

SUMMARY
Cardiac fibrosis is critically involved in the adverse remodeling accompanying dilated cardiomyopathies (DCMs), which leads to cardiac dysfunction and heart failure (HF). Connective tissue growth factor (CTGF), a profibrotic cytokine, plays a key role in this deleterious process. Some beneficial effects of IGF1 on cardiomyopathy have been described, but its potential role in improving DCM is less well characterized. We investigated the consequences of expressing a cardiac-specific transgene encoding locally acting IGF1 propeptide (muscle-produced IGF1; mIGF1) on disease progression in a mouse model of DCM [cardiac-specific and inducible serum response factor (SRF) gene disruption] that mimics some forms of human DCM. Cardiac-specific mIGF1 expression substantially extended the lifespan of SRF mutant mice, markedly improved cardiac functions, and delayed both DCM and HF. These protective effects were accompanied by an overall improvement in cardiomyocyte architecture and a massive reduction of myocardial fibrosis with a concomitant amelioration of inflammation. At least some of the beneficial effects of mIGF1 transgene expression were due to mIGF1 counteracting the strong increase in CTGF expression within cardiomyocytes caused by SRF deficiency, resulting in the blockade of fibroblast proliferation and related myocardial fibrosis. These findings demonstrate that SRF plays a key role in the modulation of cardiac fibrosis through repression of cardiomyocyte CTGF expression in a paracrine fashion. They also explain how impaired SRF function observed in human HF promotes fibrosis and adverse cardiac remodeling. Locally acting mIGF1 efficiently protects the myocardium from these adverse processes, and might thus represent a therapeutic avenue to counter DCM.
INTRODUCTION
Dilated cardiomyopathy (DCM) is the most common type of non-ischemic cardiomyopathy, and is characterized by dilation and contractile dysfunction of the left and right ventricles, which is associated with functional alterations leading to heart failure (HF). Various acquired factors affecting cardiomyocyte function and genetic mutations leading to impaired force generation or transmission can both lead to DCM. The pathological remodeling process underlying ventricular dilation involves cardiomyocytes, which undergo eccentric hypertrophy, and also interstitial cells, including resident fibroblasts and macrophages (which are critically involved in tissue fibrosis). Fibrosis contributes adversely to cardiac tissue structure and function, ultimately causing HF (Khan and Sheppard, 2006). Connective tissue growth factor (CTGF), a downstream mediator of the TGFβ pathway, plays a key role in this adverse remodeling through the promotion of fibroblast proliferation and extracellular matrix production in connective tissues (Abreu et al., 2002; Blom et al., 2002).
Despite recent advances in the pharmacological treatment of individuals with DCM, mortality remains high (Xu et al., 2010). Animal models of DCM are thus required both for testing therapeutic approaches in vivo and for investigating the underlying mechanisms. We recently developed a mouse DCM model, involving cardiac-specific and tamoxifen-inducible disruption of the serum response factor (SRF) gene by the Cre-loxP method (Parlakian et al., 2005). SRF is a MADS-box transcription factor essential for cardiac differentiation and maturation (Arsenian et al., 1998; Zhang et al., 2001; Miano et al., 2004; Parlakian et al., 2004; Niu et al., 2005). SRF binds to CarG-box DNA regulatory elements, thereby regulating several genes involved in cell growth, migration, differentiation, cytoskeleton organization and energy metabolism. Triggering SRF loss in mice results in: the downregulation of proteins involved in force generation and transmission; fibrotic development; and changes in the cytoarchitecture of cardiomyocytes without hypertrophic compensation. This leads to the development of DCM and all mice die from HF around 10 weeks after triggering SRF loss (Parlakian et al., 2005). This model thus mimics some situations observed in humans (Davis et al., 2002; Chang et al., 2003): several studies suggest that late alterations of SRF function in individuals with cardiomyopathies contribute to propelling the heart towards failure.
Insulin-like growth factor (IGF1) exerts multiple beneficial effects on the heart and can improve myocardial function in pathological situations (Duerr et al., 1995; Cittadini et al., 1996; Donath et al., 1998b; Serose et al., 2005). In particular, we described a strong protective effect in induced infarct by a cardiac-specific transgene encoding the muscle-produced IGF1 propeptide (mIGF1), which remains local to the myocardium (Santini et al., 2007). The cardiac signaling cascade induced by mIGF1 preferentially uses the PDK1-SGK1 pathway to increase protein synthesis and growth (Santini et al., 2007). We therefore investigated: (1) whether such local cardiac expression of mIGF1 counteracts some of the deleterious events linked to SRF loss, and (2) the mechanisms by which mIGF1 exerts its beneficial effects. Inducible and cardiac-specific SRF knockout mice were crossed with transgenic mice overexpressing mIGF1 in cardiomyocytes and compared with the original DCM model following deletion of the SRF gene. Local mIGF1 expression significantly lengthened the lifespan of SRF mutant mice, delayed the process leading to DCM and improved cardiac functions. These protective effects were associated with an improvement in cardiomyocyte architecture and the gene expression program, and with a massive reduction of myocardial fibrosis and concomitant amelioration of inflammation. Supplementation with mIGF1 blunted the rise in CTGF expression in cardiomyocytes that results from SRF loss, blocking fibroblast proliferation and related myocardial fibrosis. These findings reveal a key role of SRF in the modulation of cardiac fibrosis, and underscore the importance of tightly regulated SRF expression in the normal heart. They imply a mechanism whereby the mIGF1 propeptide counterbalances the effects of SRF downregulation in human DCM.
RESULTS
Cardiac-specific mIGF1 overexpression improves lifespan of SRF mutants and delays DCM development
Four experimental groups of mice were generated and analyzed after tamoxifen injection: controls, mIGF1 overexpressing, SRF mutants and mIGF1 overexpressing/SRF mutants (referred to as mIGF1/SRF mutants). We initially analyzed two groups of control mice, MHC-MerCreMer and Sf/Sf, and observed no phenotypic differences between the two (supplementary material Fig. S1). Thus, we only kept the Sf/Sf group of controls, which has been systematically included in all following experiments. All SRF mutant mice (n=12) rapidly developed DCM and died 72±1 days (minimum 52 days, maximum 92 days) after tamoxifen injection, consistent with our previous report (Parlakian et al., 2005). By contrast, mIGF1/SRF mutant mice (n=13) died 165±3 days (minimum 101 days, maximum 220 days) after tamoxifen injection. Thus, overexpression of mIGF1 in the heart clearly improved life expectancy of SRF mutant mice (Fig. 1A). Whereas SRF mutants exhibited enlargement of cardiac chambers and thinning of ventricular walls 2 months after triggering SRF loss, dilation was more moderate in the mIGF1/SRF mutants (Fig. 1B). The milder phenotype in the presence of local IGF1 expression was also reflected in heart weight to body weight ratios [4.65±0.17 (n=9) in control mice, 5.27±0.18 (n=8) in mIGF1 mice, 5.75±0.11 (n=13) in mIGF1/SRF mutants and 7.10±0.76 (n=6) in SRF mutants]. Kidney and liver weights to body weight ratios were similar in all groups (data not shown).
Fig. 1.
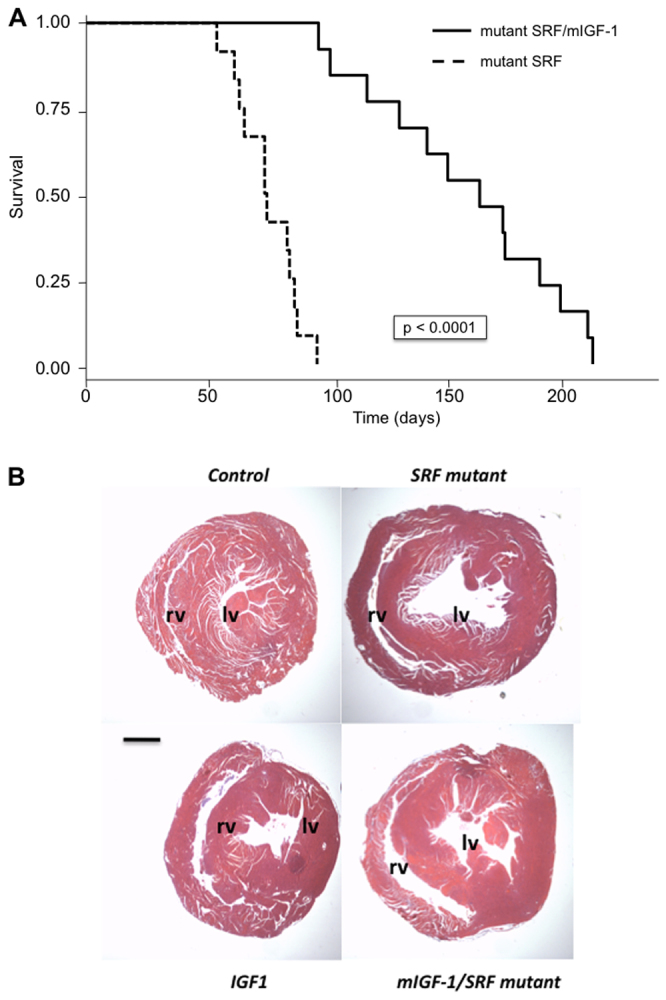
Effects of cardiac-specific mIGF1 overexpression on survival of SRF mutant mice and on cardiac dilation. (A) Kaplan-Meier curves for SRF mutant and mIGF1/SRF mutant mice; P<0.0001 with log-rank test. (B) H&E staining of paraffin-embedded heart sections. The dilation of the left (lv) and right (rv) ventricles is less marked in mIGF1/SRF than in SRF mutant samples. Scale bar: 1 mm.
mIGF1 overexpression in cardiomyocytes promotes rescue of heart function
We assessed cardiac function in the four groups of mice by serial echocardiography immediately before (0 day) and at two different times after (30 and 60 days) tamoxifen injection. At time 0, echocardiographic values were similar for control and cardiac SRF pre-mutant groups, and for cardiac mIGF1-overexpressing and cardiac mIGF1/SRF pre-mutant groups. However, this last group showed concentric left ventricular (LV) hypertrophy (Table 1). Thus, only data for controls and cardiac mIGF1-overexpressing mice are presented for time 0. At 30 days after tamoxifen treatment, the shortening velocity of circumferential fibers (Vcfc) was slightly lower in the SRF mutant group, and the mIGF1/SRF mutant group showed an intermediate phenotype with a less acute LV hypertrophy and no significant LV functional alteration (Table 1). On day 60, mIGF1/SRF mutants showed a significant improvement of cardiac function when compared with SRF mutant mice: LV contractility parameters such as Vcfc, ejection fraction (EF), and systolic velocity at mitral annulus (Sa) and posterior wall (Spw) were similar to those obtained for control and mIGF1-overexpressing mice. By contrast, a strong decrease of these four parameters was observed in SRF mutant mice (Table 1). Moderate left atrial (LA) remodeling was observed in both SRF mutant and mIGF1/SRF mutant mice, with slight LV enlargement (LV end-diastolic diameter increase; LVEDD) in the SRF mutant group. This suggests the occurrence of HF in this latter group, associated with an elevated LV filling pressure as indicated by the ratio of transmitral blood velocity to mitral annulus diastolic velocity (E/Ea; Table 1).
Table 1.
Echocardiographic measurements of control, mIGF1, SRF mutant and mIGF1/SRF mutant mice 0, 30 and 60 days after tamoxifen treatment

Cardiac cytoarchitecture is normalized in mIGF1/SRF mutant mice
Hearts of the four different genotypes were examined morphologically and histologically. Whereas hematoxylin and eosin (H&E) staining of cardiac transverse sections revealed enlarged interstitial spaces between cardiomyocytes in SRF mutant mice, these spaces were substantially smaller in mIGF1/SRF mutant mice (Fig. 2A). Cardiac cytoarchitecture was examined by confocal microscopic analysis after immunostaining for vinculin, a cytoskeletal protein, and staining for polymerized F-actin with phalloidin-TRITC. Although mIGF1/SRF mutant cardiomyocytes were still heterogeneous in size and were sometimes stretched, as in SRF mutants, their shape and alignment were more regular, without gaps between cells (Fig. 2B). In addition, intercalated discs in SRF mutant hearts were dysmorphic, whereas most intercalated discs in mIGF1/SRF mutants displayed a more normal morphology (Fig. 2B; supplementary material Fig. S2). Phalloidin staining revealed a better preservation of sarcomere organization in mIGF1/SRF than SRF mutant hearts (Fig. 2B). Cardiomyocytes from mIGF1-overexpressing mice were particularly square and thick with a regular alignment (Fig. 2B).
Fig. 2.
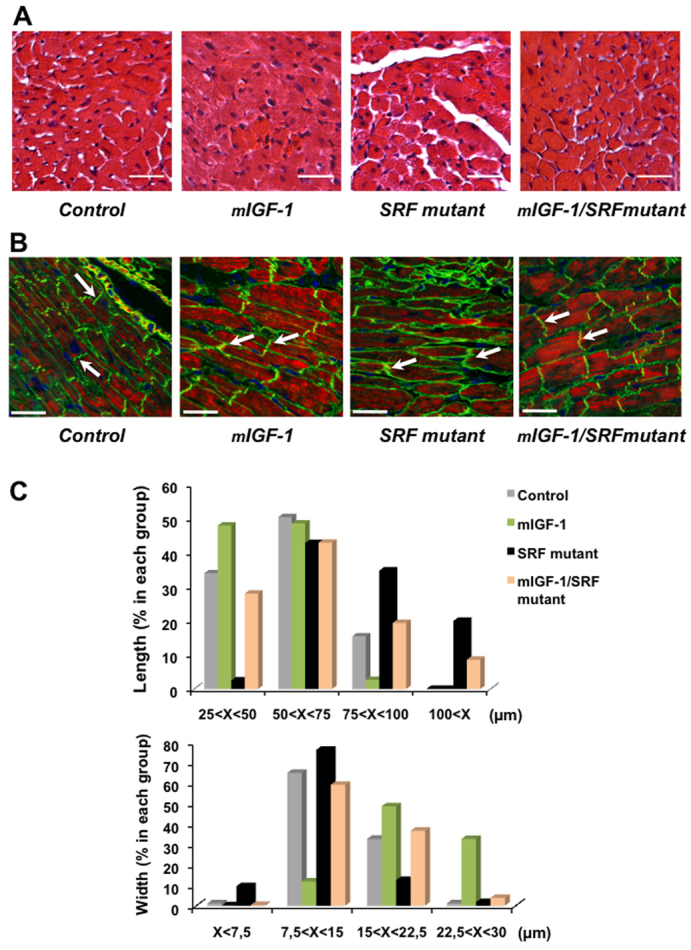
Improvement of the cardiomyocyte architecture in mIGF1/SRF mutant mice. (A) H&E staining of paraffin-embedded heart sections illustrating the amelioration of intercellular gaps in SFR mutants in the presence of mIGF1. Scale bars: 20 μm. (B) Confocal microscopy of cardiac sections labeled with Alexa-Fluor-568–phalloidin (red) for polymerized actin, anti-vinculin FITC (green) and DAPI (blue). Note the better cardiac cytoarchitecture in mIGF1/SRF mutant than SRF mutant mice: intercalated discs are substantially enlarged and irregularly shaped in SRF mutants, but mostly display almost normal morphology in mIGF1/SRF mutants (arrows). Disc sizes: min. 1 μm – max. 1.5 μm for controls and mIGF1; min. 1.5 μm – max. 3.5 μm for mIGF1/SRF mutants; and min. 4 μm – max. 6.7 μm for SRF mutants. Scale bars: 30 μm. (C) Distribution of cardiomyocyte lengths and widths in the four groups of mice; n=200 for each group (four different mice per group).
Morphometric analysis revealed that, whereas SRF mutant cardiomyocytes had increased length and decreased width compared with controls, mIGF1/SRF mutant cardiomyocyte size distribution was closer to that of controls (Fig. 2C). Noticeably, mIGF1-overexpressing cardiomyocytes were particularly short and wide (Fig. 2C). Thus, the eccentric stretching of cardiomyocytes that is characteristic of DCM is considerably attenuated when mIGF1 is overexpressed. Ultrastructural analysis of mIGF1/SRF mutants by electron microscopy confirmed that mIGF1 overexpression normalized sarcomeric organization and substructure, and the abundance and alignment of mitochondria (supplementary material Fig. S3).
mIGF1 improves the cardiac gene expression program in SRF mutant mice
The absence of SRF results in profound alterations of the cardiac gene expression program, with the downregulation of main SRF target genes and late activation of the stress-induced gene ANF (Parlakian et al., 2005). We analyzed the effects of mIGF1 overexpression, in the context of SRF loss, on the cardiac gene expression program both by real-time reverse-transcriptase PCR (RT-PCR) and northern blot analysis (Fig. 3). After induction by tamoxifen, SRF was lost from most cardiomyocytes, but a small percentage of the cardiomyocyte population continued producing SRF (∼20%; supplementary material Fig. S1). Because of this mosaic inactivation of SRF, the results (Fig. 3) only reflect global changes in gene expression, and might be different within the subpopulations of cardiac cells. Triggering SRF loss led to a similar decrease in cardiac α-actin mRNA levels irrespective of local mIGF1 expression; the abundance of skeletal α-actin mRNAs increased in SRF mutant hearts but increased significantly less in mIGF1/SRF mutant hearts (Fig. 3A,B). Skeletal α-actin immunostaining on paraffin-embedded heart sections confirmed these findings (supplementary material Fig. S4). Local mIGF1 expression led also to an attenuation of the switch in myosin heavy chain (MHC) isoform expression associated with SRF loss, with a significant increase in the level of postnatal α-MHC mRNA and a trend towards a decrease in the amount of embryonic β-MHC mRNAs (Fig. 3A). Expression of MCK, an SRF target gene involved in energy flux, remained lower in mIGF1/SRF than in SRF mutant mice (Fig. 3A). Expression of the SERCA2 and NCX1 genes, encoding proteins involved in Ca2+ reuptake and extrusion, respectively, which was weak in SRF mutant hearts, was partly restored in the presence of mIGF1 (Fig. 3). Importantly, expression of the stress-induced gene ANF was normalized in mIGF1/SRF mutant hearts, compared with elevated levels observed in the SRF mutant (Fig. 3). Elevated BNP expression in SRF mutant hearts was also normalized in the mIGF1/SRF mutant (Fig. 3A). Thus, the globally compromised cardiac gene expression pattern associated with SRF loss is improved by locally acting mIGF1.
Fig. 3.
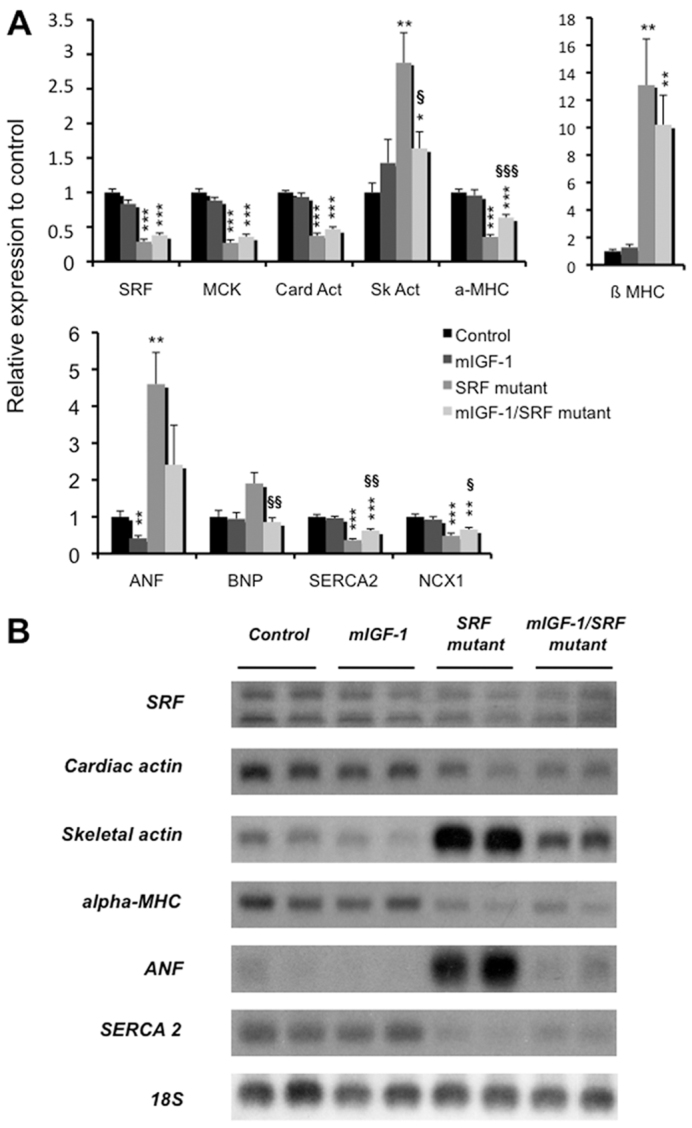
Analysis of gene expression in the hearts of mIGF1/SRF mutant and SRF mutant mice. (A) qRT-PCR analysis of control (n=7), mIGF1 (n=6), SRF mutant (n=6) and mIGF1/SRF mutant (n=10) mouse mRNA, using cyclophilin as internal control. Data are presented as means ± s.e.m. *, ** and *** indicate significant difference at P<0.05, P<0.01 and P<0.001, respectively, versus the control group, and §, §§ and §§§ indicate significant difference at P<0.05, P<0.01 and P<0.001, respectively, between the mIGF1/SRF mutant and the SRF mutant groups. (B) Northern blot analysis of gene expression in the four groups of mice. We used 18S rRNA as a loading control.
mIGF1 counters SRF-loss-related myocardial fibrosis and CTGF expression
Masson’s trichrome staining consistently revealed interstitial fibrosis in SRF mutant hearts, but no such fibrosis was detected in the presence of mIGF1 (Fig. 4A). Connexin 45 is a gap junction protein primarily associated with cardiac fibroblasts (Goldsmith et al., 2004). Connexin 45 and phalloidin co-immunostaining revealed overgrowing cardiac fibroblasts within the large spaces in the SRF mutant myocardium, but not in mIGF1/SRF mutant, control or mIGF1 hearts (Fig. 4B). These results were confirmed by immunostaining for vimentin, another marker of cardiac fibroblasts (supplementary material Fig. S5) (Camelliti et al., 2005).
Fig. 4.
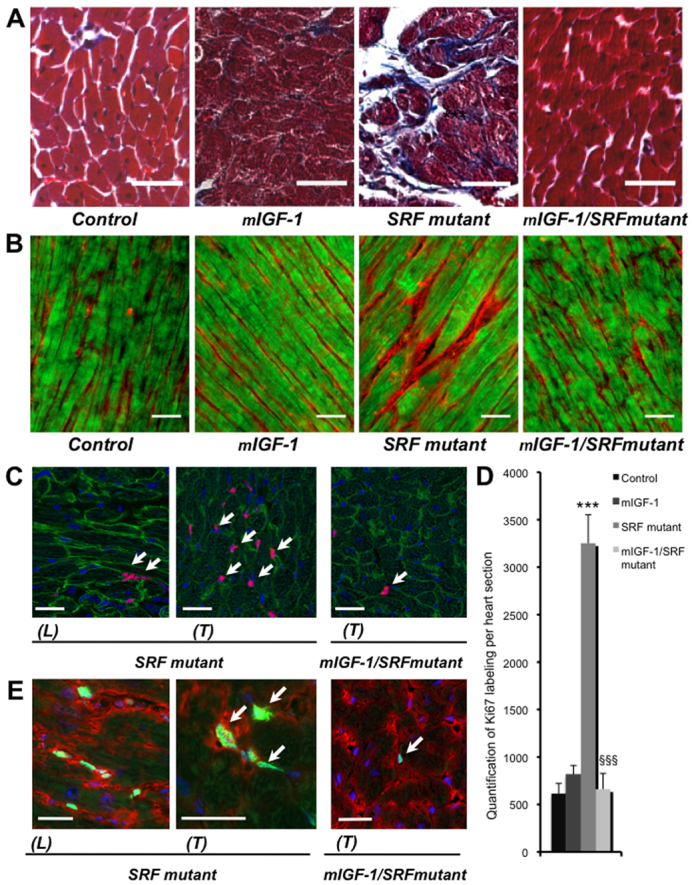
Effects of cardiac-specific mIGF1 overexpression on fibrosis and cell proliferation. (A) Histological analysis by Masson’s trichrome staining shows intensive fibrosis only in SRF mutant mouse myocardium. Scale bars: 10 μm. (B) Visualization of cardiac fibroblasts by connexin 45 staining of heart sections from control, mIGF1, SRF mutant and mIGF1/SRF mutant mice. Longitudinal frozen section showing the localization of cardiac fibroblasts stained with connexin 45 (red) and polymerized actin in myocytes labeled with Alexa-Fluor-488–phalloidin (green). Scale bars: 40 μm. These results are representative of two independent experiments. (C) Confocal microscopic view of Ki67 labeling (pink; at arrows). Labeling is stronger in the SRF mutant than the mIGF1/SRF mutant, and is exclusively located in the extracellular matrix. Longitudinal (L) and transversal (T) sections show the same tissue, with cardiomyocytes being identified by labeling with anti-vinculin FITC (green) and nuclei with DAPI (blue). Scale bars: 30 μm. (D) Quantification of Ki67 labeling per heart section reveals four times more labeling in SRF mutant hearts than all other groups. Data are presented as means ± s.e.m. ***, significant difference versus the control group, at P<0.001; §§§, significant difference between the mIGF1/SRF mutant and the SRF mutant groups, at P<0.001. These results are representative of three independent experiments. (E) Confocal microscopic view of Ki67 labeling (green; at arrows), connexin 45 staining (red) and DAPI labeling of nuclei (blue), showing that Ki67 labeling coincides with cardiac fibroblasts. Scale bars: 30 μm.
To assess whether cardiac fibroblast proliferation was involved in cardiac fibrosis, we quantified the Ki67 protein, present only in nuclei of cycling cells (Endl and Gerdes, 2000; Scholzen and Gerdes, 2000), in the various mouse genotypes. Co-immunostaining for vinculin and Ki67 indicated that the cardiomyocytes were not replicating, but that many more interstitial cells were dividing in SRF mutant mice than in the other groups of mice (Fig. 4C): there were four times more Ki67-positive cells per heart section in SRF mutants than in the mIGF1/SRF and mIGF1 mutants and controls, which all displayed similar low numbers of dividing interstitial cells (Fig. 4D). Co-immunostaining for Ki67 and connexin 45 confirmed that the larger numbers of Ki67-positive cells in SRF mutant hearts was associated with the proliferation of cardiac fibroblasts (Fig. 4E).
To investigate the impact of mIGF1 on the development of fibrosis, we quantified the expression of several genes involved in this process by RT-PCR. Increased collagen production in SRF mutant hearts was normalized in mIGF1/SRF mutant hearts, as assessed by procollagen type 1α1 but not type 3α1 transcripts (Fig. 5A). Two cardioprotective genes, adiponectin and UCP1, were induced in mIGF1/SRF mutant but not SRF mutant hearts (Fig. 5A). Cardiac CTGF expression has been documented both in cardiomyocytes and in fibroblasts. CTGF mRNA and protein levels were much higher in SRF mutant hearts than in control and mIGF1 hearts, and these high levels were not observed in mIGF1/SRF mutants (Fig. 5A,B). A similar, but not significant, trend was observed for TGFβ mRNA levels (Fig. 5A). Immunohistochemical detection of CTGF protein in heart sections was used to determine the cell specificity of these expression patterns. In control and mIGF1 hearts, CTGF immunoreactivity was extremely weak and exclusively localized outside cardiomyocytes (Fig. 6). By contrast, in SRF mutant hearts, CTGF was abundant not only in interstitial spaces corresponding to proliferating fibroblasts but also in a large number of small SRF-null cardiomyocytes. Noticeably, the few SRF-positive hypertrophic cardiomyocytes were all negative for CTGF (Fig. 6). In the presence of mIGF1, SRF mutant hearts displayed a completely normalized CTGF expression pattern, similar to that seen in controls. Taken together, these findings suggest that: (1) the increased CTGF expression observed in SRF mutant hearts stems from both a direct effect of SRF loss on CTGF gene expression within cardiomyocytes and from fibroblast proliferation, and (2) cardiac-specific mIGF1 expression counteracts the rise in CTGF production both in SRF mutant cardiomyocytes and in cardiac fibroblasts independently of SRF loss, thus attenuating cardiac fibroblast proliferation.
Fig. 5.
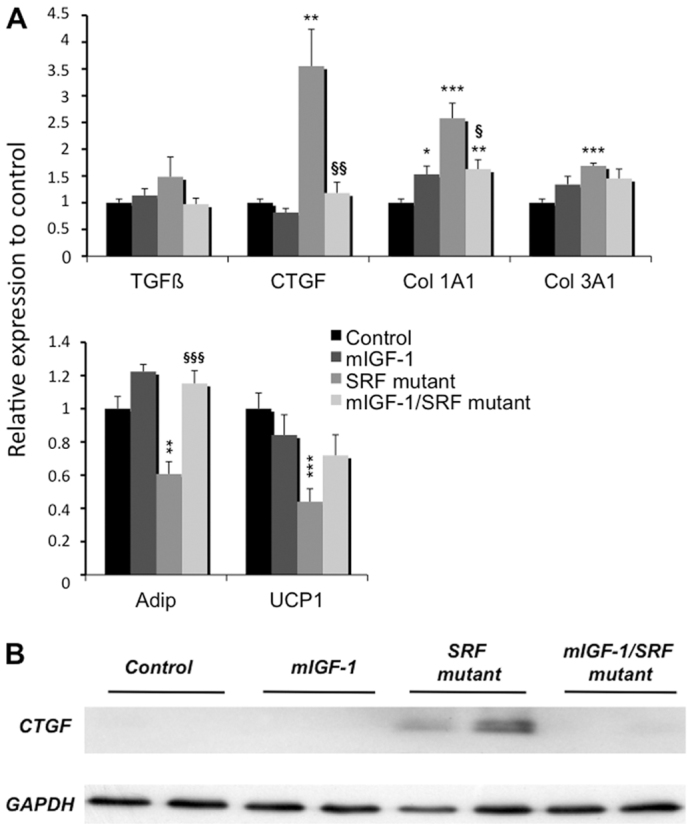
Modulation of the expression of genes involved in cardiac fibrosis by local mIGF1 overexpression. (A) qRT-PCR analysis of mRNA of genes that are markers of cardiac tissues in control (n=7), mIGF1 (n=6), SRF mutant (n=6) and mIGF1/SRF mutant (n=10) mice. Cyclophilin was used as control. Data are presented as means ± s.e.m. *, ** and ***, significant difference versus the control group, at P<0.05, P<0.01 and P<0.001, respectively; §, §§ and §§§, significant difference between the mIGF1/SRF mutant and the SRF mutant groups, at P<0.05, P<0.01 and P<0.001, respectively. (B) Western blot analysis of CTGF protein.
Fig. 6.

Effects of local mIGF1 overexpression on CTGF expression. Immunofluorescence labeling of SRF (red), CTGF (orange) and vinculin (green) on serial heart sections of control, SRF mutant and mIGF1/SRF mutant mice. The orange arrows indicate CTGF-positive/SRF-null cardiomyocytes and the white arrows indicate SRF-positive/CTGF-negative cardiomyocytes. The orange arrowheads show CTGF outside of cardiomyocytes in SRF mutant mice. These data were verified by analyzing more than 50 fields per heart section in three independent experiments. The results presented are representative of three independent experiments. Scale bar: 30 μm.
mIGF1 overexpression modulates the inflammatory response in SRF mutant mice
There is growing evidence that fibrosis and inflammation are mechanistically linked in the heart. We therefore investigated the expression of cytokines and chemokines that promote inflammation in the different mouse genotypes. Although a strong increase in proinflammatory IL-6 transcripts was observed in SRF mutant hearts, only a limited increase was observed in mIGF1/SRF mutants (Fig. 7A). The inverse profile was observed for the transcripts of the anti-inflammatory cytokine IL-4 (Fig. 7A). Transcripts encoding IL-1β, another inflammatory cytokine, and IL-10, an anti-inflammatory cytokine, were not significantly affected by mIGF1 overexpression (Fig. 7A). Expression of the monocyte chemoattractant protein 1 (MCP1) and cyclooxygenase 2 (COX2; encoding a prostaglandin-endoperoxide synthase involved in the inflammatory response) genes was significantly activated in SRF mutant mice, and this activation was attenuated in mIGF1/SRF mutants (Fig. 7A). Immunostaining with F4/80 antibody revealed macrophages in heart sections from all mouse groups; however, the signal intensity was much stronger in SRF mutant sections than in control and mIGF1/SRF mutant sections (Fig. 7B). In summary, these analyses show that local mIGF1 attenuates the inflammatory response induced by SRF loss and the concomitant cardiac remodeling.
Fig. 7.
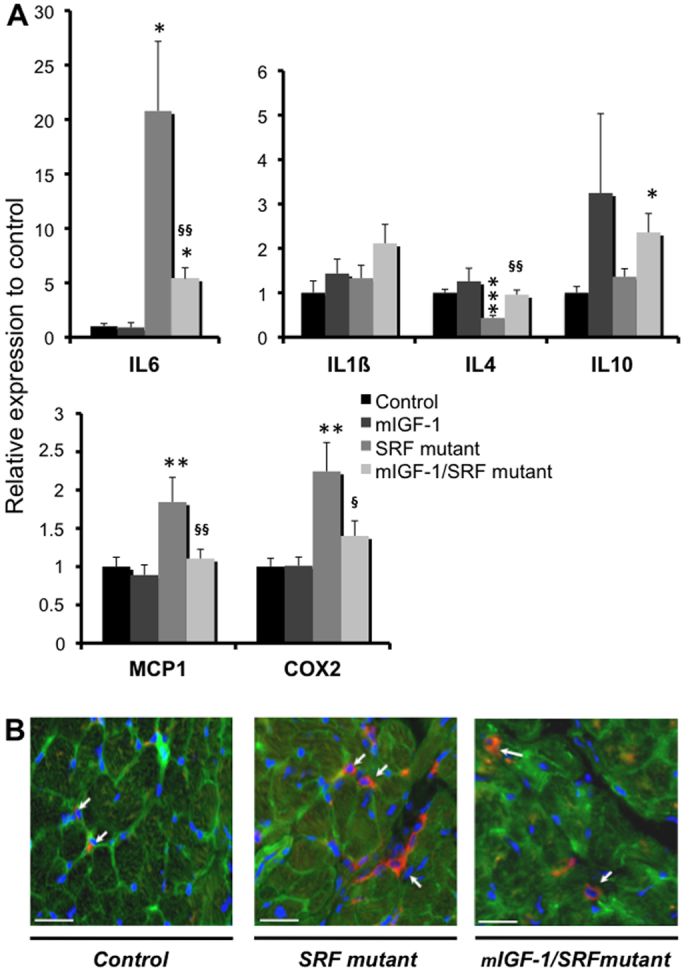
Effects of cardiac-specific mIGF1 overexpression on the inflammation profile. (A) qRT-PCR assays of IL-1β, IL-4, IL-6, IL-10, MCP1 and COX2 mRNA in cardiac tissue from control (n=7), mIGF1 (n=6), SRF mutant (n=6) and mIGF1/SRF mutant (n=10) mice. Cyclophilin mRNA was used as a control. Values are means ± s.e.m. *, ** and ***, significant difference versus the control group at P<0.05, P<0.01 and P<0.001, respectively; § and §§, significant difference between the mIGF1/SRF mutant and the SRF mutant group at P<0.05 and P<0.01, respectively. (B) Immunofluorescence labeling with F4/80 (red), vinculin (green) and DAPI (blue) on serial heart sections of control, SRF mutant and mIGF1/SRF mutant mice. The arrows indicate inflammatory cells. These results are representative of three independent experiments. Scale bars: 30 μm.
DISCUSSION
IGF1 is a peptide that acts both as a systemic growth factor and locally in an autocrine/paracrine manner, and has a key role in cardiac function and in cardiac growth (Ren et al., 1999). The locally acting mIGF1 propeptide is clearly beneficial both in enhancing repair of the heart following injury and in protecting cardiomyocytes from hypertrophic and oxidative stresses (Santini et al., 2007). Here, we report an analysis of the effects of cardiac-specific mIGF1 expression in a mouse model of DCM based on SRF gene disruption. Our in vivo findings demonstrate a crucial role of SRF in controlling CTGF expression within cardiomyocytes and thus in regulating the fibrogenic process. Cardiac supplementation with mIGF1 diminishes DCM progression, which is associated with SRF loss, at least in part by counteracting both the inflammatory response and fibrosis, mechanistically linked processes directly contributing to adverse myocardial remodeling and HF. The beneficial effects of mIGF1 in the DCM model included the blockade of induction of both CTGF and proinflammatory cytokines, and the inhibition of cardiac fibroblast proliferation.
We previously reported that triggering SRF gene disruption in adult cardiomyocytes led to DCM characterized by LV dilation, a progressive loss in contractility and fatal HF (Parlakian et al., 2005). The introduction of a cardiac-restricted mIGF1 transgene in SRF mutant mice attenuated most of these ventricular remodeling phenomena: mIGF1-associated improvements in LVEDD, Sa, Spw, Vcfc and EF activities indicate a functional preservation of the heart with a delay in cardiac dilation and the doubling of overall survival times.
Previous reports have shown that administration of the systemic IGF1 isoform improves cardiac function in humans and in animal models of DCM and HF (Duerr et al., 1995; Cittadini et al., 1996; Donath et al., 1998a; Donath et al., 1998b; Serose et al., 2005). Transgenic IGF1 overexpression also led to morphological and functional cardiac improvements in a tropomodulin-overexpressing transgenic mouse model (Welch et al., 2002). However, in this model, the IGF1-mediated correction of abnormalities was mainly attributed to myocyte regeneration due to the increased number of myocytes associated with Ki67 labeling (Welch et al., 2002). Protection in post-infarct experiments has also been suggested to be a consequence of increased cardiomyocyte proliferation induced by the mIGF1 transgene (Santini et al., 2007). By contrast, our findings do not support mIGF1 exerting its beneficial effects on DCM through the recruitment of cardiomyocyte precursor cells. Indeed, tamoxifen-induced SRF disruption is cardiomyocyte-specific and time-restricted, so any subsequent recruitment of precursor cells would have resulted in a new population of SRF-positive cardiomyocytes. At 2 months after tamoxifen injection, the abundance of the SRF protein in mIGF1/SRF mutant hearts remained low and the number of cardiomyocytes escaping SRF disruption had not increased. Accordingly, expression of downstream cardiac α-actin and MCK genes was also not elevated.
In another model, that of mosaic inactivation of SRF in the myocardium using Desmin-Cre transgenic mice, which express the Cre recombinase in only 50% of the cardiomyocytes, we could show that cardiomyocytes lacking SRF became thin and elongated, whereas cardiomyocytes still expressing SRF became hypertrophic (Gary-Bobo et al., 2008). Skeletal α-actin expression, a marker of cardiac hypertrophy, was activated in SRF-positive cardiomyocytes only (Gary-Bobo et al., 2008). Accordingly, in our model, the small percentage of the cardiomyocyte population still expressing SRF (∼20%) is expected to activate a typical concentric hypertrophic gene program to compensate for the ∼80% of cardiomyocytes depleted of SRF, which undergo eccentric elongation. Similarly, these cardiomyocytes continuing to express SRF most probably account for the reactivation of the fetal gene program, a molecular hallmark of progression towards HF, and for the increased expression of skeletal α-actin and β-MHC isoforms observed in SRF mutant hearts. Interestingly, the activation of this fetal gene program in SRF mutant hearts was partly counteracted by mIGF1. This result is consistent with the blockade of the fetal gene program by mIGF1 propeptide observed in cultured cardiomyocytes (Vinciguerra et al., 2010): in these experiments, fully processed IGF1 was ineffective, suggesting that IGF1 propeptides could have different protective effects. In our model, expression of SERCA2, NCX1 and α-MHC was restored to a certain extent in mIGF1/SRF mutants and contributed to the improved contraction rates. mIGF1 expression might therefore exert beneficial effects by limiting stress, and maintaining Ca2+ dynamics and cardiac performance. Restoration of Ca2+ dynamics by IGF1 has also been described in the tropomodulin-overexpressing transgenic mouse model (Welch et al., 2002).
The presence of the mIGF1 propeptide had several major beneficial effects at the cellular level: it was associated with relatively rectangular cardiac cell shapes, and preserved the continuity of interactions and communications between cardiomyocytes. mIGF1 also attenuated alterations in intercalated discs, which play a crucial role in force transmission and their disorganization leads to the development of DCM (Ehler et al., 2001; Ferreira-Cornwell et al., 2002). Cardiac α-actin is the major actin isoform in cardiomyocytes and constitutes the main component of the thin filament of the sarcomere (Vandekerckhove et al., 1986). Despite an abundance of cardiac α-actin transcripts in mIGF1/SRF mutant cardiomyocytes that was lower than in controls, wild-type levels of polymerized actin were maintained. Our results suggest that mIGF1 positively affects actin dynamics, either through increased translation or increased stability.
Most importantly, both the induction of the inflammatory response and cardiac fibrotic development in SRF mutant hearts were substantially attenuated in the presence of mIGF1. Indeed, mIGF1 counteracted increases, induced by loss of SRF, in the amounts of mRNA for MCP1, IL-6 and COX-2, and decreases in that for IL-4 and collagen production. Numerous studies underscore the important role played by TGFβ and one of its downstream mediators, CTGF, in the development of fibrosis (Abreu et al., 2002; Blom et al., 2002). Elevated CTGF protein levels were observed in cardiac tissue from patients with HF and correlated with the degree of myocardial fibrosis (Koitabashi et al., 2007). Consistent with these observations, levels of the transcripts of these two factors were closely correlated and higher in SRF mutant than control hearts; their abundance was completely normalized by the presence of mIGF1. CTGF regulates fibrosis by inducing fibroblast proliferation and extracellular matrix production, and its high concentration therefore probably explains the fibroblast proliferation and fibrosis in SRF mutants. Several studies also implicate CTGF in the regulation of the inflammatory process by inducing MCP1 and IL-6 in cardiomyocytes and by acting as a chemotactic factor for monocytes (Sanchez-Lopez et al., 2009; Wang et al., 2010). Thus, normalization of CTGF levels by mIGF1 contributes to decreases in both inflammation and fibrosis.
Increased CTGF expression in SRF mutants was observed both in cardiac fibroblasts and SRF-null cardiomyocytes, but never in SRF-positive cardiomyocytes. This strongly implicates SRF in repressing CTGF expression in cardiomyocytes, directly or through a repressor. Indeed, analysis of the mammalian ‘CArGome’ (CArG elements in the genome) identified CTGF as a potential SRF target, and the presence of a ‘CArG’ box in the CTGF promoter has been established (Sun et al., 2006; Muehlich et al., 2007). In the same way, SRF has been shown to control CTGF transcription in human umbilical vein endothelial cells (Muehlich et al., 2007) and to operate as a CTGF repressor in neurons (Stritt et al., 2009). Interestingly, the CTGF transcript is a target of miR133, the transcription of which is controlled by SRF (Chen et al., 2006; Niu et al., 2007; Duisters et al., 2009). Such a mechanism might also account for the SRF-mediated repression of CTGF expression. The abundance of CTGF-positive/SRF-null cardiomyocytes in SRF mutant hearts correlated with the degree of the interstitial fibrosis and increased procollagen type 1α1 and 3α1 expression, suggesting that CTGF produced and secreted by SRF-null cardiomyocytes can exert a paracrine control on cardiac fibroblast functions. Also, paracrine regulation of oligodendrocyte differentiation by SRF-directed CTGF repression in neurons was recently demonstrated (Stritt et al., 2009). In the human context, alterations of SRF function due to SRF cleavage by caspase 3 and/or aberrant splicing of SRF transcripts have been described in cardiomyocytes from severely failing hearts, accounting for altered expression of various cardiac-specific proteins that are associated with HF (Davis et al., 2002; Chang et al., 2003; Wong et al., 2012). Such impaired SRF activity might also play a crucial role in promoting fibrosis and adverse cardiac remodeling, thus propelling the heart towards failure.
How does mIGF1 counteract loss of SRF function? The dramatic rescue by the mIGF1 transgene in the DCM model could be the consequence of numerous effects: limiting cellular stress and inflammation by increasing the expression of antioxidant UCP1 and adiponectin, normalization of CTGF expression, and decreasing procollagen gene expression and subsequent fibrosis. Our results are consistent with the reduced fibrogenesis observed following IGF1 induction in activated hepatic stellate cells after liver injury and the accelerated skeletal muscle regeneration by mIGF1 transgene expression, which rapidly modulates inflammatory cytokines and chemokines (Sanz et al., 2005; Pelosi et al., 2007). The normalization of CTGF mRNA levels is in favor of IGF1 having a strong antagonistic effect on CTGF transcription. Indeed, IGF1 inhibits CTGF expression through the PI3K-Akt signaling pathway (Zhou et al., 2008). In accordance with our results in the heart, IGF1 gene transfer to cirrhotic liver induces fibrolysis and reduces fibrogenesis via the downregulation of profibrogenic molecules, including TGFβ and CTGF (Sanz et al., 2005).
In summary, our studies in vivo show that the elevated CTGF expression observed in SRF mutant hearts stems from both fibroblast proliferation and direct effects of SRF loss on CTGF gene expression within cardiomyocytes. Supplemental mIGF1 counteracts the effects of CTGF induction at both levels, blocking cardiac fibroblast proliferation, thereby counteracting both the inflammatory response and fibrosis, protecting the heart against adverse remodeling and attenuating the progression of DCM. These results advance our understanding of the mechanistic basis of HF. They also suggest that mIGF1 propeptide remaining local to the myocardium is a promising therapeutic option for HF.
METHODS
Transgenic mice
The generation and characterization of transgenic mice with inducible cardiac-specific SRF loss (α-MHC-MerCreMer: Sf/Sf) and those overexpressing mIGF1 in cardiomyocytes (α-MHC-mIGF1; abbreviated to mIGF1) have been described previously (Parlakian et al., 2005; Santini et al., 2007). The mIGF1 strain was backcrossed at least five times onto a C57BL/6N genetic background and was then crossed with mice homozygous for floxed SRF alleles (abbreviated to Sf/Sf) to generate α-MHC-mIGF1: Sf/Sf mice. Theα-MHC-mIGF1: Sf/Sf mice were crossed with α-MHC-MerCreMer: Sf/Sf mice to generate the four experimental groups of mice that were systematically included in all experiments: controls (Sf/Sf), cardiac mIGF1-overexpressing mutants (α-MHC-mIGF1: Sf/Sf), cardiac SRF pre-mutants (α-MHC-MerCreMer: Sf/Sf) and cardiac mIGF1-overexpressing/SRF pre-mutants (α-MHC-mIGF1: α-MHC-MerCreMer: Sf/Sf). Mice were selected on the basis of the results of PCR analysis of tail DNA. For all experiments, four groups of 2-month-old mice (including at least six male and six female mice per group) were given intraperitoneal tamoxifen citrate salt diluted in ethanol (20 μg/g per day; Sigma) injections daily for 4 consecutive days. All analyses were performed 2 months after the first tamoxifen injection (Parlakian et al., 2005). In our experimental conditions, no negative impact of Cre and/or tamoxifen injections was observed in our two initial groups of controls: α-MHC-MerCreMer and Sf/Sf (supplementary material Fig. S1). Thus, we only kept the Sf/Sf group of controls, which was systematically included in all experiments. Cre-mediated excision of floxed SRF alleles was verified as previously described. The resulting SRF protein loss (75–80% by comparison with the controls) was equivalent in the SRF mutant mice overexpressing or not overexpressing mIGF1 (supplementary material Fig. S6). Expression of the mIGF1 gene in the heart after triggering SRF loss was confirmed by PCR with specific primers that recognize the mRNAs for both endogenous and transgenic IGF1 isoforms. In control conditions, the heart produced small but detectable amounts of IGF1 mRNA; in mIGF1 and mIGF1/SRF mutant mice, the amounts of IGF1 mRNA were 470-times and 230-times higher than controls, respectively (supplementary material Fig. S7). This study conformed to institutional guidelines for the use of animals in research.
Echocardiography
Echocardiography was performed as previously described (Parlakian et al., 2005) with a Toshiba Powervision 6000, SSA 370A device equipped with an 8- to 14-MHz linear transducer; mice were under isoflurane anesthesia (0.75% to 1% oxygen) with spontaneous ventilation. Data were transferred online to a computer for offline analysis.
Histological analysis, immunohistochemistry and morphometric measurements
Histological analysis and immunohistochemistry using anti-SRF (1:100; Santa Cruz), anti-CTGF (1:100; Santa Cruz) and anti-skeletal actin (1:20) were performed as previously described (Parlakian et al., 2005). Fibrosis was detected by Masson’s trichrome and hematoxylin-eosin-saffron stainings. Immunofluorescence analysis of frozen sections involved the following primary antibodies: anti-SRF (1:500; Santa Cruz), anti-vinculin (1:500; hVIN-1, Sigma), anti-connexin-45 (1:400; Chemicon), anti-vimentin (1:100; Progen), anti-F4/80 (1:500; Abcam) and anti-Ki67 (1:750; Abcam) followed by incubation with Cy3- or Alexa-Fluor-488-coupled secondary antibody. Alexa-Fluor-488–phalloidin or TRITC-labeled phalloidin (0.33 μmol/l; Sigma) was used to detect polymerized actin.
The maximal length and width of cardiomyocytes were measured by confocal microscopy following vinculin-phalloidin immunofluorescence staining using ImageJ software (version 1.39u). Ten to 15 fields corresponding to more than 150 cardiac cells per group of mice were analyzed. Measurements were performed on samples from at least four individuals in each group.
Electron microscopy
Electron microscopy was performed on hearts of control, mIGF1, SRF mutant and IGF1/SRF mutant mice as previously described (Agbulut et al., 2001).
Western blot analysis
Western blotting was performed as previously described (Parlakian et al., 2004) using anti-SRF (1:800), anti-CTGF (1:250) and anti-GAPDH (1:2000) antibodies (Santa Cruz) in 5% milk.
Quantitative RT-PCR analysis
Total RNAs were extracted from hearts using the RNeasy fibrous tissue mini kit (Qiagen) and were reversed transcribed with Moloney murine leukemia virus reverse transcriptase (Invitrogen) and random hexamers (Promega) to generate cDNAs. PCR analysis was then performed with SYBR green PCR technology (ABGene). The Primer3 program (http://frodo.wi.mit.edu/primer3/) was used to select primers (available on request). Cyclophilin mRNA was used as the reference transcript.
TRANSLATIONAL IMPACT.
Clinical issue
Despite recent advances in the pharmacological treatment of individuals with dilated cardiomyopathy (DCM), mortality remains high. Cardiac fibrosis is crucially involved in the adverse remodeling process occurring in DCM that leads to cardiac dysfunction and failure. Accurate animal models of DCM are important both for understanding the mechanisms involved in the progression to failure and for testing new therapeutic approaches. Here, the authors investigate whether insulin-like growth factor 1 (IGF1) has a beneficial effect in a mouse model of DCM.
Results
The authors address this issue in a mouse model of DCM in which temporally controlled cardiac-specific inactivation of the gene encoding serum response factor (SRF; which is essential for normal cardiac differentiation and maturation) leads to progressively reduced contractility and mimics some features observed in individuals with cardiomyopathies. Cardiac expression of a locally acting IGF1 propeptide (mIGF1) limits DCM progression in this model, conferring a marked improvement in cardiac function and doubling lifespan. Beneficial effects of mIGF1 include blunting the induction of connective tissue growth factor (CTGF) expression in cardiomyocytes, an effect of SRF deficiency. This results in the blockade of myocardial fibrosis and a concomitant amelioration of inflammation.
Implications and future directions
These data uncover a key role for SRF in modulating cardiac fibrosis and repressing cardiac CTGF expression, exerting paracrine control on cardiac fibroblast function. Amelioration of fibrosis and DCM caused by SRF deficiency through expressing mIGF1 propeptide represents a promising therapeutic avenue for treating cardiac failure in humans. Some evidence suggests that SRF dysfunction in individuals with DCM contributes to heart failure. Therefore, further understanding of the underlying mechanisms in this model will help to develop treatments that counteract the adverse cardiac remodeling that is linked to alterations of SRF function in individuals with DCM.
Northern blot analysis
Northern blotting was carried out with 5 μg of total RNA as previously described (Soulez et al., 1996). The membranes were successively hybridized at 65°C with murine cDNA probes for SRF, cardiac and skeletal α-actins, α-MHC, ANF and SERCA2 labeled with [a32P]dCTP using the Megaprime DNA labeling system (Amersham). Hybridization with an 18S ribosomal probe was used for sample normalization.
Statistical analysis
Survival and morphometric results are expressed as means ± s.e.m. For the survival data, Kaplan-Meier analyses were performed using the log-rank test. For the morphometric data, the significance of differences between means was assessed with Student’s t-test. P-values of <0.05 were considered to be statistically significant.
Supplementary Material
Acknowledgments
We thank Sophie Clément for providing the skeletal α-actin antibody, and Alain Schmitt from the Electron Microscopy Facility and Etienne Audureau for their assistance (Cochin Institute, F-75014 Paris, France). We also thank Athanassia Sotiropoulos, Bénédicte Chazaud and Zhenli Li for critically reading the manuscript.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
J.-F.D., D.T. and D.D. conceived and designed the experiments. M.T., B.E., M.M., A.A., L.L., D.T. and J.-F.D. performed the experiments. M.T., B.E., M.M., D.T., D.D. and J.-F.D. contributed to data analysis. M.P.S. and N.R. provided mIGF1 transgenic mice and contributed to data analysis. M.T. and J.-F.D. wrote the manuscript. D.D. and N.R. edited the manuscript prior to submission.
FUNDING
This work was supported by the French Agence Nationale pour la Recherche [ANR-05-PCOD-003 and ANR-08-GENOPATH-038]; the British Heart Foundation (N.R. and M.P.S.); Eumodic [EU Integrated Project No. LSHG-CT-2006-037188 (to N.R. and M.P.S.)]; and Heart Repair [EU Integrated Project No. LSHM-CT-2005-018630 (to N.R. and M.P.S.)]. N.R. is an NHMRC Australia Fellow.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.009456/-/DC1
REFERENCES
- Abreu J. G., Ketpura N. I., Reversade B., De Robertis E. M. (2002). Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat. Cell. Biol. 4, 599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agbulut O., Li Z., Perie S., Ludosky M. A., Paulin D., Cartaud J., Butler-Browne G. (2001). Lack of desmin results in abortive muscle regeneration and modifications in synaptic structure. Cell Motil. Cytoskeleton 49, 51–66 [DOI] [PubMed] [Google Scholar]
- Arsenian S., Weinhold B., Oelgeschlager M., Ruther U., Nordheim A. (1998). Serum response factor is essential for mesoderm formation during mouse embryogenesis. EMBO J. 17, 6289–6299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom I. E., Goldschmeding R., Leask A. (2002). Gene regulation of connective tissue growth factor: new targets for antifibrotic therapy? Matrix Biol. 21, 473–482 [DOI] [PubMed] [Google Scholar]
- Camelliti P., Borg T. K., Kohl P. (2005). Structural and functional characterisation of cardiac fibroblasts. Cardiovasc. Res. 65, 40–51 [DOI] [PubMed] [Google Scholar]
- Chang J., Wei L., Otani T., Youker K. A., Entman M. L., Schwartz R. J. (2003). Inhibitory cardiac transcription factor, SRF-N, is generated by caspase 3 cleavage in human heart failure and attenuated by ventricular unloading. Circulation 108, 407–413 [DOI] [PubMed] [Google Scholar]
- Chen J. F., Mandel E. M., Thomson J. M., Wu Q., Callis T. E., Hammond S. M., Conlon F. L., Wang D. Z. (2006). The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 38, 228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cittadini A., Stromer H., Katz S. E., Clark R., Moses A. C., Morgan J. P., Douglas P. S. (1996). Differential cardiac effects of growth hormone and insulin-like growth factor-1 in the rat. A combined in vivo and in vitro evaluation. Circulation 93, 800–809 [DOI] [PubMed] [Google Scholar]
- Davis F. J., Gupta M., Pogwizd S. M., Bacha E., Jeevanandam V., Gupta M. P. (2002). Increased expression of alternatively spliced dominant-negative isoform of SRF in human failing hearts. Am. J. Physiol. Heart Circ. Physiol. 282, H1521–H1533 [DOI] [PubMed] [Google Scholar]
- Donath M. Y., Froesch E. R., Zapf J. (1998a). Insulin-like growth factor I and cardiac performance in heart failure. Growth Horm. IGF Res. 8, 167–170 [DOI] [PubMed] [Google Scholar]
- Donath M. Y., Sutsch G., Yan X. W., Piva B., Brunner H. P., Glatz Y., Zapf J., Follath F., Froesch E. R., Kiowski W. (1998b). Acute cardiovascular effects of insulin-like growth factor I in patients with chronic heart failure. J. Clin. Endocrinol. Metab. 83, 3177–3183 [DOI] [PubMed] [Google Scholar]
- Duerr R. L., Huang S., Miraliakbar H. R., Clark R., Chien K. R., Ross J., Jr (1995). Insulin-like growth factor-1 enhances ventricular hypertrophy and function during the onset of experimental cardiac failure. J. Clin. Invest. 95, 619–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duisters R. F., Tijsen A. J., Schroen B., Leenders J. J., Lentink V., van der Made I., Herias V., van Leeuwen R. E., Schellings M. W., Barenbrug P., et al. (2009). miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ. Res. 104, 170–178 [DOI] [PubMed] [Google Scholar]
- Ehler E., Horowits R., Zuppinger C., Price R. L., Perriard E., Leu M., Caroni P., Sussman M., Eppenberger H. M., Perriard J. C. (2001). Alterations at the intercalated disk associated with the absence of muscle LIM protein. J. Cell Biol. 153, 763–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endl E., Gerdes J. (2000). Posttranslational modifications of the KI-67 protein coincide with two major checkpoints during mitosis. J. Cell Physiol. 182, 371–380 [DOI] [PubMed] [Google Scholar]
- Ferreira-Cornwell M. C., Luo Y., Narula N., Lenox J. M., Lieberman M., Radice G. L. (2002). Remodeling the intercalated disc leads to cardiomyopathy in mice misexpressing cadherins in the heart. J. Cell Sci. 115, 1623–1634 [DOI] [PubMed] [Google Scholar]
- Gary-Bobo G., Parlakian A., Escoubet B., Franco C. A., Clement S., Bruneval P., Tuil D., Daegelen D., Paulin D., Li Z., et al. (2008). Mosaic inactivation of the serum response factor gene in the myocardium induces focal lesions and heart failure. Eur. J. Heart Fail. 10, 635–645 [DOI] [PubMed] [Google Scholar]
- Goldsmith E. C., Hoffman A., Morales M. O., Potts J. D., Price R. L., McFadden A., Rice M., Borg T. K. (2004). Organization of fibroblasts in the heart. Dev. Dyn. 230, 787–794 [DOI] [PubMed] [Google Scholar]
- Khan R., Sheppard R. (2006). Fibrosis in heart disease: understanding the role of transforming growth factor-beta in cardiomyopathy, valvular disease and arrhythmia. Immunology 118, 10–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koitabashi N., Arai M., Kogure S., Niwano K., Watanabe A., Aoki Y., Maeno T., Nishida T., Kubota S., Takigawa M., et al. (2007). Increased connective tissue growth factor relative to brain natriuretic peptide as a determinant of myocardial fibrosis. Hypertension 49, 1120–1127 [DOI] [PubMed] [Google Scholar]
- Miano J. M., Ramanan N., Georger M. A., de Mesy Bentley K. L., Emerson R. L., Balza R. O., Jr, Xiao Q., Weiler H., Ginty D. D., Misra R. P. (2004). Restricted inactivation of serum response factor to the cardiovascular system. Proc. Natl. Acad. Sci. USA 101, 17132–17137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muehlich S., Cicha I., Garlichs C. D., Krueger B., Posern G., Goppelt-Struebe M. (2007). Actin-dependent regulation of connective tissue growth factor. Am. J. Physiol. Cell Physiol. 292, C1732–C1738 [DOI] [PubMed] [Google Scholar]
- Niu Z., Yu W., Zhang S. X., Barron M., Belaguli N. S., Schneider M. D., Parmacek M., Nordheim A., Schwartz R. J. (2005). Conditional mutagenesis of the murine serum response factor gene blocks cardiogenesis and the transcription of downstream gene targets. J. Biol. Chem. 280, 32531–32538 [DOI] [PubMed] [Google Scholar]
- Niu Z., Li A., Zhang S. X., Schwartz R. J. (2007). Serum response factor micromanaging cardiogenesis. Curr. Opin. Cell Biol. 19, 618–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlakian A., Tuil D., Hamard G., Tavernier G., Hentzen D., Concordet J. P., Paulin D., Li Z., Daegelen D. (2004). Targeted inactivation of serum response factor in the developing heart results in myocardial defects and embryonic lethality. Mol. Cell. Biol. 24, 5281–5289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlakian A., Charvet C., Escoubet B., Mericskay M., Molkentin J. D., Gary-Bobo G., De Windt L. J., Ludosky M. A., Paulin D., Daegelen D., et al. (2005). Temporally controlled onset of dilated cardiomyopathy through disruption of the SRF gene in adult heart. Circulation 112, 2930–2939 [DOI] [PubMed] [Google Scholar]
- Pelosi L., Giacinti C., Nardis C., Borsellino G., Rizzuto E., Nicoletti C., Wannenes F., Battistini L., Rosenthal N., Molinaro N., et al. (2007). Local expression of IGF-1 accelerates muscle regeneration by rapidly modulating inflammatory cytokines and chemokines. FASEB J. 21, 1393–1402 [DOI] [PubMed] [Google Scholar]
- Ren J., Samson W. K., Sowers J. R. (1999). Insulin-like growth factor I as a cardiac hormone: physiological and pathophysiological implications in heart disease. J. Mol. Cell. Cardiol. 31, 2049–2061 [DOI] [PubMed] [Google Scholar]
- Sanchez-Lopez E., Rayego S., Rodrigues-Diez R., Rodriguez J. S., Rodriguez-Vita J., Carvajal G., Aroeira L. S., Selgas R., Mezzano S. A., Ortiz A., et al. (2009). CTGF promotes inflammatory cell infiltration of the renal interstitium by activating NF-kappaB. J. Am. Soc. Nephrol. 20, 1513–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini M. P., Tsao L., Monassier L., Theodoropoulos C., Carter J., Lara-Pezzi E., Slonimsky E., Salimova E., Delafontaine P., Song Y. H., et al. (2007). Enhancing repair of the mammalian heart. Circ. Res. 100, 1732–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz S., Pucilowska J. B., Liu S., Rodriguez-Ortigosa C. M., Lund P. K., Brenner D. A., Fuller C. R., Simmons J. G., Pardo A., Martinez-Chantar M. L., et al. (2005). Expression of insulin-like growth factor I by activated hepatic stellate cells reduces fibrogenesis and enhances regeneration after liver injury. Gut 54, 134–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholzen T., Gerdes J. (2000). The Ki-67 protein: from the known and the unknown. J. Cell Physiol. 182, 311–322 [DOI] [PubMed] [Google Scholar]
- Serose A., Prudhon B., Salmon A., Doyennette M. A., Fiszman M. Y., Fromes Y. (2005). Administration of insulin-like growth factor-1 (IGF-1) improves both structure and function of delta-sarcoglycan deficient cardiac muscle in the hamster. Basic Res. Cardiol. 100, 161–170 [DOI] [PubMed] [Google Scholar]
- Soulez M., Rouviere C. G., Chafey P., Hentzen D., Vandromme M., Lautredou N., Lamb N., Kahn A., Tuil D. (1996). Growth and differentiation of C2 myogenic cells are dependent on serum response factor. Mol. Cell. Biol. 16, 6065–6074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stritt C., Stern S., Harting K., Manke T., Sinske D., Schwarz H., Vingron M., Nordheim A., Knoll B. (2009). Paracrine control of oligodendrocyte differentiation by SRF-directed neuronal gene expression. Nat. Neurosci. 12, 418–427 [DOI] [PubMed] [Google Scholar]
- Sun Q., Chen G., Streb J. W., Long X., Yang Y., Stoeckert C. J., Jr, Miano J. M. (2006). Defining the mammalian CArGome. Genome Res. 16, 197–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandekerckhove J., Bugaisky G., Buckingham M. (1986). Simultaneous expression of skeletal muscle and heart actin proteins in various striated muscle tissues and cells. A quantitative determination of the two actin isoforms. J. Biol. Chem. 261, 1838–1843 [PubMed] [Google Scholar]
- Vinciguerra M., Santini M. P., Claycomb W. C., Ladurner A. G., Rosenthal N. (2010). Local IGF-1 isoform protects cardiomyocytes from hypertrophic and oxidative stresses via SirT1 activity. Aging (Albany NY) 2, 43–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., McLennan S. V., Allen T. J., Twigg S. M. (2010). Regulation of pro-inflammatory and pro-fibrotic factors by CCN2/CTGF in H9c2 cardiomyocytes. J. Cell Commun. Signal. 4, 15–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch S., Plank D., Witt S., Glascock B., Schaefer E., Chimenti S., Andreoli A. M., Limana F., Leri A., Kajstura J., et al. (2002). Cardiac-specific IGF-1 expression attenuates dilated cardiomyopathy in tropomodulin-overexpressing transgenic mice. Circ. Res. 90, 641–648 [DOI] [PubMed] [Google Scholar]
- Wong J., Zhang J., Yanagawa B., Luo Z., Yang X., Chang J., McManus B., Luo H. (2012). Cleavage of serum response factor mediated by enteroviral protease 2A contributes to impaired cardiac function. Cell Res. 22, 360–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Dewey S., Nguyen S., Gomes A. V. (2010). Malignant and benign mutations in familial cardiomyopathies: insights into mutations linked to complex cardiovascular phenotypes. J. Mol. Cell. Cardiol. 48, 899–909 [DOI] [PubMed] [Google Scholar]
- Zhang X., Chai J., Azhar G., Sheridan P., Borras A. M., Furr M. C., Khrapko K., Lawitts J., Misra R. P., Wei J. Y. (2001). Early postnatal cardiac changes and premature death in transgenic mice overexpressing a mutant form of serum response factor. J. Biol. Chem. 276, 40033–40040 [DOI] [PubMed] [Google Scholar]
- Zhou Y., Capuco A. V., Jiang H. (2008). Involvement of connective tissue growth factor (CTGF) in insulin-like growth factor-I (IGF1) stimulation of proliferation of a bovine mammary epithelial cell line. Domest. Anim. Endocrinol. 35, 180–189 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}