

Abstract
DNA barcodes are increasingly used to provide an estimate of biodiversity for small, cryptic organisms like nematodes. Nucleotide sequences generated by the barcoding process are often grouped, based on similarity, into molecular operational taxonomic units (MOTUs). In order to get a better understanding of the taxonomic resolution of a 3' 592-bp 18S rDNA barcode, we have analyzed 100 MOTUs generated from 214 specimens in the nematode suborder Criconematina. Previous research has demonstrated that the primer set for this barcode reliably amplifies all nematodes in the Phylum Nematoda. Included among the Criconematina specimens were 25 morphologically described species representing 12 genera. Using the most stringent definition of MOTU membership, where a single nucleotide difference is sufficient for the creation of a new MOTU, it was found that an MOTU can represent a subgroup of a species (e.g. Discocriconemella limitanea), a single species (Bakernema inaequale), or a species complex (MOTU 76). A maximum likelihood phylogenetic analysis of the MOTU dataset generated four major clades that were further analyzed by character-based barcode analysis. Fourteen of the 25 morphologically identified species had at least one putative diagnostic nucleotide identified by this character-based approach. These diagnostic nucleotides could be useful in biodiversity assessments when ambiguous results are encountered in database searches that use a distance-based metric for nucleotide sequence comparisons. Information and images regarding specimens examined during this study are available online.
Keywords: Criconematidae, DNA taxonomy, phylogeny, barcode analysis, plant parasitic nematodes, nematode diversity
The estimation of nematode biodiversity exemplifies the challenges in exploring a taxon with a major percentage of its diversity undescribed. In the phylum Nematoda, it is probably an overestimate to suggest that the approximately 27,000 described species represent 5-10% of the existing nematode taxa on the planet (Hugot et al., 2001; Creer et al., 2010; Fonseca et al., 2010). This “well-acknowledged biodiversity identification gap”, the ratio of known species (described) to unknown species (not yet described), has been attributed to the small size of nematodes, their simple morphology, intraspecific variation, and the lack of nematode taxonomists (Creer et al., 2010). One study of nematode diversity in a tropical forest in Cameroon estimated that 6,000 scientist-hours of labor were required to sort and catalogue 431 morphologically identified nematode species, for a survey in which over 90% of the specimens could not be assigned to known species (Bloemers et al., 1997). It is no wonder that molecular approaches that can possibly expedite the process of species discovery and description have been actively pursued (Blaxter, 2004; Markmann and Tautz, 2005; Bhadury et al., 2006; Donn et al., 2008; Porazinska et al., 2009, 2010a, 2010b; Powers et al., 2009; Da Silva et al., 2010; Abebe et al., 2011).
Ironically, this identification gap will likely widen as molecular approaches increase in their application. With the advent of high throughput, next generation sequencing, an entire community of nematodes can be rapidly reduced to a single set of sequences (Creer et al. 2010; Porazinska et al., 2010b). These sequences, if they are derived from a common gene following PCR of pooled DNA from the nematode community, may be considered as a set of MOTUs (molecular operational taxonomic units) (Floyd et al., 2002; Blaxter et al., 2005; Caron et al., 2009; Creer et al. 2010; Jones et al. 2011). An MOTU can be defined as a cluster of sequences that fall within a designated cutoff value of sequence identity, the cutoff value being established by the author (Caron et al., 2009). The cutoff value could require 100% sequence identity, in which case each unique sequence is considered a separate MOTU. The taxonomic significance of a given MOTU depends on a number of factors such as the genetic region under analysis, the rate of evolution of that region, experimental error, and the congruence of gene trees and species trees. Since these factors are seldom completely understood, it is not a trivial question to ask, “What does an MOTU represent?”
In this study we explore the performance of a 3' 592 bp 18S barcode as a tool to generate MOTUs and assess nematode diversity. The term barcode in this case refers to the specific region of the 18S gene amplified by the 18S1.2a/18Sr2b PCR primer set, and the nucleotide sequence between them, but not including the primer sequence itself. The barcode was selected due to its evolutionarily conserved nature, exhibiting a balance between phylogenetic breadth and taxonomic resolution. It has previously been used in a phylum-wide molecular survey of nematode communities within a lowland Costa Rican rain forest (Powers et al., 2009), a metagenetic analysis of artificially constructed nematode communities (Porazinska et al., 2009) and a multi-phyla metagenetic survey replicating the aforementioned Costa Rican rain forest study (Porazinska et al., 2010a). MOTUs derived from 18S have the advantage of comparison to the 18S-based Nematode Tree of Life which in its most current published form includes 1215 taxa (van Megen et al., 2009). We assume that MOTUs that link with morphologically and taxonomically characterized entities are a richer source of systematic information and maximize information content from studies employing MOTUs unlinked from taxonomically characterized entities. Therefore, a second purpose of this study is to enlarge the reference database in order to facilitate future systematic studies.
To address the question of MOTU representation, we apply the 3' 592 bp 18S barcode to an analysis of a single, globally distributed suborder of plant-parasitic nematodes. The suborder Criconematina Siddiqi, 1980 ranges from the humid tropics to arctic and alpine habitats. There are an estimated 750 described species in the suborder (Subbotin et al., 2005). They are found on a wide range of hosts feeding on plants as diverse as hardwoods, conifers, bromeliads, grasses and moss. They are believed to have a high level of endemicity and are some of the most abundant soil-dwelling plant parasites in tropical forests (Wouts, 2006). Their high endemicity, poor dispersal capabilities, and apparent lack of specialized survival stages make them a potential subject for biogeographic analysis (Bernard and Schmitt, 2005; Wouts, 2006). They are potential indicators for soil disturbance (Bernard, 1992). While a few species appear to be adapted to disturbances associated with agricultural production, the vast majority are confined to native habitats with a relatively stable soil structure (Hoffman and Norton, 1976; Bernard, 1982; Peneva et al., 2000) and tend to disappear when these habitats are disrupted. It is widely believed that Criconematina constitutes a monophyletic group, although the relationships and composition of sub-groups are generally considered to be “taxonomically opaque” and in a perpetual state of taxonomic turmoil (Siddiqi, 2000; Subbotin et al., 2005, 2006; Bert et al., 2008; Hunt, 2008). Monophyly of the suborder has been supported by both molecular and morphological analysis (Holterman et al., 2006; Subbotin et al., 2006; Bert et al., 2008; van Megen et al., 2009).
The nematodes in this study have been obtained through a series of collections spanning the years 1999 to 2010 (Table 1). Nematodes were individually isolated from soil samples, many digitally photographed (most often while alive), measured, processed for PCR, amplified and sequenced for the 18S barcode. Collection localities included non-cultivated as well as cultivated soils, with approximately one-third of the specimens recovered from Costa Rica and the remaining specimens from the United States, Mexico, and Europe. An additional 21 sequences from GenBank were added to the analysis.
Table 1.
MOTUs and individual specimens used in this study.

The specific objective of this study is to apply a phylogenetic and a character-based barcode analysis to a 100-MOTU dataset of Criconematina specimens. This dataset includes 25 a priori identified species, recognized by traditional morphological analysis. The dataset also includes specimens that could not be identified a priori to species with confidence. The unknown specimens may represent new species or specimens that do not provide sufficient information for an accurate identification. We attempt to determine if there exists any nucleotide sequence support for the morphologically identified taxa. This analysis should provide insight into the taxonomic resolution of the 18S barcode, which in turn should enhance studies of nematode biodiversity.
Materials and Methods
Nematode collections: The earliest collected specimens in this study, those collected between 1999 and 2005, tend to have less associated morphological data, as methods were being developed to obtain both molecular and morphological information from an individual specimen. Two biodiversity surveys contributed a significant number of specimens to this study; a 1999 nematode survey of Konza Prairie, a designated Long Term Ecological Reserve, and a 2005 survey of La Selva Biological Research Station operated by the Organization of Tropical Studies (NSF DEB 0640807) (NSF DEB 9806439). The geographic coverage in this study includes specimens from Atlantic and Pacific coasts, and Central Valley of Costa Rica. North American specimens were collected from 21 U.S. states and a single state in Mexico. Twenty one GenBank accessions were added to the analysis, all of which represent European collections. Four sampling sites represent type localities from which the targeted species was obtained.
Nematode morphological identification: Nematodes were observed by differential interference microscopy on a Leica DMLB microscope, images recorded by a Leica DC300 video camera, and measurements obtained using an eyepiece micrometer at 1000x magnification. Observations were made on living nematodes whenever possible. In some cases such as Bakernema specimens, the elaborate cuticular ornamentation is more visible in living than dead or fixed specimens. After nematode measurement, the slide was carefully dismantled by removing the cover slip, the nematode recovered using a fine insect pin pick, added to an 18ul drop of sterile water, and then smashed on a cover slip with a clear, sterile micropipette tip. Nematode residue was stored in PCR reaction tubes in a -20oC freezer until PCR amplification.
DNA amplicon characteristics, terminology and assumptions: The 18S1.2a/18Sr2b primer set typically amplifies a 635-bp region of the 18S ribosomal gene, with the 3'-most primer located 180-bp from the first internal transcribed spacer (ITS1). The primer set, 18S1.2a: 5'-CGATCAGATACCGCCCTAG-3' (forward) and 18Sr2b: 5'-TACAAAGGGCAGGGACGTAAT-3' (reverse) will amplify nematodes throughout the phylum and will amplify some non-nematode taxa. The term barcode in this study applies to that specific region of the 18S gene bounded by those primers. This barcode is distinct and does not overlap with the 5'-18S barcode region analyzed by Floyd et al., (2002). In this study, a single nucleotide difference is sufficient to designate a new MOTU. Both strands of the amplified product were sequenced in these analyses by direct sequencing at the University of Arkansas Medical Center Sequencing Facility.
We assume that each individual specimen is represented by a single 3'-18S barcode sequence. We know this is not the case among all nematodes species, as in the polyploid species of Meloidogyne and other select species (Abad et al., 2008; Lunt, 2008). Several specimens in this study produced nucleotide sequences that indicated heterogeneity within the barcode of that individual. Those specimens are noted in Tables 2 and 3. All sequences used in this study have been added to GenBank (Table 1).
Table 2.
Clade A polymorphic and diagnostic nucleotide positions with diagnostic characters. Diagnostic nucleotides are those shared by all individuals of a species and not found in any other species. The diagnostic nucleotide characters are shaded with a bold outline. Synapomorphic characters are shaded without a bold outline. Numbers after taxon labels refer to the number of specimens examined. GB refers to sequences obtained from GenBank. Numbering of the nucleotide position starts with 1 which is the first nucleotide following primer 18S1.2a.

Table 3.
Clade B polymorphic and diagnostic nucleotide positions with diagnostic characters. Diagnostic nucleotides are those shared by all individuals of a species and not found in any other species. The diagnostic nucleotide characters are shaded with a bold outline. Numbers after taxon labels refer to the number of specimens examined. GB refers to sequences obtained from GenBank. Numbering of the nucleotide position starts with 1 which is the first nucleotide following primer 18S1.2a.

Each specimen is supplied with a voucher identification number or Nematode ID (NID) number. These numbers have been applied sequentially and chronologically. In some cases NID numbers were applied retroactively. When multiple amplifications are made from a single specimen, a unique amplification number is associated with the NID number. MOTU designations were applied following the pooling of redundant sequences by the Redundant Taxa tool in Maclade.
DNA preparation, sequence alignment, phylogenetic and character-based analysis: DNA was amplified and sequenced as previously described (Powers et al., 2010). 18S sequences were edited and assembled using CodonCode Aligner (CodonCode Corp, Dedham, Massachusetts), DNA aligned by MUSCLE 3.7 (Edgar, 2004) and maximum likelihood analysis generated by PHYML 3.0 using approximate likelihood-ratio tests for the estimation of branch support (Anisimova et al., 2006). The FASTA file for the MOTU dataset is available in Dryad (DOI-pending).
Character-based barcode analysis of nucleotide sequences is an alternative approach to species diagnosis using DNA barcodes (DeSalle et al., 2005; Sarkar et al., 2008). It differs from the more traditional method of barcode analysis in that it is not a distance-based approach, but rather treats the nucleotide sites in a DNA sequence as characters and the different character states, A,T,C,G, are referred to as character attributes (CA) (Sarkar et al., 2002) or nucleotide diagnostics (ND) (Wong et al., 2009). A nucleotide diagnostic can be designated simple and pure when a particular nucleotide is fixed for a particular species, and found in all members of that species and no others. Compound nucleotide diagnostics consist of several nucleotide sites where the combination of nucleotides at those sites is only found in one species. In this study only pure, simple nucleotide diagnostics are analyzed. In large datasets, the first step in character-based barcode analysis is the generation of a phylogenetically derived guide tree which is subsequently examined node by node for the presence of diagnostic nucleotides. The computer program CAOS (Characteristic Attributes Organization System) is an automated method for the discovery of nucleotide diagnostics (Sarkar et al., 2008). The 100-MOTU 3'-18S barcode dataset was simple enough to conduct a manual analysis of nucleotide diagnostics using the maximum likelihood tree and its major clades as a guide tree.
Online access: Images and measurements of terminal taxa from the barcode tree are available online (http://nematode.unl.edu/CriconematidProject_Trees.htm). Individual specimens are listed by their NID numbers in Table 1.
Results
Barcode characteristics: This dataset is comprised of 100 18S barcode MOTUs derived from 214 sequences from nematodes in the suborder Criconematina (Table 1). The ClustalW alignment is 602 nucleotides in length which includes 10 hypothesized sites of nucleotide insertion or deletion (indels). There are 470 (78%) invariant and 132 polymorphic nucleotide sites in the dataset. Among the polymorphic nucleotide sites, 56 (42%) are singletons, positions where a single MOTU has a nucleotide not shared by any others in the dataset.
Barcode species analysis: The dataset includes 25 nominal species identified by the authors through microscopic examinations of morphological characteristics. A maximum likelihood tree for the 100 MOTUs is presented in Figure 1. Four clusters with moderate support values (0.80-0.93) have been identified and were labeled A-D for character-based barcode analysis.
Fig. 1.

Maximum likelihood tree of Criconematina 18S 3’ MOTUs. Shaded clades A-D were analyzed separately by character-based barcode analysis. Species binomials followed by GB were sequences added to the analyses from GenBank. Images and measurements of terminal taxa can be seen at http://nematode.unl.edu/CriconematidProject_Trees.htm Approximate likelihood-ratio test support values above 0.80 identify nodes of relatively strong support.
Within clade A, there are nine morphologically identified nominal species not considering GenBank entries (Fig. 1, Table 2). Included in this clade are species that morphologically fall within the genera Ogma, Xenocriconemella, Criconema, and Hemicriconemoides. Five of the Ogma species possessed morphological characters that permitted assignment to known species. However, neither phylogenetic analysis nor character-based barcode analysis recognized all Ogma MOTUs as collectively comprising a natural group exclusive of the other genera in the clade. Ogma decalineatum and O. octangulare shared a T at nucleotide 67 to the exclusion of all other MOTUs in clade A (Table 2). Another nucleotide character (C) at position 391 provides evidence for relatedness of these two species to O. seymouri. The O. menzeli MOTU from Tennessee (M72) differs by two nucleotides from the European O. menzeli in GenBank (M73). M76 is a broadly distributed MOTU, one of only two MOTUs found in both Costa Rica and the United States. Additionally, it shares 100% identity with GenBank accession EU669918, an O. cobbi reported from Europe. Morphologically, the adult females that represent M76 include a range of phenotypes, particularly in the arrangement of scales on the adult female cuticle.
Xenocriconemella macrodora is represented by 12 specimens and three MOTUs (M98, M99, M100) collected from five U.S. states. There are four diagnostic nucleotide sites, including two insertions, which are observed in every specimen of this species. These are found at nucleotide positions 349, 352, 363, and 364. Hemicriconemoides wessoni was collected at two sites in Florida, one site within 60 miles of the type locality. Three MOTUs were observed for this species, each diagnosable by nucleotides T and G at positions 362 and 365 respectively. Criconema permistum and C. sphagni were represented by one and three specimens respectively, each containing a single, unique fixed nucleotide. Other Criconema species in clade A are not united by shared derived characters, reflecting a lack of phylogenetic support for the genus.
Clade B, with the exception of a single MOTU (M4), is exclusively represented by Discocriconemella limitanea from Costa Rica (Table 3). The clade is well-supported phylogenetically. D. limitanea is represented by 12 specimens and 11 MOTUs which break into two discrete subgroups. There are six nucleotide sites that separate the two subgroups. Morphologically, however, there are no characters that appear to discriminate between the subgroups, and both subgroups are found in Las Cruces and La Selva Biological Research Stations, geographically distinct rainforest habitats of Costa Rica. MOTU M4 was recovered from cultivated passionfruit in Costa Rica and conforms morphologically to Mesocriconema crenatum (Loof, 1964) De Grisse & Loof, 1965.
Clade C includes six nominal species identified by morphology (Table 4). Both phylogenetic analysis and character-based barcode analysis support Mesocriconema rusticum and M. curvatum as diagnosable species within this clade. Nucleotide sites at 472 and 488 diagnose M. rusticum, and an additional two synapomorphic characters at sites 46 and 503 support a sister group relationship with M. ornatum. Mesocriconema rusticum was represented by 6 specimens and two MOTUs collected from 5 U.S. states. Four nucleotide sites, 31, 34, 56, and 59 diagnose M. curvatum. MOTU M63 was represented by 18 specimens and includes two morphologically identifiable species M. xenoplax and Discocriconemella inarata. A previous paper has addressed the more detailed taxonomy of these two species (Powers et al., 2010). There are no diagnosable characters in this 18S barcode for discrimination between M. xenoplax and D. inarata. Three additional MOTUs, M65, M66 and M67 from GenBank have been identified as M. xenoplax from Europe. Mesocriconema discus (M57) was collected at its type locality in South Dakota, however there were no discrete nucleotide characters that could be considered as diagnosable nucleotide sites.
Table 4.
Clade C polymorphic and diagnostic nucleotide positions with diagnostic characters. Diagnostic nucleotides are those shared by all individuals of a species and not found in any other species. The diagnostic nucleotide characters are shaded with a bold outline. Synapomorphic characters are shaded without a bold outline. Numbers after taxon labels refer to the number of specimens examined. GB refers to sequences obtained from GenBank. Numbering of the nucleotide position starts with 1 which is the first nucleotide following primer 18S1.2a.

Clade D was largely comprised of Hemicycliophora species, the two related sheath genera Hemicaloosia and Loofia, Lobocriconema, and Criconemoides species (Table 5). Criconemoides annulatus (M16), represented by two specimens from the Rocky Mountains in Colorado, possessed three diagnostic nucleotide sites. Lobocriconema thornei (M51) and a closely related Lobocriconema species (M50) had four synapomorphic sites, and each was diagnosable by a single autapomorphic site. Among the sheath genera, two synapomorphic nucleotide sites at 43 and 47 united all specimens. Hemicycliophora gracilis, represented by a single MOTU collected in Colorado and Nebraska, possessed five autapomorphic diagnostic sites. Hemicycliophora typica and the two species from GenBank in this dataset did not possess diagnosable nucleotides in the 18S barcode.
Table 5.
Clade D polymorphic and diagnostic nucleotide positions with diagnostic characters. Diagnostic nucleotides are those shared by all individuals of a species and not found in any other species. The diagnostic nucleotide characters are shaded with a bold outline. Synapomorphic characters are shaded without a bold outline. Numbers after taxon labels refer to the number of specimens examined. GB refers to sequences obtained from GenBank. Numbering of the nucleotide position starts with 1 which is the first nucleotide following primer 18S1.2a.

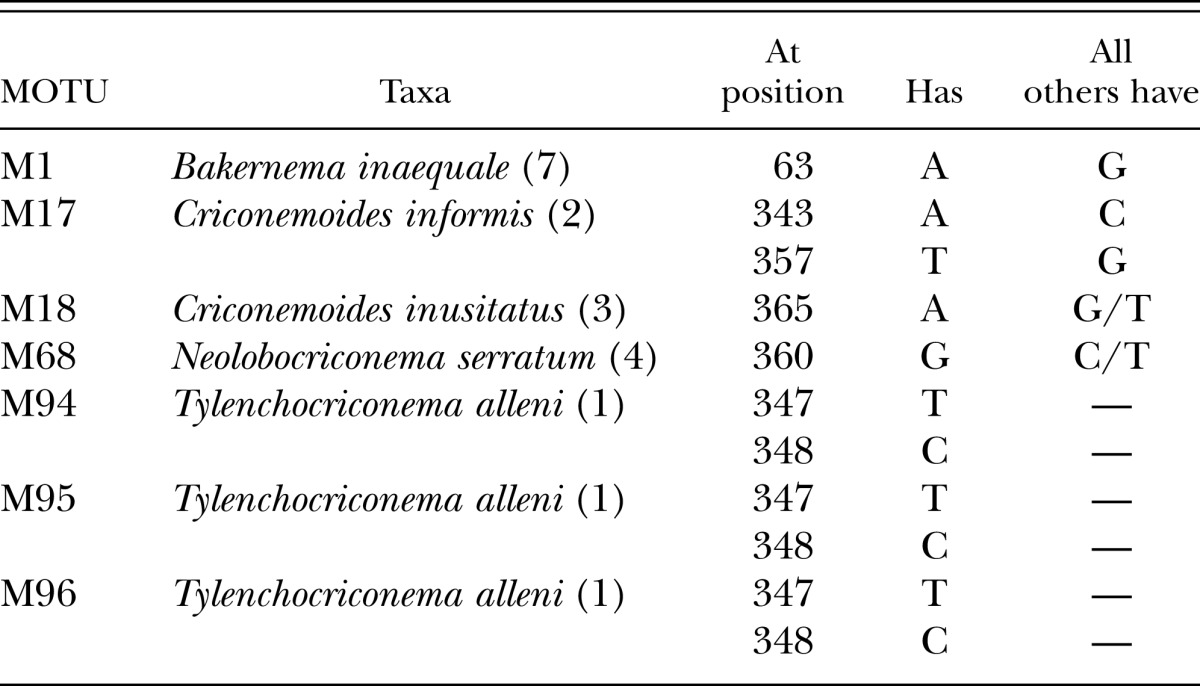
Five notable, diagnosable species in the 100-MOTU dataset did not fall within clades A-D (Table 6). Bakernema inaequale is a species endemic to North America and immediately recognizable by its irregularly arranged, membranous cuticular scales. Seven specimens from Tennessee and Connecticut shared a single MOTU (M1) and were diagnosable in the full dataset by an A at nucleotide position 63. Criconemoides informis (M17) had two diagnostic nucleotides: an A and T at positions 343 and 357, respectively. Criconemoides inusitatus (M18), collected from the type locality in Ames, IA and from Delaware, had a single diagnostic nucleotide site at position 365. A species morphologically conforming to Neolobocriconema serratum (M68) collected from Missouri and Nebraska, had a single diagnostic site at position 360. Three MOTUs (M94, M95, M96) represented the unusual criconematid nematode Tylenchocriconema alleni, a species known solely from epiphytic bromeliads in the new world tropics (Raski and Siddiqui, 1975). Two nucleotide sites at 347 and 348 were diagnostic for the three MOTUs.
Table 6.
Diagnostic nucleotide positions for species not included in clades A-D. These species compared against the entire Criconematina data set. Numbers after taxon labels refer to number of specimens examined. Numbering of the nucleotide position starts with 1 which is the first nucleotide following primer 18S1.2a.
Discussion
The small number of phylogenetically informative nucleotide sites (76) and the relatively few well-supported clades observed in the maximum likelihood tree indicate that limited phylogenetic inference can be derived from this 3' region of 18S. None of the well-supported clades could be interpreted as support for the existing morphologically-based classification of Criconematina sensu Siddiqi (2000). Conversely, there is not strong support for alternative groupings of MOTUs. Simply there are not enough phylogenetically informative sites in this 18S barcode to construct a robust phylogeny. Subbotin et al., (2005, 2006) arrived at a similar conclusion with analysis of the D2/D3 region of 28S rDNA. Those studies included 23 nominal taxa from 11 genera. The 38 samples analyzed exhibited a geographic coverage that included two specimens from North America, 12 from Venezuela, and the remaining specimens from Europe. According to the authors, “none of the phylogenetic analyses of the D2-D3 dataset allowed resolution of the relationships between main lineages.”
Lack of phylogenetic resolution does not mean that the 3'-18S barcode does not have value as a measure of biodiversity or as an aid in diagnostics. A major advantage of the primer set is that PCR amplification is consistent and reliable across the entire nematode phylum. That consistency allows for an unbiased comparison of nematode community composition. Within the suborder Criconematina, barcode discrimination is at multiple taxonomic levels. In some cases a single MOTU clearly identified a complex of species. MOTU 76, for example, corresponded to a group of Ogma species that have scales arranged singularly in longitudinal rows along the length of the body, or arranged in rows consisting of clusters of 4-6 scales, or with scales densely packed on the annules forming a continuous elongated fringe. Similarly MOTU 63 consists of geographically wide-spread North American isolates that conform to Mesocriconema xenoplax and Discocriconemella inarata, a grassland species that appears to have secondarily lost the submedian lobes (Powers et al., 2010). In other cases, multiple MOTUs seem to correspond to a morphologically conserved species complex. Discocriconemella limitanea is comprised of multiple MOTUs with no indication of corresponding morphological change. The nucleotide variability within the barcode identifies subgroups that may suggest the existence of cryptic species. Here the barcode analysis has provided initial evidence in the species discovery process and should be followed by a complete taxonomic analysis to resolve the taxonomic status of the subgroups.
The absence of a direct correspondence between MOTUs as defined in this study (1 bp cutoff) and morphologically identified species suggest that the MOTUs generated by the 3'-18S barcode should not be uncritically considered as proxies for species. The relationship between MOTUs and species can be evaluated by character-based DNA barcode analysis, which is a method to discover diagnostic characters in species where the delimitation step has already been established (DeSalle et al., 2005; DeSalle 2006; Kelly et al., 2007; Rach et al., 2008; Wong et al., 2009; Naro-Maciel et al., 2010). As a character-based approach it is compatible with traditional morphological identification systems in its recognition of diagnostic characteristics based on the assumption that members of established taxonomic groups share attributes that are absent from comparable groups (Sarkar et al., 2002; Rach et al. 2008). Bakernema inaequale, for example, is diagnosable by the presence of irregularly spaced membranous scales on the cuticle and an A at nucleotide position 63 in the 3'-18S barcode. Xenocriconemella macrodora is diagnosable by an approximately 100 um flexible stylet, an A and G substitution at positions 349 and 352 respectively, plus a TC insertion at position 363-364. In the 100-MOTU Criconematina dataset, 14/25 a priori identified species had at least one diagnostic character. Moreover, in several cases, while no diagnosable nucleotide characters were recognized at the species level, a synapomorphic character was present that indicated grouping at a higher taxonomic level (e.g. Ogma decalineatum, O. octangulare, O. seymouri). Given the evolutionarily conserved nature of the 3' portion of the 18S gene, it is surprising that over 50% of the known species would possess putative diagnostic nucleotides. Alternative explanations for the apparent diagnostic signal could be attributed to sequencing error, insufficient sampling of species and populations, or misidentification of the nominal species. The validation of these results will require increased sampling of species throughout their known range. These caveats notwithstanding, from a biodiversity and biogeographic perspective the application of this barcode to a comparison of nematode communities could hasten the effort to describe the pattern of nematode diversity as it currently exists at the landscape scale. Also the characterization of new MOTUs will identify gaps in the taxonomic knowledge and lead to species discovery. Furthermore, it is important to emphasize that barcode approaches, whether they target individual specimens or an entire community of specimens, are still dependent on reference databases to convey meaningful taxonomic information, with the recognition that sequences alone, apart from their biological context, are limited in their systematic value (Hajibabaei et al., 2007; Stevens et al., 2011).
Literature Cited
- Abad P, Gouzy J, Aury J, Castagnone-Sereno P, Danchin EGJ, Deleury E, Perfus-Barbeoch L, Anthouard V, Artiguenave F, Blok VC, Caillaud M, Coutinho PM, Dasilva C, De Luca F, Deau F, Esquibet M, Flutre T, Goldstone JV, Hamamouch N, Hewezi T, Jaillon O, Jubin C, Leonetti P, Magliano M, Maier TR, Markov GV, McVeigh P, Pesole G, Poulain J, Robinson-Rechavi M, Sallet E, Segurens B, Steinbach D, Tytgat T, Ugarte E, van Ghelder C, Veronico P, Baum TJ, Blaxter M, Bleve-Zacheo T, Davis EL, Ewbank JJ, Favery B, Grenier E, Henrissat B, Jones JT, Laudet V, Maule AG, Quesneville H, Rosso M, Schiex T, Smant G, Weissenbach J, Wincker P. Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nature Biotechnology. 2008;26:909–915. doi: 10.1038/nbt.1482. [DOI] [PubMed] [Google Scholar]
- Abebe E, Mekete T, Thomas WK. A critique of current methods in nematode taxonomy. African Journal of Biotechnology. 2011;10:312–323. [Google Scholar]
- Anisimova M, Gascuel O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Systematic Biology. 2006;55:539–552. doi: 10.1080/10635150600755453. [DOI] [PubMed] [Google Scholar]
- Bernard EC. Soil nematode biodiversity. Biology and Fertility of Soils. 1992;14:99–103. [Google Scholar]
- Bernard EC. Criconematina (Nematoda: Tylenchida) from the Aleutian Islands. Journal of Nematology. 1982;14:323–331. [PMC free article] [PubMed] [Google Scholar]
- Bernard EC, Schmitt DP. Nematode assemblages in native plant communities of Molokai, Hawaii. Journal of Nematology. 2005;37:242–248. [PMC free article] [PubMed] [Google Scholar]
- Bert W, Leliaert F, Vierstraete AR, Vanfleteren JR, Borgonie G. Molecular phylogeny of the Tylenchina and evolution of the female gonoduct (Nematoda: Rhabditida) Molecular Phylogenetics and Evolution. 2008;48:728–744. doi: 10.1016/j.ympev.2008.04.011. [DOI] [PubMed] [Google Scholar]
- Bhadury P, Austen MC, Bilton DT, Lambshead PJ, Rogers AD, Smerdon GR. Development and evaluation of a DNA-barcoding approach for the rapid identification of nematodes. Marine Ecology Progress Series. 2006;320:1–9. [Google Scholar]
- Blaxter ML. The promise of a DNA taxonomy. Philosophical Transactions of the Royal Society of London B Biological Sciences. 2004;359:669–679. doi: 10.1098/rstb.2003.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaxter M, Mann J, Chapman T, Thomas F, Whitton C, Floyd R, Abebe E. Defining operational taxonomic units using DNA barcode data. Philosophical Transactions of the Royal Society of London B Biological Sciences. 2005;360:1935–1943. doi: 10.1098/rstb.2005.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloemers GF, Hodda M, Lambshead PJD, Lawton JH, Wanless FR. The effects of forest disturbance on diversity of tropical soil nematodes. Oecologia (Berlin) 1997;111:575–582. doi: 10.1007/s004420050274. [DOI] [PubMed] [Google Scholar]
- Caron DA, Countway PD, Savai P, Gast RJ, Schnetzer A, Moorthi SD, Dennett MR, Moran DM, Jones AC. Defining DNA-based operational taxonomic units for microbial-eukaryote ecology. Applied and Environmental Microbiology. 2009;75:5797–5808. doi: 10.1128/AEM.00298-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creer S, Fonseca VG, Porazinska DL, Giblin-Davis R, Sung W, Power DM, Packer M, Carvalho GR, Blaxter ML, Lambshead PJD, Thomas WK. Ultrasequencing of the meiofaunal biosphere: Practice, pitfalls and promises. Molecular Ecology. 2010;19:4–20. doi: 10.1111/j.1365-294X.2009.04473.x. [DOI] [PubMed] [Google Scholar]
- Da Silva NRR, Da Silva MC, Genevois VF, Esteves AM, Ley P, Decraemer W, Rieger TT, Correia MT. Marine nematode taxonomy in the age of DNA: The present and future of molecular tools to assess their biodiversity. Nematology. 2010;12:661–672. [Google Scholar]
- DeSalle R. What's in a character? Journal of Biomedical Informatics. 2006;39:6–17. doi: 10.1016/j.jbi.2005.11.002. [DOI] [PubMed] [Google Scholar]
- DeSalle R, Egan MG, Siddall M. The unholy trinity: Taxonomy, species delimitation and DNA barcoding. Philosophical Transactions of the Royal Society of London B Biological Sciences. 2005;360:1905–1916. doi: 10.1098/rstb.2005.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donn S, Griffiths BS, Neilson R, Daniell TJ. DNA extraction from soil nematodes for multi-sample community studies. Applied Soil Ecology. 2008;38:20–26. [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd R, Abebe E, Papert A, Blaxter M. Molecular barcodes for soil nematode identification. Molecular Ecology. 2002;11:839–850. doi: 10.1046/j.1365-294x.2002.01485.x. [DOI] [PubMed] [Google Scholar]
- Fonseca VG, Carvalho GR, Sung W, Johnson HF, Power DM, Neill SP, Packer M, Blaxter ML, Lambshead PJ, Thomas WK, Creer S. Second-generation environmental sequencing unmasks marine metazoan biodiversity. Nature Communications. 2010;1:98. doi: 10.1038/ncomms1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajibabaei M, Singer GAC, Hebert PDN, Hickey DA. DNA barcoding: How it complements taxonomy, molecular phylogenetics and population genetics. Trends in Genetics. 2007;23:167–172. doi: 10.1016/j.tig.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Hoffman JK, Norton DC. Distribution patterns of some Criconematinae in different forest associations. Journal of Nematology. 1976;8:32–35. [PMC free article] [PubMed] [Google Scholar]
- Holterman M, van der Wurff A, van den Elsen S, van Megen H, Bongers T, Holovachov O, Bakker J, Helder J. Phylum-wide analysis of SSU rDNA reveals deep phylogenetic relationships among nematodes and accelerated evolution toward crown clades. Molecular Biology and Evolution. 2006;23:1792–1800. doi: 10.1093/molbev/msl044. [DOI] [PubMed] [Google Scholar]
- Hugot JP, Baujard P, Morand S. Biodiversity in helminths and nematodes as a field of study: An overview. Nematology. 2001;3:199–208. [Google Scholar]
- Hunt DJ. Fauna of New Zealand 55. 2008. Wim M. Wouts. Criconematina (Nematoda: Tylenchida) Manaaki Whenua Press, Landcare Research, Lincoln, New Zealand, 2006 Nematology 10:781. [Google Scholar]
- Jones M, Ghoorah A, Blaxter M. jMOTU and Taxonerator: Turning DNA barcode sequences into annotated operational taxonomic units. PloS ONE. 2011;6(4):e19259. doi: 10.1371/journal.pone.0019259. doi:10.1371/journal.pone.0019259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RP, Sarkar IN, Eernisse DJ, Desalle R. DNA barcoding using chitons (genus Mopalia) Molecular Ecology Notes. 2007;7:177–183. [Google Scholar]
- Loof PAA. Four new species of Criconemoides from The Netherlands.Versl. Plantenz. Ziek. Dierst. 1964;141:160–168. [Google Scholar]
- Lunt DH. Genetic tests of ancient asexuality in root knot nematodes reveal recent hybrid origins. BMC Evolutionary Biology. 2008;8:194–209. doi: 10.1186/1471-2148-8-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markmann M, Tautz D. Reverse taxonomy: An approach towards determining the diversity of meiobenthic organisms based on ribosomal RNA signature sequences. Philosophical Transactions of the Royal Society of London B Biological Sciences. 2005;360:1917–1924. doi: 10.1098/rstb.2005.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naro-Maciel E, Reid B, Fitzsimmons NN, Le M, Desalle R, Amato G. DNA barcodes for globally threatened marine turtles: A registry approach to documenting biodiversity. Molecular Ecology Resources. 2010;10:252–263. doi: 10.1111/j.1755-0998.2009.02747.x. [DOI] [PubMed] [Google Scholar]
- Peneva V, Neilson R, Boag B, Brown DJF. Criconematidae (Nematoda) from oak forests in two nature reserves in Russia. Systematic Parasitology. 2000;46:191–201. doi: 10.1023/a:1006338019502. [DOI] [PubMed] [Google Scholar]
- Porazinska DL, Giblin-Davis R, Esquivel A, Powers TO, Sung W, Thomas WK. Ecometagenetics confirms high tropical rainforest nematode diversity. Molecular Ecology. 2010a;19:5521–5530. doi: 10.1111/j.1365-294X.2010.04891.x. [DOI] [PubMed] [Google Scholar]
- Porazinska DL, Giblin-Davis R, Faller L, Farmerie W, Kanzaki N, Morris K, Powers TO, Tucker AE, Sung W, Thomas WK. Evaluating high-throughput sequencing as a method for metagenomic analysis of nematode diversity. Molecular Ecology Resources. 2009;9:1439–1450. doi: 10.1111/j.1755-0998.2009.02611.x. [DOI] [PubMed] [Google Scholar]
- Porazinska DL, Giblin-Davis R, Sung W, Thomas WK. Linking operational clustered taxonomic units (OCTUs) from parallel ultra sequencing (PUS) to nematode species. Zootaxa. 2010b;2427:55–63. [Google Scholar]
- Powers TO, Harris T, Higgins R, Sutton L, Powers KS. Morphological and molecular characterization of Discocriconemella inarata, an endemic nematode from north american native tallgrass prairies. Journal of Nematology. 2010;42:35–45. [PMC free article] [PubMed] [Google Scholar]
- Powers TO, Neher DA, Mullin P, Esquivel A, Giblin-Davis R, Kanzaki N, Stock SP, Mora MM, Uribe-Lorio L. Tropical nematode diversity: Vertical stratification of nematode communities in a Costa Rican humid lowland rainforest. Molecular Ecology. 2009;18:985–996. doi: 10.1111/j.1365-294X.2008.04075.x. [DOI] [PubMed] [Google Scholar]
- Rach J, DeSalle R, Sarkar IN, Schierwater B, Hadrys H. Character-based DNA barcoding allows discrimination of genera, species and populations in Odonata. Proceedings of the Royal Society Biological Sciences Series B. 2008;275:237–247. doi: 10.1098/rspb.2007.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raski DJ, Siddiqui IA. Tylenchocriconema alleni n.g. n.sp. from Guatemala (Tylenchocriconematidae n.fam.; Tylenchocriconematoidea n. superfam.; Nematoda) Journal of Nematology. 1975;7:247–251. [PMC free article] [PubMed] [Google Scholar]
- Sarkar IN, Thornton JW, Planet PJ, Figurski DH, Schierwater B, DeSalle R. An automated phylogenetic key for classifying homeoboxes. Molecular Phylogenetics and Evolution. 2002;24:388–399. doi: 10.1016/s1055-7903(02)00259-2. [DOI] [PubMed] [Google Scholar]
- Sarkar IN, Planet PJ, Desalle R. CAOS software for use in character-based DNA barcoding. Molecular Ecology Resources. 2008;8:1256–1259. doi: 10.1111/j.1755-0998.2008.02235.x. [DOI] [PubMed] [Google Scholar]
- Siddiqi MR. Tylenchida parasites of plants and insects. 2000. Second Edition CABI, Wallingford, UK. [Google Scholar]
- Stevens MI, Porco D, D'Haese CA, Deharveng L. Comment on "Taxonomy and the DNA barcoding enterprise" by Ebach (2011) Zootaxa. 2011:85–88. [Google Scholar]
- Subbotin SA, Sturhan D, Chizhov VN, Vovlas N, Baldwin JG. Phylogenetic analysis of Tylenchida Thorne, 1949 as inferred from D2 and D3 expansion fragments of the 28S rRNA gene sequences. Nematology. 2006;8:455–474. [Google Scholar]
- Subbotin SA, Vovlas N, Crozzoli R, Sturhan D, Lamberti F, Moens M, Baldwin JG. Phylogeny of Criconematina Siddiqi, 1980 (Nematoda: Tylenchida) based on morphology and D2-D3 expansion segments of the 28S-rRNA gene sequences with application of a secondary structure model. Nematology. 2005;7:927–944. [Google Scholar]
- van Megen H, van den Elsen S, Holterman M, Karssen G, Mooyman P, Bongers T, Holovachov O, Bakker J, Helder J. A phylogenetic tree of nematodes based on about 1200 full-length small subunit ribosomal DNA sequences. Nematology. 2009;11:927–950. [Google Scholar]
- Wong EH-K, Shivji MS, Hanner RH. Identifying sharks with DNA barcodes: Assessing the utility of a nucleotide diagnostic approach. Molecular Ecology Resources. 2009;9:243–256. doi: 10.1111/j.1755-0998.2009.02653.x. [DOI] [PubMed] [Google Scholar]
- Wouts WM. Fauna of New Zealand 55. 2006. Criconematina (Nematoda: Tylenchida) Manaaki Whenua Press, Canterbury, New Zealand. [Google Scholar]