

The idiopathic inflammatory myopathies (IIM) are a group of autoimmune disorders, including polymyositis (PM), dermatomyositis (DM), inclusion body myositis, and myositis associated with malignancy and other connective tissue diseases. Both DM and PM can have associated interstitial lung disease (ILD).1,2
A number of autoantibodies, called myositis-specific autoantibodies, have been described. These include antibodies to aminoacyl-tRNA synthetases, to signal recognition particle, and to the nuclear helicase Mi-2. The most common of these is the anti Jo-1 antibody directed against the antihistidyl–tRNA synthetase. It is detectable in approximately 15% to 30% of myositis patients overall, and is more common in PM.3 The presence of these antibodies has been associated with a clinical subset characterized by myositis, ILD, arthritis, fever, Raynaud phenomenon and mechanic’s hands, referred to as the antisynthetase syndrome.2
There is a well-established association of antisynthetase antibodies and the presence of ILD. Patients may present first with ILD and then later develop myositis. One series reported 10 patients with antisynthetase antibodies and ILD with no clinical evidence of myositis. Of these patients, 2 had anti Jo-1 antibodies. In patients with the antisynthetase syndrome, the lung involvement usually determines the prognosis of the disease.3 In another series, 3 patients with Jo-1 antibodies developed fatal acute respiratory distress syndrome.4 While not all patients develop rapidly progressive fatal lung disease, the presence of antisynthetase antibodies has been associated with a poor prognostic outcome.2– 4
Despite the limited number of randomized controlled studies, the mainstay of therapy for PM and DM is corticosteroids plus either methotrexate or azathioprine.5 Other agents such as cyclosporine, tacrolimus, cyclophosphamide, intravenous immunoglobins, and rituximab have been used with some success.5– 8 Frequently, weakness improves more than pulmonary symptoms following treatment.2,9
The clinical characteristics of African American (AA) patients with anti-Jo-1 antibody ILD and/or myositis have not been well described in the literature. We describe the clinical characteristics of our patients with anti-Jo-1 antibody disease, more than half of whom are AA reflecting the demographics of our medical center.
PATIENTS AND METHODS
We identified 15 patients with Jo-1 positive antibodies who were seen in our rheumatology outpatient clinic between 1991 and 2007. A retrospective chart review was performed to determine demographic information, clinical characteristics, treatment, and outcome. The protocol was approved by our institutional review board.
Available pulmonary function tests (PFT) were reviewed at disease presentation and follow-up. Improvement in forced vital capacity (FVC) was defined as a 10% or greater increase above the baseline value. Deterioration was defined as a 10% or greater decrease in the FVC below the baseline value. Stability was defined as any change less than 10% of the baseline.
Available chest CT scans were reviewed for nonspecific interstitial pneumonia, usual interstitial pneumonia, and cryptogenic organizing pneumonia pattern. The chest CT scans were also reviewed for fibrosis, which was defined as the presence of traction bronchiectasis or honeycombing. Categorical data were compared using Fisher exact test and quantitative variables were analyzed using nonparametric Mann-Whitney U test.
RESULTS/DISCUSSION
Fifteen patients with Jo-1 positive ILD and/or myositis were included in this case series. The main clinical characteristics of all the patients are summarized in Table 1 (see Table, Supplemental Digital Content 1, http://links.lww.com/RHU/A2) (Fig. 1). Follow-up ranged from 5 months to 13 years. One patient had an associated thymoma diagnosed 2 months before the diagnosis of PM. Another patient had breast cancer diagnosed 18 years before the diagnosis of ILD. The remainder of the patients had no evidence of malignancy.
FIGURE 1.
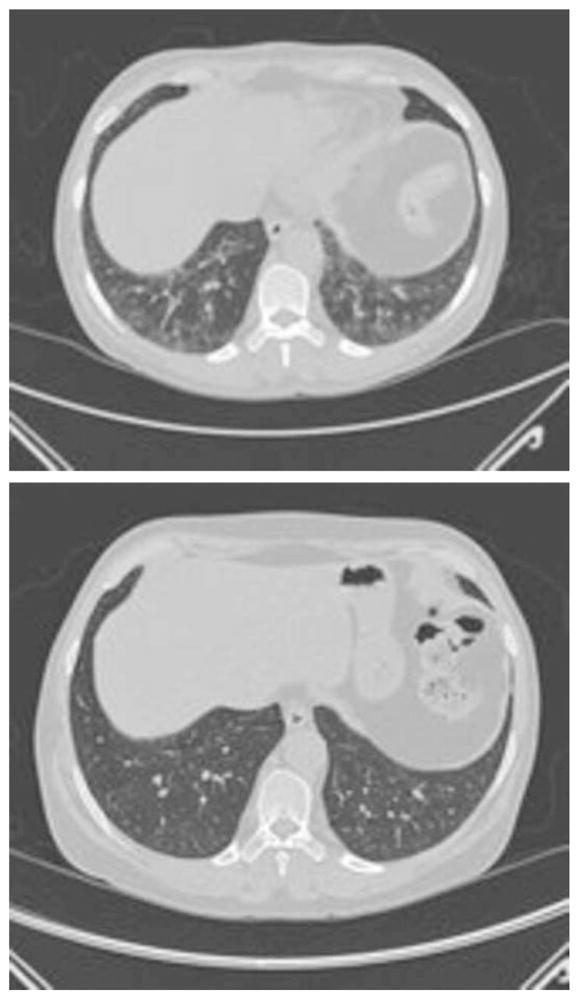
Chest CT scans in patient (see online) 15 before and after treatment with prednisone and azathioprine.
The presence of anti-Jo-1 antibodies is known to be associated with increased rates of ILD in PM and DM patients; however, their prognostic role is uncertain.10,11 We noted from our series that 14 of 15 patients had ILD as determined by PFTs and chest CTs, 10 of whom had ILD at disease onset. We found that the AA patients who presented with pulmonary involvement had lower initial mean FVCs than the white patients (49% predicted in AA, 75% predicted in whites, P = 0.17). This did not appear to be dependent on the time to obtaining the initial FVC, as this was comparable between the 2 groups.
We noted the lack of fibrosis on the initial CT scan of the chest in the patients who did not have anti-Ro/SSA antibodies; 0/4 SSA negative patients had fibrosis compared with 6/8 SSA positive patients, P = 0.03. Anti-Ro/SSA is a myositis-associated autoantibody known to be coexistent in some antisynthetase-positive myositis patients.12 It has been postulated that in patients with anti-Jo-1 positive antisynthetase syndrome, the presence of anti-Ro/SSA antibodies causes a more severe ILD as measured by high resolution CT scan of the chest.12 Anti-Ro/SSA antibodies have also been associated with lupus pneumonitis.13
All of our patients who presented with weakness had both symptomatic and biochemical improvement with treatment. While 7 of 14 patients had new or worse fibrosis on CT, 10 of 14 patients with pulmonary involvement either improved or remained stable as measured by FVC in follow up. One patient died of respiratory failure due to progression of acute interstitial pneumonia. The presence of ILD in PM and DM has been reported to be a poor prognostic indicator. Mortality in various reports has ranged from 30% to 52% over 2 to 5 years2,14; mortality was lower in our patients. Although conclusions about treatment can not be made in our small retrospective case series, we attribute our good outcomes to the early institution of combination immunosuppressive therapy.
Our report is limited by missing data and the small number of patients, which makes it difficult to draw firm conclusions. PFTs were not corrected for racial variation in normal predicted values, but this would have only a small effect. Negative inspiratory force, looking for respiratory muscle weakness, was not performed in all patients.
Our AA patients appeared to present with more severe pulmonary disease. Anti-Ro/SSA antibodies may be a marker of pulmonary fibrosis. The optimal treatment regimen is not yet defined and requires well-designed prospective studies. Given that ILD is a major contributor to the morbidity and mortality of patients with myositis, a multidisciplinary approach to treatment is best.
Supplementary Material
Footnotes
Supplemental digital content is available through direct URL citations in the HTML and PDF versions of this article (www.jclinrheum.com).
References
- 1.Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344–347. doi: 10.1056/NEJM197502132920706. [DOI] [PubMed] [Google Scholar]
- 2.Love LA, Leff RL, Fraser DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991;70:360–374. doi: 10.1097/00005792-199111000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Friedman AW, Targoff IN, Arnett FC. Interstitial lung disease with autoantibodies against aminoacyl-tRNA synthetases in the absence of clinically apparent myositis. Semin Arthritis Rheum. 1996;26:459– 467. doi: 10.1016/s0049-0172(96)80026-6. [DOI] [PubMed] [Google Scholar]
- 4.Clawson K, Oddis CV. Adult respiratory distress syndrome in polymyositis patients with the anti-Jo-1 antibody. Arthritis Rheum. 1995;38:1519–1523. doi: 10.1002/art.1780381020. [DOI] [PubMed] [Google Scholar]
- 5.Oddis CV. Idiopathic inflammatory myopathies: a treatment update. Curr Rheum Rep. 2003;5:431– 436. doi: 10.1007/s11926-003-0053-1. [DOI] [PubMed] [Google Scholar]
- 6.Wilkes MR, Sereika SM, Fertig N, et al. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum. 2005;52:2439–2446. doi: 10.1002/art.21240. [DOI] [PubMed] [Google Scholar]
- 7.Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601– 607. doi: 10.1002/art.20849. [DOI] [PubMed] [Google Scholar]
- 8.Dalakas MC, Illa I, Dambrosia JM, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med. 1993;329:1993–2000. doi: 10.1056/NEJM199312303292704. [DOI] [PubMed] [Google Scholar]
- 9.Spath M, Schroder M, Schlotter-Weigel B, et al. The long-term outcome of anti-Jo-1-positive inflammatory myopathies. J Neurol. 2004;251:859– 864. doi: 10.1007/s00415-004-0449-5. [DOI] [PubMed] [Google Scholar]
- 10.Marie I, Hachulla E, Chérin P, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum. 2002;47:614– 622. doi: 10.1002/art.10794. [DOI] [PubMed] [Google Scholar]
- 11.Fathi M, Vikgren J, Boijsen M, et al. Interstitial lung disease in polymyositis and dermatomyositis: longitudinal evaluation by pulmonary function and radiology. Arthritis Rheum. 2008;59:677– 685. doi: 10.1002/art.23571. [DOI] [PubMed] [Google Scholar]
- 12.La Corte R, Lo Mo Naco A, Locaputo A, et al. In patients with antisynthetase syndrome the occurance of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity. 2006;39:249–253. doi: 10.1080/08916930600623791. [DOI] [PubMed] [Google Scholar]
- 13.Boulware DW, Hedgpeth MT. Lupus pneumonitis and anti-SSA(Ro) antibodies. J Rheumatol. 1989;16:479– 481. [PubMed] [Google Scholar]
- 14.Arsura EL, Greenberg AS. Adverse impact of interstitial pulmonary fibrosis on prognosis in polymyositis and dermatomyositis. Semin Arthritis Rheum. 1988;18:29–37. doi: 10.1016/0049-0172(88)90032-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.