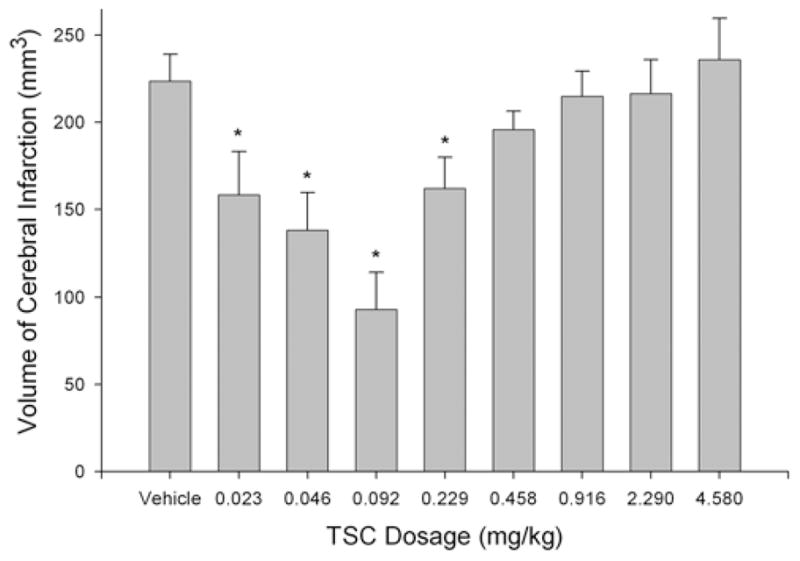
Effect of TSC on cerebral infarct volume in a model of permanent (24-hour) focal ischemia. The bar graph shows group means and SEs for the vehicle- and TSC-treated animals (7 rats per group). The volume of cerebral infarction was reduced by TSC in a dose-dependent manner, characterized by a U-shaped curve. This protective action achieved statistical significance at TSC dosages ranging from 0.023 to 0.229 mg/kg, with a maximal protective effect being achieved at 0.092 mg/kg. * p < 0.05, 1-way ANOVA and the LSD post hoc test.
Fig. 2.

Examples of cerebral infarcts in vehicle-treated and TSC-treated animals. Serial coronal sections obtained from a vehicle-treated animal and a TSC-treated animal (0.092 mg/kg) are shown. Sections were stained with tetrazolium chloride for the detection of infarcted tissue. Areas of tissue infarction appear white in these sections, while the healthy tissue appears red. The infarct size is substantially smaller inthe animal from the TSC-treated group.
TABLE 1.
Physiological parameters in animals from the permanent (24-hour) ischemia experiment (Experiment 1)*
| Dosage & Timing of Measurements | MABP (mm Hg) | HR (beats/min) | pH | PCO2 (mm Hg) | PO2 (mm Hg) |
|---|---|---|---|---|---|
| vehicle
| |||||
| before 3-VO | 122.9 ± 4.2 | 353.4 ± 8.8 | 7.453 ± 0.020 | 29.1 ± 2.8 | 168.0 ± 12.3 |
| during 3-VO & infusion | 122.8 ± 2.8 | 377.2 ± 11.4 | 7.446 ± 0.022 | 29.2 ± 2.5 | 175.8 ± 10.6 |
| after infusion | 125.8 ± 2.2 | 385.5 ± 6.5 | 7.427 ± 0.026 | 31.3 ± 2.2 | 176.1 ± 9.2 |
|
| |||||
| 0.023 mg/kg TSC
| |||||
| before 3-VO | 121.1 ± 3.7 | 376.6 ± 7.2 | 7.470 ± 0.019 | 28.2 ± 1.8 | 178.6 ± 8.1 |
| during 3-VO & infusion | 119.6 ± 4.0 | 394.7 ± 10.8 | 7.474 ± 0.011 | 31.9 ± 0.7 | 178.5 ± 7.9 |
| after infusion | 124.0 ± 5.1 | 392.2 ± 10.2 | 7.458 ± 0.018 | 28.5 ± 2.0 | 179.3 ± 9.4 |
|
| |||||
| 0.046 mg/kg TSC
| |||||
| before 3-VO | 121.6 ± 3.2 | 369.1 ± 5.6 | 7.455 ± 0.016 | 32.0 ± 2.6 | 190.2 ± 7.1 |
| during 3-VO & infusion | 123.6 ± 3.4 | 386.3 ± 12.5 | 7.457 ± 0.017 | 31.2 ± 2.3 | 176.8 ± 6.3 |
| after infusion | 119.5 ± 1.7 | 377.3 ± 14.4 | 7.460 ± 0.017 | 32.1 ± 2.8 | 168.6 ± 7.5 |
|
| |||||
| 0.092 mg/kg TSC
| |||||
| before 3-VO | 125.0 ± 6.8 | 364.6 ± 8.0 | 7.480 ± 0.015 | 31.5 ± 2.6 | 192.5 ± 7.0 |
| during 3-VO & infusion | 124.4 ± 4.1 | 386.4 ± 13.9 | 7.451 ± 0.016 | 30.6 ± 1.5 | 183.3 ± 6.8 |
| after infusion | 122.8 ± 3.3 | 376.4 ± 15.8 | 7.426 ± 0.007 | 33.8 ± 1.6 | 168.2 ± 8.8 |
|
| |||||
| 0.230 mg/kg TSC
| |||||
| before 3-VO | 116.3 ± 5.4 | 378.3 ± 9.8 | 7.474 ± 0.013 | 30.3 ± 1.5 | 185.7 ± 9.8 |
| during 3-VO & infusion | 121.8 ± 4.3 | 394.7 ± 4.0 | 7.461 ± 0.017 | 30.2 ± 1.5 | 190.6 ± 9.8 |
| after infusion | 131.3 ± 6.3 | 397.5 ± 12.1 | 7.453 ± 0.016 | 30.4 ± 2.3 | 187.6 ± 6.4 |
|
| |||||
| 0.458 mg/kg TSC
| |||||
| before 3-VO | 117.5 ± 3.4 | 339.7 ± 17.6 | 7.457 ± 0.014 | 30.5 ± 2.1 | 167.5 ± 12.0 |
| during 3-VO & infusion | 128.5 ± 6.7 | 357.3 ± 18.7 | 7.433 ± 0.020 | 29.4 ± 2.6 | 171.9 ± 11.4 |
| after infusion | 125.2 ± 6.2 | 355.2 ± 5.9 | 7.436 ± 0.006 | 29.7 ± 1.9 | 174.4 ± 11.2 |
|
| |||||
| 0.916 mg/kg TSC
| |||||
| before 3VO | 113.7 ± 4.4 | 347.3 ± 6.7 | 7.432 ± 0.012 | 31.4 ± 2.3 | 177.1 ± 7.7 |
| during 3-VO & infusion | 115.9 ± 4.0 | 370.1 ± 10.5 | 7.447 ± 0.015 | 32.2 ± 1.7 | 182.2 ± 6.8 |
| after infusion | 110.7 ± 2.3 | 364.9 ± 7.1 | 7.444 ± 0.012 | 31.6 ± 1.5 | 184.8 ± 4.9 |
|
| |||||
| 2.290 mg/kg TSC
| |||||
| before 3-VO | 118.8 ± 6.7 | 347.2 ± 7.9 | 7.451 ± 0.021 | 31.7 ± 1.8 | 177.4 ± 6.3 |
| during 3-VO & infusion | 119.6 ± 5.3 | 359.8 ± 8.6 | 7.435 ± 0.007 | 32.0 ± 2.2 | 178.2 ± 4.5 |
| after infusion | 116.8 ± 4.7 | 356.4 ± 9.1 | 7.439 ± 0.005 | 31.6 ± 1.6 | 182.8 ± 4.6 |
|
| |||||
| 4.580 mg/kg TSC
| |||||
| before 3-VO | 110.7 ± 2.7 | 357.0 ± 12.3 | 7.452 ± 0.017 | 27.6 ± 2.6 | 178.8 ± 8.2 |
| during 3-VO & infusion | 113.1 ± 2.5 | 374.3 ± 11.1 | 7.456 ± 0.015 | 30.6 ± 2.0 | 175.4 ± 6.7 |
| after infusion | 111.1 ± 2.3 | 362.4 ± 12.0 | 7.443 ± 0.013 | 31.6 ± 2.5 | 179.9 ± 4.8 |
Values shown are means ± SEs. No significant intergroup differences were observed for MABP, heart rate, blood pH, blood PCO2, or blood PO2.
Abbreviations: HR = heart rate; pH = blood pH; PCO2 = blood PCO2; PO2 = blood PO2.
Experiment 2: Effect of TSC on Cerebral Infarction in a Model of Transient Focal Ischemia
The effect of TSC was next tested in a model of ischemia-reperfusion involving 2 hours of ischemia followed by 22 hours of reperfusion. The dosage of TSC providing an optimal neuroprotective effect in Experiment 1 (0.092 mg/kg) was used, with treatment being initiated 10 minutes after the onset of ischemia. Treatment with TSC significantly reduced the volume of cerebral infarction. The volume of infarction in the TSC-treated group was reduced by approximately 45% as compared with that in the vehicle-treated group (Fig. 3). Physiological parameters monitored 1) before ischemia, 2) during ischemia and TSC treatment, and 3) after reperfusion are shown in Table 2. No significant differences among groups were observed for the physiological parameters.
Fig. 3.

Bar graph showing effect of TSC on cerebral infarct volume after transient focal ischemia. The graph shows group means and SEs for cerebral infarct volume in vehicle-treated and TSC-treated (0.092 mg/kg) animals (6 rats per group). The cerebral infarct volume was reduced significantly in the TSC-treated group. ** p < 0.01, Student t-test.
TABLE 2.
Physiological parameters in animals from the transient (2-hour) ischemia experiment (Experiment 2)*
| Dosage & Timing of Measurements | MABP (mm Hg) | HR (beats/min) | pH | PCO2 (mm Hg) | PO2 (mm Hg) |
|---|---|---|---|---|---|
| vehicle
| |||||
| before 3-VO | 108.7 ± 4.5 | 348.6 ± 14.7 | 7.440 ± 0.024 | 31.3 ± 1.9 | 164.5 ± 3.0 |
| during 3-VO & infusion | 107.0 ± 4.2 | 373.1 ± 19.1 | 7.436 ± 0.024 | 29.3 ± 1.8 | 176.4 ± 7.7 |
| after infusion | 102.4 ± 2.9 | 377.3 ± 14.7 | 7.425 ± 0.016 | 30.1 ± 2.7 | 174.2 ± 8.3 |
|
| |||||
| 0.092 mg/kg TSC
| |||||
| before 3-VO | 106.0 ± 3.2 | 368.6 ± 18.7 | 7.418 ± 0.020 | 32.9 ± 1.9 | 172.3 ± 7.5 |
| during 3-VO & infusion | 106.1 ± 3.3 | 395.8 ± 17.3 | 7.417 ± 0.018 | 31.8 ± 1.7 | 184.1 ± 9.4 |
| after infusion | 103.9 ± 3.3 | 391.4 ± 14.6 | 7.431 ± 0.014 | 31.4 ± 1.1 | 170.3 ± 15.9 |
Values shown are means ± SEs. No significant intergroup differences were observed for MABP, heart rate, blood pH, blood PCO2, or blood PO2.
Experiment 3: Effect of TSC on Brain Tissue Oxygenation in a Model of Transient Focal Ischemia
An enhancement of brain tissue oxygenation by TSC has been demonstrated previously under nonpathological circumstances in the presence of elevated FiO2;40 however, the impact of TSC on tissue oxygenation in ischemic brain has not been evaluated. In Experiment 3, tissue oxygenation was monitored in the ischemic penumbra of the 3-VO model in animals treated with TSC or vehicle. Animals underwent transient focal ischemia as in the previous experiment and tissue oxygen levels were monitored with a Licox probe. Tissue oxygen levels were reduced substantially in both groups of animals in response to vascular occlusion (Fig. 4). Treatment with TSC (0.092 mg/kg) beginning 10 minutes after the onset of ischemia resulted in a progressive and significant increase in tissue oxygenation, as compared with vehicle-treated controls. Within 10 minutes of administering TSC, the levels of penumbral tissue oxygenation began to rise, and during the course of the recordings TSC reversed the intensity of oxygen deprivation by approximately 50%. It is also noteworthy that upon reflow the oxygen overshoot (reperfusion hyperoxia) was less pronounced in the TSC-treated group. Thus, the intensity of hypoxia during vascular occlusion was reduced, while reperfusion hyperoxia was attenuated in the TSC-treated group. As in the previous experiment, physiological parameters did not differ between groups (data not shown).
Fig. 4.

Graph showing brain tissue partial oxygen levels during ischemia-reperfusion. Values in each animal were normalized to a preischemia baseline and this baseline is expressed as a value of 1.00. Group means and SEs are shown for vehicle-treated animals (7 rats, filled circles) and TSC-treated animals (5 rats, filled triangles). The timings of bolus and infusion treatments of vehicle or TSC are indicated on the graph. Treatment with TSC produced a progressive increase in tissue oxygen levels during ischemia. Upon reperfusion, a substantial hyperoxia response was observed in the vehicle-treated group; this effect was blunted in the TSC-treated group. Note that the y axis is interrupted on the graph above the baseline level to facilitate group comparisons both during and after ischemia. * p < 0.05, compared with corresponding time points for the 2 groups.
Discussion
Therapeutic strategies designed to attenuate ischemic injury to the nervous system have generally focused on limiting the induction and/or progression of cellular mechanisms of injury. These strategies have taken various forms, but they commonly involve either the suppression of metabolic activity or the inhibition of specific injury cascades. An alternative approach has been to enhance metabolic supply to areas of partial perfusion to sustain essential cellular functions. This approach typically entails an increase in systemic oxygen supply (hyperoxygenation) under hyperbaric and more recently normobaric conditions. Each of these strategies holds considerable promise and warrants continued and careful development.
The current study describes a novel therapeutic approach that is related conceptually to hyperoxygenation therapy. Trans-sodium crocetinate is a modified carotenoid compound that is capable of enhancing the diffusivity of small molecules in aqueous solutions. This compound and its parent compound crocetin have previously been shown to increase tissue oxygenation in multiple organ systems.40,45,48 In a model of hemorrhagic shock, TSC both improved oxygen consumption and increased survival.45 Observations presented herein support these previous findings by demonstrating that TSC treatment increases tissue oxygen levels in an area of partial perfusion in the brain. To our knowledge, this is the first evidence of a medicinally induced increase in tissue oxygenation in the penumbra of a focal ischemic event. While the mechanistic underpinnings of enhanced tissue oxygenation were not the subject of the current study, previous studies demonstrate that TSC increases hydrogen bonding among water molecules, leading to a reduction in chaos in the aqueous solution.30 This process is known as “structure-building” and in aqueous solutions (for example, plasma and interstitial fluids) increased structure can facilitate the diffusivity of small molecules, including oxygen and glucose. Inasmuch as the plasma phase is a critical site of resistance for the delivery of blood-borne oxygen to tissue,26,27,65 it is postulated that enhanced diffusivity of metabolic substrates from the vasculature contributes to the TSC-induced increase in tissue oxygenation in the ischemic penumbra.
A central goal of the current study was to evaluate the potential protective actions of TSC against ischemic injury to the brain. In a rat model of permanent focal ischemia, TSC was effective in reducing infarct volume when treatment was initiated soon after the onset of ischemia. This effect was dose dependent, with significant protection in the dosage range of 0.023–0.229 mg/kg and optimal protection at a dosage of 0.092 mg/kg TSC. The mechanism underlying the reduced efficacy of TSC at the highest dosages is at present unknown and remains a matter for future investigation. The protective effect of TSC at its optimal dosage represented a reduction in infarct volume of approximately 60%, which rivals maximal protective effects of other interventions (for example, hypothermia) observed in the 3-vessel model of focal ischemia.18
A key a priori concern about any therapy involving increased oxygenation is that elevated levels of tissue oxygen could aggravate outcomes during the reperfusion phase that follows transient ischemia. Considerable evidence supports the concept that an oxidative burst and free radical generation play critical roles in ischemia-reperfusion injury. Reperfusion in the presence of elevated tissue oxygen could conceivably amplify damaging effects of oxidative injury. This issue was evaluated using 2 approaches. First, the hyperoxia response during postischemic reperfusion was examined in TSC-treated animals. Second, the impact of TSC on cerebral infarction was assessed subsequent to ischemia-reperfusion. Rather than potentiating postischemic hyperoxia, treatment with TSC actually blunted postischemic hyperoxia. We speculate that this reduction in the reperfusion-hyperoxia reflects a healthier state of the penumbral tissue, owing to metabolic substrate supplementation produced by TSC during ischemia. Consistent with the “healthier tissue” concept is the observation that TSC significantly reduced postischemic injury after ischemia-reperfusion. Treatment with TSC reduced cerebral infarct volume by approximately 45% when initiated 10 minutes after the onset of ischemia. Thus, even though the levels of tissue oxygen were elevated at the onset of reperfusion, tissue hyperoxia was blunted during reperfusion, and tissue survival was improved by TSC.
It is important to consider the possibility that TSC could exert its protective actions via mechanism(s) other than the facilitation of small molecule diffusion. For instance, some carotenoid compounds possess free radical scavenging activity, an effect that could provide a protective influence against ischemic neural injury. While TSC is capable of scavenging free radicals, this effect only achieves significance at dosages much higher than those required for TSC to enhance diffusivity and improve survival.56 Other mechanisms by which TSC could theoretically exert protection include effects on blood flow and/or cellular metabolism. These possibilities have been examined previously with the structurally similar parent compound of TSC, crocetin. However, no significant effects on blood flow,24 oxygen solubility in blood,17 oxyhemoglobin saturation,17 or oxidative phosphorylation17 were observed in response to treatment with crocetin. Although the effect of TSC on blood viscosity has not been studied, it has been shown to produce a slight increase in the viscosity of water,56 which would not be predicted to increase blood flow. Thus, the evidence available to date is inconsistent with the concept that an alternative mechanism to enhanced diffusivity is responsible for the protective actions provided by TSC.
Taken together, the present findings indicate that TSC could be of value for limiting intraoperative ischemic injury or other anticipated forms of ischemic challenge. Based on its presumptive mechanism of action, TSC may be capable of inhibiting a broad range of neural injury mechanisms by reducing both the intensity of the metabolic challenge during partial ischemia and by attenuating reperfusion-induced oxidative injury.
Conclusions
The protective actions of TSC represent a novel therapy based on the concept of “metabolic reflow,” which can operate under conditions of partial vascular perfusion. It will be of interest for future studies to evaluate the potential efficacy of this compound when applied in a delayed manner after the onset of ischemia. If TSC were to provide a similar protective effect under such conditions, it might also prove to be of value in the context of other forms of ischemic injury, such as transient ischemic attacks and stroke.
Acknowledgments
This study was supported by grant no. NS057168 from the NIH/NINDS to Dr. Lee and grant no. T32 GM08328 from the NIH/ NIGMS to Ryon Clarke and Dr. Lee.
The authors thank Mark Fitzgerald, Yi Wang, and Yu Cai for critical reading of the manuscript.
Abbreviations used in this paper
- BP
blood pressure
- CCA
common carotid artery
- LSD
least significant difference
- MABP
mean arterial BP
- MCA
middle cerebral artery
- NIGMS
National Institute of General Medical Sciences
- NIH
National Institutes of Health
- NINDS
National Institute of Neurological Disorders and Stroke
- TSC
trans-sodium crocetinate
- 3-VO
3-vessel occlusion
Footnotes
Disclosure
Dr. Gainer was a faculty member in the Department of Chemical Engineering at the University of Virginia during the performance of the studies described herein. He is now the chief scientific consultant for Diffusion Pharmaceuticals, LLC, which is developing trans-sodium crocetinate for clinical use.
Portions of this work were presented in abstract form at the Society for Neuroscience Annual Meeting, San Diego, 2007.
References
- 1.Alternative Therapy Evaluation Committee for the Insurance Corporation of British Columbia. A review of the scientific evidence on the treatment of traumatic brain injuries and strokes with hyperbaric oxygen. Brain Inj. 2003;17:225–236. doi: 10.1080/0269905021000030814. [DOI] [PubMed] [Google Scholar]
- 2.Al-Waili NS, Butler GJ, Beale J, Abdullah MS, Hamilton RWB, Lee BY, et al. Hyperbaric oxygen in the treatment of patients with cerebral stroke, brain trauma, and neurologic disease. Adv Ther. 2005;22:659–678. doi: 10.1007/BF02849960. [DOI] [PubMed] [Google Scholar]
- 3.Anderson DC, Bottini AG, Jagiella WM, Westphal B, Ford S, Rockswold GL, et al. A pilot study of hyperbaric oxygen in the treatment of human stroke. Stroke. 1991;22:1137–1142. doi: 10.1161/01.str.22.9.1137. [DOI] [PubMed] [Google Scholar]
- 4.Badr AE, Yin W, Mychaskiw G, Zhang JH. Dual effect of HBO on cerebral infarction in MCAO rats. Am J Physiol Regul Integr Comp Physiol. 2001;280:R766–R770. doi: 10.1152/ajpregu.2001.280.3.R766. [DOI] [PubMed] [Google Scholar]
- 5.Bennett MH, Wasiak J, Schnabel A, Kranke P, French C. Hy-perbaric oxygen therapy for acute ischaemic stroke. Cochrane Database Syst Rev. 2005;3:CD004954. doi: 10.1002/14651858.CD004954.pub2. [DOI] [PubMed] [Google Scholar]
- 6.Beynon C, Sun L, Marti HH, Heiland S, Veltkamp R. Delayed hyperbaric oxygenation is more effective than early prolonged normobaric hyperoxia in experimental focal cerebral ischemia. Neurosci Lett. 2007;425:141–145. doi: 10.1016/j.neulet.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Bramlett HM, Dave KR, Prado R, Perez-Pinzon MA, Torbatti D, Alonso OF, et al. Hyperbaric oxygen treatment provides histopathological protection following fluid percussion brain injury and secondary hypoxia. J Cereb Blood Flow Metab. 2001;21(1 Suppl):S-190. [Google Scholar]
- 8.Bullock MR. Hyperoxia. J Neurosurg. 2008;109:421–423. doi: 10.3171/JNS/2008/109/9/0421. (Letter) [DOI] [PubMed] [Google Scholar]
- 9.Burt JT, Kapp JP, Smith RR. Hyperbaric oxygen and cerebral infarction in the gerbil. Surg Neurol. 1987;28:265–268. doi: 10.1016/0090-3019(87)90304-1. [DOI] [PubMed] [Google Scholar]
- 10.Carson S, McDonagh M, Russman B, Helfand M. Hyperbaric oxygen therapy for stroke: a systematic review of the evidence. Clin Rehabil. 2005;19:819–833. doi: 10.1191/0269215505cr907oa. [DOI] [PubMed] [Google Scholar]
- 11.Diringer MN. Hyperoxia. good or bad for the injured brain? Curr Opin Crit Care. 2008;14:167–171. doi: 10.1097/MCC.0b013e3282f57552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diringer MN, Aiyagari V, Zazulia AR, Videen TO, Powers WJ. Effect of hyperoxia on cerebral metabolic rate for oxygen measured using positron emission tomography in patients with acute severe head injury. J Neurosurg. 2007;106:526–529. doi: 10.3171/jns.2007.106.4.526. [DOI] [PubMed] [Google Scholar]
- 13.Diringer MN, Powers WJ, Zazulia AR, Videen TO. Hyperoxia and traumatic brain injury. J Neurosurg. 2009;110:607–610. [Google Scholar]
- 14.Eschenfelder CC, Krug R, Yusofi AF, Meyne JK, Herdegen T, Koch A, et al. Neuroprotection by oxygen in acute transient focal cerebral ischemia is dose dependent and shows superiority of hyperbaric oxygenation. Cerebrovasc Dis. 2008;25:193–201. doi: 10.1159/000113856. [DOI] [PubMed] [Google Scholar]
- 15.Fehlings MG, Baker A. Is there a role for hyperoxia in the management of severe traumatic brain injury? J Neurosurg. 2007;106:525. doi: 10.3171/jns.2007.106.4.525. (Letter) [DOI] [PubMed] [Google Scholar]
- 16.Flynn EP, Auer RN. Eubaric hyperoxemia and experimental cerebral infarction. Ann Neurol. 2002;52:566–572. doi: 10.1002/ana.10322. [DOI] [PubMed] [Google Scholar]
- 17.Gainer JL, Rudolph DB, Caraway DL. The effect of crocetin on hemorrhagic shock in rats. Circ Shock. 1993;41:1–7. [PubMed] [Google Scholar]
- 18.Goto Y, Kassell NF, Hiramatsu K, Soleau SW, Lee KS. Effects of intraischemic hypothermia on cerebral damage in a model of reversible focal ischemia. Neurosurgery. 1993;32:980–985. doi: 10.1227/00006123-199306000-00017. [DOI] [PubMed] [Google Scholar]
- 19.Helms AK, Whelan HT, Torbey MT. Hyperbaric oxygen therapy of acute ischemic stroke. Stroke. 2007;38:1137–1139. doi: 10.1161/01.STR.0000259832.57823.83. [DOI] [PubMed] [Google Scholar]
- 20.Helms AK, Whelan HT, Torbey MT. Hyperbaric oxygen therapy of cerebral ischemia. Cerebrovasc Dis. 2005;20:417–426. doi: 10.1159/000088979. [DOI] [PubMed] [Google Scholar]
- 21.Henninger N, Bouley J, Nelligan JM, Sicard KM, Fisher M. Normobaric hyperoxia delays perfusion/diffusion mismatch evolution, reduces infarct volume, and differentially affects neuronal cell death pathways after suture middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab. 2007;27:1632–1642. doi: 10.1038/sj.jcbfm.9600463. [DOI] [PubMed] [Google Scholar]
- 22.Henninger N, Küppers-Tiedt L, Sicard KM, Günther A, Sch-neider D, Schwab S. Neuroprotective effect of hyperbaric oxygen therapy monitored by MR-imaging after embolic stroke in rats. Exp Neurol. 2006;201:316–323. doi: 10.1016/j.expneurol.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 23.Hiramatsu K, Kassell NF, Goto Y, Soleau S, Lee KS. A reproducible model of reversible, focal, neocortical ischemia in Sprague-Dawley rat. Acta Neurochir (Wien) 1993;120:66–71. doi: 10.1007/BF02001472. [DOI] [PubMed] [Google Scholar]
- 24.Holloway GM, Gainer JL. The carotenoid crocetin enhances pulmonary oxygenation. J Appl Physiol. 1988;65:683–686. doi: 10.1152/jappl.1988.65.2.683. [DOI] [PubMed] [Google Scholar]
- 25.Huang ZX, Kang ZM, Gu GJ, Peng GN, Yun L, Tao HY, et al. Therapeutic effects of hyperbaric oxygen in a rat model of endothelin-1-induced focal cerebral ischemia. Brain Res. 2007;1153:204–213. doi: 10.1016/j.brainres.2007.03.061. [DOI] [PubMed] [Google Scholar]
- 26.Huxley VH, Kutchai H. Effect of diffusion boundary layers on the initial uptake of O2 by red cells. Theory versus experiment. Microvasc Res. 1983a;26:89–107. doi: 10.1016/0026-2862(83)90058-4. [DOI] [PubMed] [Google Scholar]
- 27.Huxley VH, Kutchai H. The effect of red cell membrane and a diffusion boundary layer on the rate of oxygen uptake by human erythrocytes. J Physiol. 1981;316:75–83. doi: 10.1113/jphysiol.1981.sp013773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawamura S, Yasui N, Shirasawa M, Fukasawa H. Therapeutic effects of hyperbaric oxygenation on acute focal cerebral ischemia in rats. Surg Neurol. 1990;34:101–106. doi: 10.1016/0090-3019(90)90104-w. [DOI] [PubMed] [Google Scholar]
- 29.Kim HY, Singhal AB, Lo EH. Normobaric hyperoxia extends the reperfusion window in focal cerebral ischemia. Ann Neu-rol. 2005;57:571–575. doi: 10.1002/ana.20430. [DOI] [PubMed] [Google Scholar]
- 30.Laidig KE, Daggett V, Gainer JL. Altering diffusivity in biological solutions via change of solution structure and dynamics. J Am Chem Soc. 1998;120:9394–9395. [Google Scholar]
- 31.Liu W, Sood R, Chen Q, Sakoglu U, Hendren J, Cetin O, et al. Normobaric hyperoxia inhibits NADPH oxidase-mediated matrix metalloproteinase-9 induction in cerebral microvessels in experimental stroke. J Neurochem. 2008;107:1196–1205. doi: 10.1111/j.1471-4159.2008.05664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lou M, Eschenfelder CC, Herdegen T, Brecht S, Deuschl G. Therapeutic window for use of hyperbaric oxygenation in focal transient ischemia in rats. Stroke. 2004;35:578–583. doi: 10.1161/01.STR.0000111599.77426.A0. [DOI] [PubMed] [Google Scholar]
- 33.Lou M, Zhang H, Wang J, Wen SQ, Tang ZQ, Chen YZ, et al. Hyperbaric oxygen treatment attenuated the decrease in regional glucose metabolism of rats subjected to focal cerebral ischemia: a high resolution positron emission tomography study. Neuroscience. 2007;146:555–561. doi: 10.1016/j.neuroscience.2007.01.046. [DOI] [PubMed] [Google Scholar]
- 34.Menzel M, Doppenberg EMR, Zauner A, Soukup J, Reinert MM, Bullock R. Increased inspired oxygen concentration as a factor in improved brain tissue oxygenation and tissue lactate levels after severe human head injury. J Neurosurg. 1999;91:1–10. doi: 10.3171/jns.1999.91.1.0001. [DOI] [PubMed] [Google Scholar]
- 35.Menzel M, Doppenberg EMR, Zauner A, Soukup J, Reinert MM, Clausen T, et al. Cerebral oxygenation in patients after severe head injury: monitoring and effects of arterial hyper-oxia on cerebral blood flow, metabolism and intracranial pressure. J Neurosurg Anesthesiol. 1999;11:240–251. doi: 10.1097/00008506-199910000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Nighoghossian N, Trouillas P. Hyperbaric oxygen in the treatment of acute ischemic stroke: an unsettled issue. J Neurol Sci. 1997;150:27–31. doi: 10.1016/s0022-510x(97)05398-7. [DOI] [PubMed] [Google Scholar]
- 37.Nighoghossian N, Trouillas P, Adeleine P, Salord F. Hyperbar-ic oxygen in the treatment of acute ischemic stroke. A double-blind pilot study. Stroke. 1995;26:1369–1372. doi: 10.1161/01.str.26.8.1369. [DOI] [PubMed] [Google Scholar]
- 38.Neubauer RA, End E. Hyperbaric oxygenation as an adjunct therapy in strokes due to thrombosis. A review of 122 patients. Stroke. 1980;11:297–300. doi: 10.1161/01.str.11.3.297. [DOI] [PubMed] [Google Scholar]
- 39.Nortje J, Coles JP, Timofeev I, Fryer TD, Aigbirhio FI, Smielewski P, et al. Effect of hyperoxia on regional oxygenation and metabolism after severe traumatic brain injury: preliminary findings. Crit Care Med. 2008;36:273–281. doi: 10.1097/01.CCM.0000292014.60835.15. [DOI] [PubMed] [Google Scholar]
- 40.Okonkwo DO, Wagner J, Melon DE, Alden T, Stone JR, Helm GA, et al. Trans-sodium crocetinate increases oxygen delivery to brain parenchyma in rats on oxygen supplementation. Neu-rosci Lett. 2003;352:97–100. doi: 10.1016/j.neulet.2003.08.044. [DOI] [PubMed] [Google Scholar]
- 41.Ostrowski RP, Colohan AR, Zhang JH. Mechanisms of hy-perbaric oxygen-induced neuroprotection in a rat model of subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2005;25:554–571. doi: 10.1038/sj.jcbfm.9600048. [DOI] [PubMed] [Google Scholar]
- 42.Qin Z, Karabiyikoglu M, Hua Y, Silbergleit R, He Y, Keep RF, et al. Hyperbaric oxygen-induced attenuation of hemorrhagic transformation after experimental focal transient cerebral ischemia. Stroke. 2007;38:1362–1367. doi: 10.1161/01.STR.0000259660.62865.eb. [DOI] [PubMed] [Google Scholar]
- 43.Reinert M, Barth A, Rothen HU, Schaller B, Takala J, Seiler RW. Effects of cerebral perfusion pressure and increased fraction of inspired oxygen on brain tissue oxygen, lactate and glucose in patients with severe head injury. Acta Neurochir (Wien) 2003;145:341–350. doi: 10.1007/s00701-003-0027-0. [DOI] [PubMed] [Google Scholar]
- 44.Rockswold SB, Rockswold GL, Vargo JM, Erickson CA, Sut-ton RL, Bergman TA, et al. Effects of hyperbaric oxygenation therapy on cerebral metabolism and intracranial pressure in severely brain injured patients. J Neurosurg. 2001;94:403–411. doi: 10.3171/jns.2001.94.3.0403. [DOI] [PubMed] [Google Scholar]
- 45.Roy JW, Graham MC, Griffin AM, Gainer JL. A novel fluid resuscitation therapy for hemorrhagic shock. Shock. 1998;10:213–217. doi: 10.1097/00024382-199809000-00010. [DOI] [PubMed] [Google Scholar]
- 46.Rusyniak DE, Kirk MA, May JD, Kao LW, Brizendine EJ, Welch JL, et al. Hyperbaric oxygen therapy in acute ischemic stroke: results of the Hyperbaric Oxygen in Acute Ischemic Stroke Trial Pilot Study. Stroke. 2003;34:571–574. doi: 10.1161/01.str.0000050644.48393.d0. [DOI] [PubMed] [Google Scholar]
- 47.Schäbitz WR, Schade H, Heiland S, Kollmar R, Bardutzky J, Henninger N, et al. Neuroprotection by hyperbaric oxygenation after experimental focal cerebral ischemia monitored by MRI. Stroke. 2004;35:1175–1179. doi: 10.1161/01.STR.0000125868.86298.8e. [DOI] [PubMed] [Google Scholar]
- 48.Seyde WC, McKernan DJ, Laudeman T, Gainer JL, Long-necker DE. Carotenoid compound crocetin improves cerebral oxygenation in hemorrhaged rats. J Cereb Blood Flow Me-tab. 1986;6:703–707. doi: 10.1038/jcbfm.1986.126. [DOI] [PubMed] [Google Scholar]
- 49.Shin HK, Dunn AK, Jones PB, Boas DA, Lo EH, Moskowitz MA, et al. Normobaric hyperoxia improves cerebral blood flow and oxygenation, and inhibits peri-infarct depolarizations in experimental focal ischaemia. Brain. 2007;130:1631–1642. doi: 10.1093/brain/awm071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singhal AB. A review of oxygen therapy in ischemic stroke. Neurol Res. 2007;29:173–183. doi: 10.1179/016164107X181815. [DOI] [PubMed] [Google Scholar]
- 51.Singhal AB, Benner T, Roccatagliata L, Koroshetz WJ, Schaefer PW, Lo EH, et al. A pilot study of normobaric oxygen therapy in acute ischemic stroke. Stroke. 2005;36:797–802. doi: 10.1161/01.STR.0000158914.66827.2e. [DOI] [PubMed] [Google Scholar]
- 52.Singhal AB, Dijkhuizen RM, Rosen BR, Lo EH. Normobaric hyperoxia reduces MRI diffusion abnormalities and infarct size in experimental stroke. Neurology. 2002;58:945–952. doi: 10.1212/wnl.58.6.945. [DOI] [PubMed] [Google Scholar]
- 53.Singhal AB, Wang X, Sumii T, Mori T, Lo EH. Effects of nor-mobaric hyperoxia in a rat model of focal cerebral ischemia-reperfusion. J Cereb Blood Flow Metab. 2002;22:861–868. doi: 10.1097/00004647-200207000-00011. [DOI] [PubMed] [Google Scholar]
- 54.Solenski N, Kwan A, Yanamoto H, Bennett J, Kassell NF, Lee K. Differential elevation of hydroxyl radical in core and penumbra regions during focal reversible cerebral ischemia. Stroke. 1997;28:2545–2551. doi: 10.1161/01.str.28.12.2545. [DOI] [PubMed] [Google Scholar]
- 55.Stennett AK, Dempsey GL, Gainer JL. Trans-sodium croceti-nate and diffusion enhancement. J Phys Chem B. 2006;110:18078–18080. doi: 10.1021/jp064308+. [DOI] [PubMed] [Google Scholar]
- 56.Stennett AK, Murray RJ, Roy JW, Gainer JL. Trans-sodium crocetinate and hemorrhagic shock. Shock. 2007;28:339–344. doi: 10.1097/shk.0b013e3180487b2d. [DOI] [PubMed] [Google Scholar]
- 57.Sunami K, Takeda Y, Hashimoto M, Hirakawa M. Hyperbaric oxygen reduces infarct volume in rats by increasing oxygen supply to the ischemic periphery. Crit Care Med. 2000;28:2831–2836. doi: 10.1097/00003246-200008000-00025. [DOI] [PubMed] [Google Scholar]
- 58.Tisdall MM, Tachtsidis I, Leung TS, Elwell CE, Smith M. Increase in cerebral aerobic metabolism by normobaric hyperoxia after traumatic brain injury. J Neurosurg. 2008;109:424–432. doi: 10.3171/JNS/2008/109/9/0424. [DOI] [PubMed] [Google Scholar]
- 59.Tolias CM, Kumaria A, Bullock MR. Hyperoxia and traumatic brain injury. J Neurosurg. 2009;110:607–609. doi: 10.3171/JNS.2009.110.3.0607a. [DOI] [PubMed] [Google Scholar]
- 60.Tolias CM, Reinert M, Seiler R, Gilman C, Scharf A, Bullock MR. Normobaric hyperoxia—induced improvement in cerebral metabolism and reduction in intracranial pressure in patients with severe head injury: a prospective historical cohort-matched study. J Neurosurg. 2004;101:435–444. doi: 10.3171/jns.2004.101.3.0435. [DOI] [PubMed] [Google Scholar]
- 61.Veltkamp R, Siebing DA, Sun L, Heiland S, Bieber K, Marti HH, et al. Hyperbaric oxygen reduces blood-brain barrier damage and edema after transient focal cerebral ischemia. Stroke. 2005;36:1679–1683. doi: 10.1161/01.STR.0000173408.94728.79. [DOI] [PubMed] [Google Scholar]
- 62.Veltkamp R, Sun L, Herrmann O, Wolferts G, Hagmann S, Sie-bing DA, et al. Oxygen therapy in permanent brain ischemia: potential and limitations. Brain Res. 2006;1107:185–191. doi: 10.1016/j.brainres.2006.05.108. [DOI] [PubMed] [Google Scholar]
- 63.Veltkamp R, Warner DS, Domoki F, Brinkhous AD, Toole JF, Busija DW. Hyperbaric oxygen decreases infarct size and behavioral deficit after transient focal cerebral ischemia in rats. Brain Res. 2000;853:68–73. doi: 10.1016/s0006-8993(99)02250-7. [DOI] [PubMed] [Google Scholar]
- 64.Weinstein PR, Anderson GG, Telles DA. Results of hyperbaric oxygen therapy during temporary middle cerebral artery occlusion in unanesthetized cats. Neurosurgy. 1987;20:518–524. doi: 10.1227/00006123-198704000-00002. [DOI] [PubMed] [Google Scholar]
- 65.Yamaguchi K, Nguyen-Phu D, Scheid P, Piiper J. Kinetics of O2 uptake and release by human erythrocytes studied by a stopped-flow technique. J Appl Physiol. 1985;58:1215–1224. doi: 10.1152/jappl.1985.58.4.1215. [DOI] [PubMed] [Google Scholar]
- 66.Yin D, Zhang JH. Delayed and multiple hyperbaric oxygen treatments expand therapeutic window in rat focal cerebral ischemic model. Neurocrit Care. 2005;2:206–211. doi: 10.1385/NCC:2:2:206. [DOI] [PubMed] [Google Scholar]
- 67.Yin W, Badr AE, Mychaskiw G, Zhang JH. Down regulation of COX-2 is involved in hyperbaric oxygen treatment in a rat transient focal cerebral ischemia model. Brain Res. 2002;926:165–171. doi: 10.1016/s0006-8993(01)03304-2. [DOI] [PubMed] [Google Scholar]
- 68.Zhang JH, Lo T, Mychaskiw G, Colohan A. Mechanisms of hyperbaric oxygen and neuroprotection in stroke. Pathophys-iology. 2005;12:63–77. doi: 10.1016/j.pathophys.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 69.Zhang JH, Singhal AB, Toole JF. Oxygen therapy in ischemic stroke. Stroke. 2003;34:e152–e155. doi: 10.1161/01.STR.0000087098.30644.E1. [DOI] [PubMed] [Google Scholar]