

SUMMARY
The size of the pancreas is determined by intrinsic factors, such as the number of progenitor cells, and by extrinsic signals that control the fate and proliferation of those progenitors. Both the exocrine and endocrine compartments of the pancreas undergo dramatic expansion after birth and are capable of at least partial regeneration following injury. Whether the expansion of these lineages relies on similar mechanisms is unknown. Although we have shown that the Wnt signaling component β-catenin is selectively required in mouse embryos for the generation of exocrine acinar cells, this protein has been ascribed various functions in the postnatal pancreas, including proliferation and regeneration of islet as well as acinar cells. To address whether β-catenin remains important for the maintenance and expansion of mature acinar cells, we have established a system to follow the behavior and fate of β-catenin-deficient cells during postnatal growth and regeneration in mice. We find that β-catenin is continuously required for the establishment and maintenance of acinar cell mass, extending from embryonic specification through juvenile and adult self-renewal and regeneration. This requirement is not shared with islet cells, which proliferate and function normally in the absence of β-catenin. These results make distinct predictions for the relative role of Wnt–β-catenin signaling in the etiology of human endocrine and exocrine disease. We suggest that loss of Wnt–β-catenin activity is unlikely to drive islet dysfunction, as occurs in type 2 diabetes, but that β-catenin is likely to promote human acinar cell proliferation following injury, and might therefore contribute to the resolution of acute or chronic pancreatitis.
INTRODUCTION
Of fundamental importance to developmental biology and medicine is the question of whether postnatal growth or regeneration of a tissue recapitulates the molecular mechanisms of its embryonic development. The answers are complex and variable across different tissues. For example, both bone formation and fracture repair require BMP-Smad signaling, although the relevant BMP ligands themselves differ between these processes (Rosen, 2009). By contrast, although the development of skeletal muscle progenitor cells requires Pax3 and Pax7, these transcription factors are dispensable for regeneration by adult muscle stem cells (Lepper et al., 2009).
Addressing these questions in the pancreas, a bifunctional organ comprising anatomically distinct endocrine and exocrine cell types, has implications for several devastating diseases, including diabetes, pancreatitis and pancreatic cancer. Lineage-tracing studies in the mouse have recently converged upon the finding that, although endocrine islet cells are generated de novo in the embryonic pancreas, their postnatal maintenance and replenishment relies instead on the proliferation of differentiated cells (Dor et al., 2004; Kopinke and Murtaugh, 2010; Solar et al., 2009). With respect to mechanism, several genes that are dispensable for embryonic islet development but are required for the expansion of insulin-producing β-cells after birth have been identified in mice (Chen et al., 2009; Georgia and Bhushan, 2004; Kushner et al., 2005; Rane et al., 1999; Zhang et al., 2006). A further distinction between physiological and regenerative growth is suggested by the regeneration-specific β-cell defect observed in mice lacking the Glp1r gene (De Leon et al., 2003). Such a distinction might also apply to exocrine acinar cells, in which genes including Notch1 and the hedgehog signaling component Smo are dispensable for normal development and homeostasis but are required for regeneration following caerulein-induced pancreatitis (Fendrich et al., 2008; Siveke et al., 2008). In the present study, we address the postnatal role of a gene that is required for acinar cell development, β-catenin (Ctnnb1) (Murtaugh et al., 2005; Wells et al., 2007).
Using the Cre-loxP system, we and others have previously shown that deletion of β-catenin, an essential component of the Wnt signaling pathway, abrogates acinar cell specification and development in mice (Murtaugh et al., 2005; Wells et al., 2007). We have further shown that this gene is dispensable for the survival and phenotypic maintenance of adult acinar cells, as well as for the function of adult insulin-producing β-cells (Murtaugh et al., 2005). Additional studies of Wnt–β-catenin signaling – using distinct methodologies – suggest a more complicated and context-dependent role for β-catenin. For example, other investigators have reported a pancreatitis-like phenotype in postnatal β-catenin knockout pancreata (Dessimoz et al., 2005; Morris et al., 2010; Wells et al., 2007), suggesting that this gene is required not only for differentiation of acinar cells but also for their normal maintenance. Depending on the experimental design, hyperactivation of Wnt–β-catenin signaling can cause pancreatic agenesis, acinar cell hyperplasia or islet cell hyperplasia (Heiser et al., 2006; Rulifson et al., 2007; Strom et al., 2007). Finally, inhibiting Wnt signaling in β-cells specifically is reported to impair their proliferation and glucose homeostasis function (Dabernat et al., 2009; Rulifson et al., 2007). These seemingly contradictory results suggest that the phenotype of β-catenin deletion in the pancreas is surprisingly dependent on the precise spatiotemporal domain of Cre activity.
With respect to the role of β-catenin in postnatal acinar cells, most previous studies have used Cre transgenes that are active throughout the developing pancreas, raising the possibility of non-cell-autonomous effects. By contrast, our own previous finding that β-catenin is dispensable in differentiated acinar cells was made in the uninjured adult pancreas (Murtaugh et al., 2005), where basal levels of proliferation and apoptosis are so low that alterations due to loss of β-catenin could go undetected. In the present study, we address the postnatal requirements for β-catenin under conditions of rapid as well as gradual cellular turnover and expansion. We find that β-catenin is indispensable for physiological and regenerative proliferation of acinar cells, but dispensable for their survival and phenotypic maintenance. Together, this work identifies a continuous requirement for β-catenin in acinar development, extending from specification in the embryo to self-renewal in the juvenile and regeneration in the adult.
RESULTS
Deletion of β-catenin in the adult pancreas does not compromise acinar cell maintenance
To establish a system in which the fate of β-catenin-deficient acinar cells could be directly and quantitatively compared to those of corresponding wild-type cells, we crossed mice carrying both a null allele of β-catenin and a deleter transgene, Elastase-CreERT (Murtaugh et al., 2005; Stanger et al., 2005) (Ctnnb1Δ/+; ElaCreERT mice), to mice that were double-homozygous for a conditional allele of β-catenin (Brault et al., 2001) and a Cre-dependent EYFP reporter construct (Srinivas et al., 2001) (Ctnnb1lox/lox; R26REYFP/EYFP mice). The ElaCreERT-containing offspring of this cross are genetically matched except for β-catenin: they are heterozygous either for the conditional and wild-type alleles (Ctnnb1lox/+; R26REYFP/+; ElaCreERT) or for the conditional and null alleles (Ctnnb1Δ/lox; R26REYFP/+; ElaCreERT). The former genotype permits tamoxifen-inducible EYFP labeling of acinar cells without β-catenin deficiency and is henceforth referred to as ‘control’. The latter permits simultaneous deletion of β-catenin and EYFP marking, and is henceforth referred to as ‘ABKO’, for acinar-specific β-catenin knockout. In principle, by comparing identically treated control and ABKO mice, we should detect a requirement for β-catenin in proliferation or survival as a relative decrease in the EYFP labeling indices of ABKO pancreata compared with those of controls.
To assess the functionality of this approach, we administered 10 mg tamoxifen to adult control or ABKO mice (‘pulse’) and analyzed EYFP labeling after 10 days (‘chase’). As described in the Methods, we used quantitative immunofluorescence to compare the EYFP labeling index, between genotypes, of amylase-expressing acinar cells. We found widespread EYFP expression (>50%) by acinar cells of both control and ABKO mice (Fig. 1A,B), with indistinguishable labeling indices between the genotypes (Fig. 1C). Immunofluorescence confirmed that EYFP-positive (EYFP+) acinar cells of ABKO pancreata were negative for β-catenin (Fig. 1D–G), allowing us to use EYFP as a surrogate marker for the deletion of β-catenin. Pancreata from control and ABKO mice that did not receive tamoxifen [harvested at postnatal day 60 (P60)] exhibited a low amount of acinar cell EYFP labeling [control (n=3): 4.8±2.3%; ABKO (n=3): 4.0±0.8%; P=0.8], resulting from leakiness of the ElaCreERT transgene. As expected from our previous work (Murtaugh et al., 2005), EYFP-labeled ABKO acinar cells were outwardly indistinguishable from controls, with normal expression of the epithelial junction protein E-cadherin (Fig. 1H–K) and normal polarity, as indicated by the apical localization of amylase-containing secretory granules (Fig. 1L–O).
Fig. 1.

Establishing a system to trace the fate of wild-type and β-catenin-deficient acinar cells. Control (Ctnnb1lox/+; R26REYFP/+; ElaCreERT) and acinar-specific β-catenin knockout (ABKO: Ctnnb1Δ/lox; R26REYFP/+; ElaCreERT) mice received tamoxifen at 2 months of age and were analyzed 10 days later by immunofluorescence on frozen sections. (A,B) EYFP (green) is widely expressed in amylase+ acinar cells (red), indicating successful recombination. Scale bar: 100 μm. (C) Overall EYFP labeling of acinar cells does not differ between genotypes, 10 days post-tamoxifen (n=2 mice per genotype; P=0.48). (D–G) In controls, β-catenin localizes normally to the cell membrane of EYFP+ acinar cells, whereas, in ABKO pancreata, EYFP marks cells that have lost β-catenin expression. (H–K) E-cadherin exhibits identical membrane localization in EYFP+ acinar cells of control and ABKO mice, suggesting that loss of β-catenin does not perturb cell-cell adhesion. (L–O) Expression and apical localization of amylase is identical in control and β-catenin-deficient cells, indicating that loss of β-catenin does not affect acinar polarity or differentiation state.
These results indicate that our experimental design can be used to compare acinar cell maintenance between genotypes, in terms of both number and phenotype. They also confirm that β-catenin is dispensable for the survival of acinar cells and the maintenance of their differentiated state, contrasting with its essential requirement during development (Murtaugh et al., 2005).
β-catenin-deficient acinar cells do not contribute to postnatal expansion of the exocrine compartment
The adult mouse pancreas is a highly quiescent organ, and very few acinar cells would be expected to divide during the course of the preceding experiment (≤2% per day) (Magami et al., 2002; Teta et al., 2007). Acinar cells divide much more rapidly shortly after birth, as the entire organism grows in mass and cellularity (Magami et al., 2002), and we sought to investigate the role of β-catenin signaling during this expansion phase. To this end, we performed neonatal knockout experiments by administering tamoxifen (10 mg) to nursing mothers of newborn (P0) control and ABKO pups (Kopinke and Murtaugh, 2010), and analyzed the distribution of EYFP label in P7 infants and P30 juveniles.
The pancreata of neonatal ABKO mice were noticeably smaller than those of controls at P7 (Fig. 2A,B) but not at P30 (data not shown). This might indicate that loss of β-catenin affects acinar cell expansion only during the first few days of life, or else that compensatory growth of unrecombined acinar cells eventually makes up for the deficiency imposed by β-catenin deletion. The latter possibility was supported by quantification of EYFP-labeled acinar cells, which revealed a twofold reduction in labeled ABKO acinar cells at P7, compared with control littermates (Fig. 2C,D), increasing to a fivefold reduction by P30 (Fig. 2E–G). At the latter stage, most acinar cells in ABKO pancreata remained EYFP negative and retained β-catenin expression (Fig. 3A,B). These data indicate that β-catenin function is required for the normal expansion of postnatal acinar cell mass.
Fig. 2.
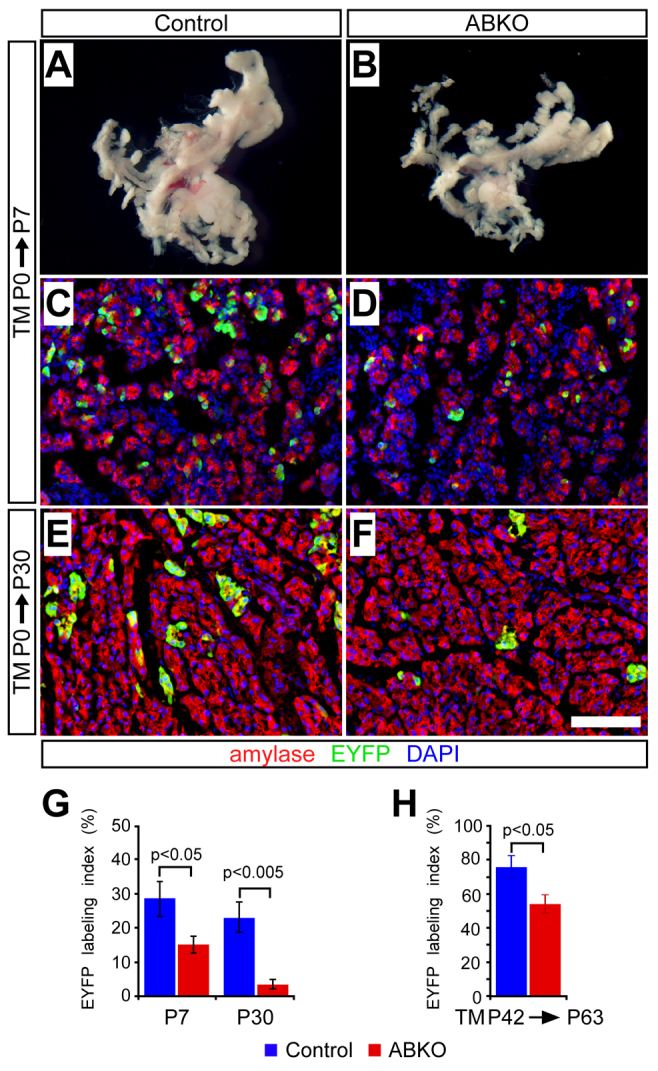
β-catenin is required for physiological expansion of acinar cells. Control or ABKO mice received tamoxifen (TM) either on the day of birth, via maternal gavage, or as adults (P42), and were analyzed as neonates (P7), juveniles (P30) and adults (P63) (n=4–7 mice per genotype per time point). (A,B) At P7, pancreata of TM-treated ABKO mice are visibly smaller than those of control littermates (photographs taken at identical magnification). (C–F) Staining for EYFP lineage labeling (green) of amylase+ acinar cells (red) reveals relatively fewer EYFP+ cells in ABKO pancreata at P7 (C,D) and P30 (E,F), compared with controls. Scale bar: 100 μm. (G) The EYFP labeling index of amylase+ acinar cells is decreased at both time points in young ABKO mice, indicating that β-catenin-deficient acinar cells do not efficiently contribute to postnatal expansion of the exocrine pancreas. (H) Control and ABKO mice received tamoxifen at 6 weeks of age (P42) and were assayed for EYFP expression after a 3-week chase (P63). The EYFP labeling index of amylase+ acinar cells is decreased in ABKO pancreata, consistent with a requirement for β-catenin in the slow homeostatic expansion of acinar cells.
Fig. 3.
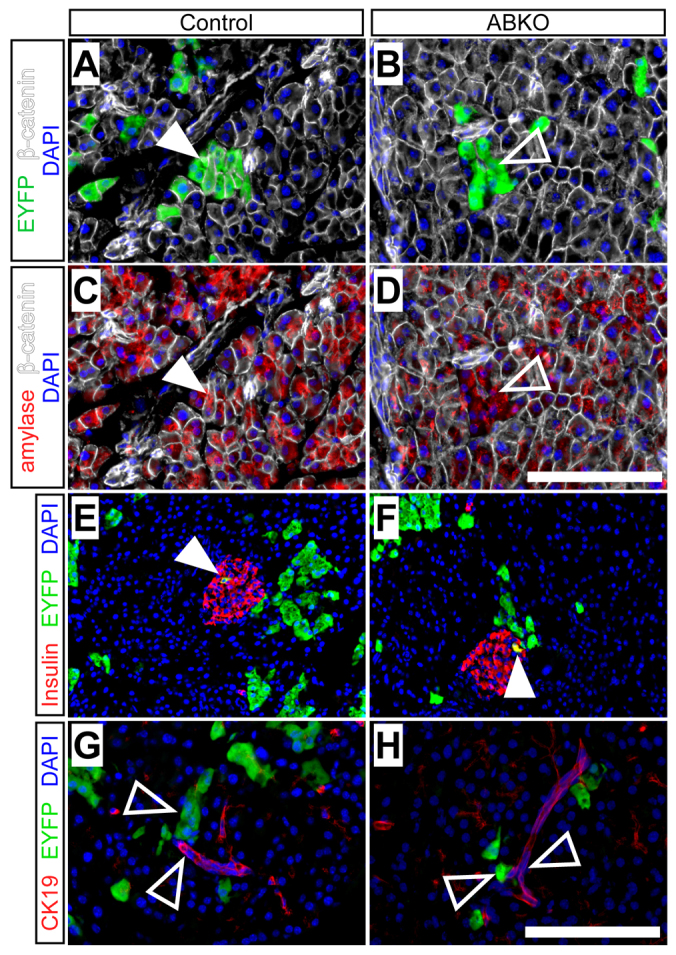
Acinar cells maintain normal differentiation status in the absence of β-catenin. Control or ABKO mice received tamoxifen at birth and were analyzed as juveniles (P30). (A,B) At P30, EYFP+ cells (green) co-express β-catenin (white) in control but not ABKO pancreata (closed and open arrowheads, respectively). (C,D) Both control and β-catenin-deficient ABKO pancreata maintain normal cytoplasmic amylase expression (red). (E,F) Leaky recombination within insulin+ β-cells (red) by ElaCreERT produces similar islet labeling in control and ABKO pancreata (arrowheads). (G,H) EYFP+ cells of control and ABKO pancreata do not co-express the duct marker CK19 (red). Arrowheads indicate adjacent, non-overlapping EYFP+ and CK19+ cells. Scale bars: 100 μm.
The relative deficiency of EYFP+ acinar cells, in the ABKO background, could be caused by any of several cellular defects, including loss of differentiation state, increased cell death and decreased proliferation. Although, under normal physiological conditions, adult acinar cells do not transdifferentiate into duct or islet cells (Desai et al., 2007; Strobel et al., 2007), they can adopt a duct-like phenotype during cell culture (De Lisle and Logsdon, 1990; Means et al., 2005), and in response to oncogenic and mitogenic signals in vivo (Blaine et al., 2010; De La O et al., 2008; Habbe et al., 2008; Ji et al., 2009; Morris et al., 2010). However, this did not seem to be the case for neonatal acinar cells that had lost β-catenin: as was found in the short-term adult labeling experiment described above (Fig. 1), EYFP expression in P30 juvenile knockout pancreata identified cells that lacked β-catenin but retained normal expression of the acinar marker amylase (Fig. 3A–D). Furthermore, although ElaCreERT induces a low level of ectopic recombination in insulin+ β-cells (Strobel et al., 2007), we found no significant difference in the fraction of EYFP-labeled β-cells between control and ABKO pancreata [control (n=2): 3.4±0.3% EYFP labeling of insulin+ cells; ABKO (n=2): 5.1±0.4% labeling of insulin+ cells; P=0.09], suggesting that β-catenin-deficient acinar cells had not transdifferentiated to islets (Fig. 3E,F). Finally, EYFP+ cells did not express the ductal marker CK19 in either control or ABKO backgrounds (Fig. 3G,H). These data indicate that, although β-catenin is needed for acinar development in utero, its deletion after birth does not divert acinar cells to an alternative fate, but instead prevents their normal expansion during juvenile growth.
β-catenin is required for proliferation of neonatal acinar but not islet cells
To determine whether the requirement for β-catenin in acinar expansion was manifested at the level of cell division, we compared control and β-catenin-deficient acinar cells for expression of the proliferation marker Ki67. Immunostaining revealed a fourfold relative decrease, compared with controls, in the fraction of EYFP-labeled acinar cells staining for Ki67 in ABKO pancreata at P7 (Fig. 4). Although a compensatory increase in proliferation might be expected among those acinar cells that retain β-catenin in the ABKO pancreas, we found no significant change in the Ki67+ fraction of EYFP-negative (EYFP–) acinar cells between control (30.3±2.6%; n=5) and ABKO (30.5±1.4%; n=7; P=0.9) pancreata at this stage. It will be interesting to determine whether β-catenin-retaining acinar cells of ABKO pancreata exhibit a higher proliferation index at later stages, when overall proliferation declines in controls. In contrast to the dramatic proliferation phenotype of β-catenin-deficient cells, we found that EYFP+ acinar cells in ABKO pancreata were not subject to increased rates of apoptosis when compared with controls, as indicated by a similar (and very low) proportion of EYFP+ acinar cells co-expressing the apoptosis marker cleaved caspase-3 (cCasp3) at P7 [control (n=2): 0.5±0.05%; ABKO (n=2): 0.7±0.4%; P=0.7]. Because decreased cell division, even without increased death, would suffice to explain the impaired expansion of ABKO cells, we conclude that proliferation represents the major requirement for β-catenin in neonatal acinar cells.
Fig. 4.

Neonatal and adult acinar cell proliferation requires intact β-catenin function. To determine whether β-catenin is required for acinar cell proliferation in the neonatal and adult pancreas, we pulsed mice with tamoxifen on the day of birth, via maternal gavage, or as adults (P42), and analyzed neonatal (P7) and adult (P63) pancreata, respectively. Prior to sacrifice, adult mice received a 7-day BrdU pulse to label proliferating cells. (A,B) Staining for EYFP+ acinar cells (green) reveals a reduction in β-catenin-deficient acini in ABKO pancreata. (C,D) The fraction of proliferating EYFP+ acinar cells, marked by anti-Ki67 antibody labeling (white arrowheads), is markedly decreased in ABKO mice (open arrowhead indicates Ki67-negative/EYFP+ acinar cells) compared with controls. Scale bar: 100 μm. (E) Quantification of the Ki67 labeling index of EYFP+ cells reveals a significant reduction in the proliferative capacity of β-catenin-deficient acini compared with controls (n=5–7 mice per genotype). (F) Staining for BrdU revealed an approximately tenfold reduction in the fraction of BrdU-labeled EYFP+ cells in ABKO mice compared with controls, indicating a loss of proliferative ability in the resting adult pancreas (n=4–5 mice per genotype).
Although β-catenin is needed to generate acinar cells in the embryonic pancreas, it is dispensable for islet cell specification (Murtaugh et al., 2005). As noted in the Introduction, however, various studies have suggested a role for Wnt–β-catenin in postnatal islet growth or function (Dabernat et al., 2009; Liu and Habener, 2008; Rulifson et al., 2007). To determine whether islet and acinar cells share a requirement for β-catenin in proliferation, we deleted β-catenin in endocrine precursors specifically using a neurogenin-3 (Ngn3)-Cre BAC transgene (Schonhoff et al., 2004). The resulting islet-specific β-catenin knockout (IsBKO) mice were recovered at weaning in normal mendelian ratios, and exhibited neither impaired survival nor glucose intolerance through ≥1 year of age (supplementary material Fig. S1). We analyzed control and IsBKO pancreata for proliferation at P7, when islet as well as acinar cells undergo rapid proliferative expansion (Magami et al., 2002). Staining of IsBKO pancreata at this stage revealed normal organization of insulin+ β-cells and glucagon+ α-cells into islets devoid of β-catenin protein (Fig. 5A–H). Furthermore, β-catenin-deficient islet cells proliferated indistinguishably from controls, as indicated by BrdU labeling (Fig. 5I–K). These results support our prior finding that pan-pancreatic β-catenin deletion did not impair adult islet function (Murtaugh et al., 2005) and suggest that, as is the case in utero, the major functional requirement for β-catenin after birth is specific to acinar cells. This specificity also indicates that β-catenin does not represent a ‘housekeeping gene’ for cell cycle progression that is required in all cell types.
Fig. 5.

Islet cells develop and proliferate independently of β-catenin status. Neonatal (P7) control (Ctnnb1lox/+; Ngn3-Cre) and islet-cell-specific β-catenin knockout (IsBKO: Ctnnb1Δ/lox; Ngn3-Cre) mice were administered BrdU 1 hour prior to sacrifice and analyzed by immunofluorescence. (A–D) Islets of control mice exhibit clear β-catenin staining (red) in both insulin+ β-cells (green) and glucagon+ α-cells (white). Dashed lines in A indicate the area magnified in B–D. White and yellow arrowheads indicate insulin+ and glucagon+ cells, respectively, expressing β-catenin (closed; B–D) or lacking β-catenin (open; F–H). (E–H) Islet morphology and marker expression is normal in IsBKO pancreata, despite loss of β-catenin in β- and α-cells. (I,J) Anti-BrdU staining (red) reveals a similar distribution of proliferating cells among control and IsBKO endocrine cells. Arrowheads denote BrdU+/insulin+ β-cells. (K) Quantification reveals no significant difference in BrdU labeling of β-cells between control and IsBKO mice (n=2 mice per genotype; P=0.97). Scale bar: 100 μm.
β-catenin is required for homeostatic proliferation of acinar cells in the adult pancreas
Whereas the juvenile pancreas grows rapidly, in parallel with the organism, the adult pancreas grows slowly and exhibits little turnover. Nonetheless, rare division of acinar cells can be observed in the adult (Magami et al., 2002; Teta et al., 2007), raising the question of whether this low level of proliferation also depends on β-catenin function. To determine whether β-catenin is required for acinar cell proliferation in the normal adult pancreas, we pulsed control and ABKO mice with tamoxifen at 6 weeks of age and assayed EYFP expression and proliferation at 9 weeks of age. To maximize the detection of rare S-phase events, we administered BrdU via the drinking water for 7 days prior to sacrifice, thereby capturing a week of cell proliferation in the adult pancreas (Teta et al., 2007). We found a modest but significant decrease in the fraction of EYFP+ acinar cells in ABKO pancreata compared with controls (Fig. 2H), suggesting that loss of β-catenin prevents normal expansion of acinar cells. More dramatically, staining for BrdU revealed a tenfold reduction in the fraction of BrdU+ EYFP+ β-catenin-deficient acinar cells compared with controls (Fig. 4F). Thus, we conclude that β-catenin is required not only for rapid acinar expansion in the juvenile pancreas, but also for steady-state proliferation in the adult.
Loss of β-catenin does not sensitize adult acinar cells to pancreatitis-associated injury
The pancreas is capable of at least partial regeneration from a variety of injuries, including acute pancreatitis induced by supraphysiological levels of the acinar secretagogue caerulein (Willemer et al., 1992). Regeneration in this model is preceded by transient expression of progenitor-like markers, including Pdx1 (Fendrich et al., 2008; Jensen et al., 2005; Morris et al., 2010), suggesting that re-establishment of acinar cell mass involves a genetic program similar to that of embryonic acinar development. Indeed, it is reported that pancreas-specific β-catenin knockout mice (generated using a Ptf1aCre deleter strain) exhibit an almost complete loss of acinar cells after caerulein treatment (Morris et al., 2010). To determine the cell-autonomous requirement for β-catenin during injury and regeneration, we induced a pulse of acinar-specific recombination by tamoxifen treatment (10 mg) of adult control and ABKO mice, waited 1 week, and induced pancreatitis by a 2-day course of caerulein treatment (Jensen et al., 2005).
Whereas saline-treated pancreata appeared normal at all time points, pancreata of mice that were administered caerulein exhibited clear abnormalities at 2 days post-treatment, including fibroblast and leukocyte infiltration (Fig. 6A–D), and acinar-ductal metaplasia indicated by co-expression of amylase with the duct marker CK19 (supplementary material Fig. S2). Previous studies have demonstrated that acinar cells undergoing pancreatitis-induced metaplasia upregulate Pdx1, a transcription factor normally expressed in progenitor cells, while maintaining expression of Ptf1a, a master regulator of acinar cell identity (Jensen et al., 2005; Molero et al., 2007). Both of these phenomena were observed identically between control and β-catenin-deficient acinar cells, confirming that loss of β-catenin does not disrupt the differentiation state of regenerating acinar cells (supplementary material Fig. S3). Importantly, there was also no difference in the fraction of EYFP+ acinar cells between ABKO and control mice at the 2-day post-caerulein time point, indicating that loss of β-catenin did not affect the ability of acinar cells to survive pancreatitis-associated injury (Fig. 6E–I).
Fig. 6.

β-catenin-deficient acinar cells are not preferentially injured during caerulein-induced pancreatitis. Control and ABKO mice received tamoxifen at 2 months of age and were treated with caerulein 1 week later to induce acute pancreatitis. The extent of acinar cell regeneration was analyzed histologically on wax sections, and quantitatively by immunofluorescence on frozen sections. (A–D) 2 days after the injection series was complete, H&E staining revealed no abnormalities in the pancreata of saline-treated mice, and a similar appearance of overall injury between caerulein-treated control and ABKO mice. At this stage, acute pancreatitis is manifested by infiltrating fibroblasts and inflammatory cells (black arrowheads) and the presence of dilated, metaplastic acini (open arrowheads). is, islet; du, duct. (E–H) A similar extent and distribution of EYFP labeling (green) is observed among amylase+ acinar cells (red) of saline- and caerulein-treated mice, regardless of genotype. (I) Quantification reveals no significant difference in the percentage of R26REYFP-labeled acinar cells, between treatments or genotypes (n=4–5 mice per genotype per treatment group). (J–L) At 2 days after caerulein treatment, double-staining for EYFP (green) and the apoptosis marker cleaved caspase-3 (red) reveals no increase in apoptosis among R26REYFP-labeled acinar cells of control and ABKO mice (n=3 mice per genotype). White arrowheads indicate cCasp3+ cells, co-expressing EYFP (closed) or unlabeled (open). Scale bars: 100 μm.
As an independent indicator of pancreatic injury, we assayed serum amylase levels before and after treatment, and found that caerulein-treated mice of both genotypes exhibited a transient increase in serum amylase 1 hour after the last caerulein injection, with a return to baseline 1 day later (supplementary material Fig. S4). Finally, staining for the apoptotic marker cCasp3 revealed quantitatively similar overlap with EYFP+ acinar cells in control and ABKO pancreata, indicating that pancreatitis-associated injury was not more likely to trigger apoptosis of β-catenin-deficient acinar cells than in control cells (Fig. 6J–L). Taken together, our results indicate that pancreatitis is not aggravated by deletion of β-catenin specifically in acinar cells, in contrast to a previous study of mice with pan-pancreatic β-catenin deficiency (Morris et al., 2010).
β-catenin is required for acinar cell proliferation during regeneration
Previous studies have found substantially complete acinar regeneration within 7 days following caerulein treatment (Fendrich et al., 2008; Jensen et al., 2005). To look specifically at regeneration, we analyzed control and ABKO pancreata 14 days after saline or caerulein administration, from mice treated in parallel to those analyzed at the 2-day time point. The appearance of these pancreata was grossly indistinguishable between genotype and treatment groups, and the histology of 14-day post-caerulein ABKO pancreata was similar to that of caerulein-treated controls (Fig. 7A–D). This result differs from the almost complete involution and fatty replacement of acinar tissue found after caerulein treatment of mice with a pan-pancreatic deletion of β-catenin (using Ptf1aCre) (Morris et al., 2010).
Fig. 7.

β-catenin is required for regenerative expansion of acinar cells. (A–D) 14 days after treatment with saline or caerulein, H&E staining reveals comparable regeneration and resolution of fibrosis in control and ABKO mice subjected to acute pancreatitis. is, islet; du, duct. (E–I) EYFP labeling (green) is similar between saline and caerulein-treated control mice, whereas the fraction of EYFP+ acinar cells is dramatically reduced in ABKO pancreata (n=5–7 mice per genotype and treatment group; P<0.0006). (J–L) Control and ABKO mice were administered a 1-hour BrdU pulse, at 2 days post-caerulein treatment, sacrificed and analyzed for labeling. The fraction of BrdU+ R26REYFP-labeled cells is significantly reduced in ABKO pancreata, indicating decreased acinar cell proliferation following acute pancreatitis (n=3 mice per genotype; P<0.05). Scale bars: 100 μm.
Examination of EYFP labeling at the 14-day time point, however, revealed a dramatic decrease in the labeling index of ABKO acinar cells following regeneration (Fig. 7E–I). As in the neonatal labeling experiments, residual EYFP+ acinar cells were β-catenin deficient, yet exhibited a normal differentiated acinar phenotype (supplementary material Fig. S5).
In both genotypes, normalization of organ morphology was accompanied by resolution of metaplasia, as indicated by downregulation of the duct marker CK19 in EYFP+ acinar cells (supplementary material Fig. S2). As noted above, β-catenin-deficient and control acinar cells exhibited similar levels of apoptosis following caerulein administration, suggesting that cell death did not prevent mutant cells from contributing to regeneration. Instead, we found a major defect in proliferation: administering BrdU 1 hour prior to sacrifice revealed a dramatic decrease in S-phase cells among EYFP+/β-catenin-deficient acinar cells at 2 days post-caerulein treatment (Fig. 7J–L). As in juvenile mice, we did not observe evidence for a compensatory increase in the proliferation index of EYFP-negative acinar cells [control + caerulein (n=4): 1.8±0.26%; ABKO + caerulein (n=4): 1.6±0.35%; P=0.6]. The fraction of wild-type acinar cells incorporating BrdU was relatively low at this early time point, leaving open the possibility that an additional defect in survival, manifested at an intermediate time point not yet analyzed, could have contributed to the ultimate decrease in ABKO acinar cells at 14 days post-caerulein. Alternatively, the window of wild-type acinar cell proliferation might be extended when a substantial fraction of the pancreas comprises ABKO cells, such that even a relatively low wild-type proliferation rate could produce dramatic out-competition of ABKO cells. In any event, the overall agreement between these results and our findings in neonatal and uninjured adult mice indicate a continuous requirement for β-catenin in acinar cell proliferation, during both physiological and regenerative growth.
DISCUSSION
During postnatal growth and regeneration of the pancreas, new acinar cells are generated almost exclusively from the division of pre-existing acinar cells (Desai et al., 2007; Strobel et al., 2007). β-catenin has an established role in the embryonic development of acinar cells (Murtaugh et al., 2005; Wells et al., 2007); however, whether this requirement applies after birth has not been addressed. Here we demonstrate a continuous requirement for β-catenin in the establishment and maintenance of acinar cell mass, extending from organogenesis in the embryo through expansion and homeostasis in the juvenile and adult organ, as well as regeneration following injury.
To our knowledge, this is the first study to interrogate the specific requirement for β-catenin in adult acinar cells. In several respects, our findings extend those of others and ourselves. First, we and others have shown that β-catenin deletion during organogenesis reduces acinar cell number at birth and beyond (Morris et al., 2010; Murtaugh et al., 2005; Wells et al., 2007). Here, we demonstrate a continued requirement for β-catenin after birth, operating at the level of proliferation rather than specification or differentiation. Recent studies indicate that acinar-like ‘tip cells’ of the embryonic pancreas represent multipotent progenitors that contribute to islets and ducts for the first several days of pancreatic organogenesis (Zhou et al., 2007). It is tempting to speculate that immature tip cells also require β-catenin for proliferative expansion, the failure of which partly explains the embryonic β-catenin deletion phenotype.
As well as acinar cells, we investigated the role of β-catenin in postnatal islet cells; none was detectable. This result agrees with prior studies of adult, pan-pancreatic β-catenin knockout mice, which exhibit normal glucose homeostasis (Dessimoz et al., 2005; Murtaugh et al., 2005; Wells et al., 2007), but contradicts the emerging hypothesis that β-catenin promotes proliferation or function of insulin-producing β-cells (Liu and Habener, 2010; Welters and Kulkarni, 2008). Evidence supporting this hypothesis includes β-cell hypoplasia following misexpression of the β-catenin inhibitor Axin (Rulifson et al., 2007), and perinatal lethality observed when β-catenin is deleted with an insulin-promoter-driven Cre transgene (RIP-Cre) (Dabernat et al., 2009). Both of these experiments have the potential for non-specific effects, however. In addition to β-catenin, Axin has been found to inhibit activin–TGF-β signaling through Smad3 (Guo et al., 2008), the latter of which is required for postnatal β-cell expansion (Smart et al., 2006). The RIP-Cre transgene, meanwhile, has been shown to induce widespread recombination in the brain (Wicksteed et al., 2010) and to confer metabolic phenotypes independent of deletion in β-cells (Lee et al., 2006). In our hands, Ngn3-Cre drives highly efficient deletion of β-catenin in islet cells without apparent phenotype, suggesting that results obtained with RIP-Cre must be interpreted cautiously.
Human genetics provides indirect evidence linking Wnt–β-catenin signaling to islet function, because non-coding single-nucleotide polymorphisms (SNPs) in TCF7L2, a transcription factor partner of β-catenin, are associated with type-2 diabetes risk (Florez, 2007). Because distinct TCF7L2 isoforms can promote or inhibit Wnt signaling, including within islet cells (Le Bacquer et al., 2011; Tang et al., 2008; Vacik et al., 2011), it is hard to predict how TCF7L2 polymorphisms might modulate Wnt pathway output, and whether the effects relevant to diabetes are played out in β-cells or in peripheral tissues. Although our results indicate that Wnt–β-catenin signaling is not essential to β-cell function, it will be important to determine whether β-catenin-deficient islet cells adapt normally to metabolic stressors implicated in type-2 diabetes.
Our finding that β-catenin is required differentially in mouse acini and islets emphasizes its cell-type-dependent role in proliferation. Gain-of-function studies similarly indicate that hyperactivation of β-catenin signaling promotes acinar but not islet expansion (Heiser et al., 2006; Strom et al., 2007). Although information on growth control in the human pancreas remains scarce, autopsy-based studies suggest that β-cell and exocrine mass expand with distinct kinetics during childhood (Meier et al., 2008), supporting the hypothesis that proliferation in these compartments is controlled by different mechanisms. Although some aspects of organ growth control might be species specific, our finding that β-catenin is required for all phases of mouse acinar proliferation, from neonatal growth to adult homeostasis and regeneration, suggests that this action of β-catenin is fundamental and ancestral. Studies of rare pancreatic cancer subtypes provide direct evidence for a conserved and acinar-specific role of β-catenin in humans: mutational activation of β-catenin signaling is observed in acinar cell carcinomas but not insulinomas (Abraham et al., 2002), suggesting that the former cancer exploits the proliferation control circuits of its untransformed precursors.
The cell of origin for acinar carcinoma remains unknown, and the activation of β-catenin in this cancer could reflect a role for this pathway in promoting acinar cell identity rather than proliferation per se. Our studies, however, indicate that β-catenin is not required for the phenotypic maintenance of acinar cells, even following injury. During experimental pancreatitis, in particular, we find that β-catenin-deficient and control acinar cells behave similarly in all respects except proliferation. By contrast, Morris et al. found that mice with pan-pancreatic β-catenin deletion, driven by Ptf1aCre, exhibit a more severe response to experimental pancreatitis, including sustained loss of acinar cells (Morris et al., 2010). The difference between this model and ours could reflect the fact that the Ptf1aCre produces complete β-catenin ablation in acinar cells, whereas a considerable fraction of acinar cells retains β-catenin in ABKO mice. Alternatively, the more severe phenotypes observed with Ptf1aCre could reflect recombination in non-acinar cells, including ducts (Kawaguchi et al., 2002). Intact duct cell function is required for maintenance of the entire exocrine compartment, as indicated by deletion of the ciliogenesis regulator Kif3a (Cano et al., 2006). Although primary cilia are found only on duct cells, their ablation produces a pancreatitis-like phenotype that includes non-cell-autonomous acinar cell death. Interestingly, Kif3a knockout mice exhibit fat infiltration into the exocrine pancreas, a phenotype that is frequently seen in severe human pancreatitis (Bockman, 1997) and which can be observed even in uninjured, pan-pancreatic β-catenin knockout mice (Morris et al., 2010; Wells et al., 2007) (L.C.M., unpublished data). We have never observed this phenotype in ABKO mice, suggesting that it reflects a role of β-catenin in multipotent progenitor or mature duct cells, rather than a cell-autonomous function in maintaining acinar survival or differentiation.
The downstream effectors of β-catenin in acinar cells remain undefined, although the Wnt–β-catenin target gene Myc is a strong candidate because its deletion abolishes the exocrine hyperplasia of Apc knockout mice (Strom et al., 2007). The apparently reciprocal phenotypes observed upon deletion and activation of β-catenin suggest that it acts as a signaling molecule in the pancreas, although we note that β-catenin can function independently of Wnt ligands (Nelson and Nusse, 2004). Although additional experiments will be required to determine the precise role of Wnt proteins per se in acinar proliferation, small-molecule modulators of β-catenin signaling are increasingly available (Rey and Ellies, 2010) and could be clinically useful. Human pancreatitis, both acute and chronic, is associated with acinar cell proliferation (Ebert et al., 1999; Zimmermann et al., 2002). By enhancing proliferation, pharmacological β-catenin agonists might enhance regeneration and improve the outcome of acute pancreatitis. By contrast, clinical observations indicate that the pain of chronic pancreatitis will occasionally decline in parallel with a decline in acinar cell function, possibly due to the ‘burn-out’ of residual acinar cells (Sakorafas et al., 2007). By inhibiting regeneration, β-catenin antagonists might accelerate this phenomenon, and perhaps limit overall disease severity. Given that Wnt–β-catenin signaling is active and important in other adult tissues, such as the intestine, effective interventions might require identifying pancreas-specific effectors of this pathway. Nonetheless, the widely used psychiatric agent lithium is now recognized as a potent activator of β-catenin, owing to its inhibition of the GSK3 kinase (O’Brien and Klein, 2009). The fact that patients tolerate long-term lithium treatment suggests that a degree of Wnt–β-catenin modulation can be achieved without prohibitive side effects.
We note that the specific and circumscribed phenotypes of ABKO mice contrast with those of knockouts in other developmental signaling cascades, including Notch and Hedgehog. Inhibiting these pathways, in the context of pancreatitis, causes increased cell death and prevents the resolution of metaplasia (Fendrich et al., 2008; Siveke et al., 2008), implying that small molecules targeting Notch or Hedgehog will have pleiotropic effects, including the alteration of differentiation states. We and others have shown that acinar cells transdifferentiate during Kras-induced initiation of pancreatic ductal adenocarcinoma, and that this process is accelerated by pancreatitis (Carriere et al., 2009; De La O et al., 2008; De La O and Murtaugh, 2009; Habbe et al., 2008; Ji et al., 2009; Morris et al., 2010), emphasizing the importance of studying the mechanisms controlling growth and maintenance of this cell type. Understanding the cell-autonomous and non-autonomous roles of β-catenin in acinar cell expansion, injury and regeneration might shed light on the linked etiologies of pancreatitis and pancreatic cancer, as well as identify a molecular pathway that could be harnessed in treating these generally intractable conditions.
METHODS
Mouse breeding and genetic manipulation
All mouse experiments were performed according to a protocol approved by the University of Utah IACUC. The following mice were obtained from the Jackson Laboratories: floxed and germline β-catenin loss-of-function mice (Ctnnb1tm2Kem/J and Ctnnb1tm2.1Kem; referred to here as Ctnnb1lox and Ctnnb1Δ, respectively) (Brault et al., 2001); the Cre-dependent EYFP reporter strain Gt(ROSA)26Sortm1(EYFP)Cos (Srinivas et al., 2001), referred to here as R26REYFP; and Ngn3Cre BAC transgenic mice (Schonhoff et al., 2004). Pdx1Cre and Elastase-CreERT (ElaCreERT) transgenic mice (Gu et al., 2002; Murtaugh et al., 2005; Stanger et al., 2005) were provided by Doug Melton (Harvard University, MA). Ctnnb1Δ and Cre transgenic lines were maintained by outcrossing to CD-1 wild-type mice, whereas the Ctnnb1lox and R26REYFP alleles were maintained by inbreeding on a mixed CD-1 × C57BL/6 background. Unless otherwise noted, experiments were performed in young adult mice, 8–12 weeks in age at the outset of any treatment. To activate recombination by ElaCreERT, we administered tamoxifen (Sigma), dissolved in corn oil, by oral gavage.
Glucose tolerance tests
Mice were fasted overnight with access to water and injected with D-glucose at 2 mg/g body weight. Blood was drawn from a tail incision before glucose injection and at indicated time points post-injection, and read with an Ascencia Contour glucometer (Bayer). Net area-under-curve (AUC) values were calculated by the trapezoidal rule. Results are reported as mean ± s.e.m.
Caerulein treatment
We induced acute pancreatitis by caerulein treatment of 2-month-old male and female mice, conditionally null for β-catenin (Ctnnb1Δ/lox; R26REYFP/+; ElaCreERT) as well as control littermates (Ctnnb1+/lox; R26REYFP/+; ElaCreERT). Following an established protocol (Jensen et al., 2005), mice received repeated intraperitoneal (i.p.) injections of caerulein (Bachem; 0.1 μg/g in 0.8% NaCl) eight times daily over 2 days (16 injections total). Negative controls were injected in parallel with saline alone. We refer to the last day of injections as ‘day 0’, such that mice sacrificed 48 hours after the last injection would constitute the 2-day post-treatment group. To monitor serum amylase levels, cheek bleeds of approximately 100μl were drawn, chilled on ice and clarified by centrifugation. Serum samples were diluted 1:4 with PBS, and 7 μl of diluted serum mixed with 280 μl Infinity serum amylase reagent (Thermo-Fisher) prior to analysis on a VMax Kinetic microplate reader (Molecular Devices).
Tissue processing and staining
Mice were euthanized with isoflurane, and tissues were dissected into ice-cold PBS for further processing. BrdU labeling was used to identify proliferating cells, either by injecting mice with BrdU (50 μg/g body weight) at 1 hour prior to sacrifice, or by administering it in the drinking water (1 mg/ml) over 7 days. Pancreata were dissected into multiple fragments and fixed either for paraffin sections [zinc-buffered formalin (Polysciences), overnight] or frozen sections (4% paraformaldehyde/PBS; 4°C 2–4 hours), followed by processing as previously described (Kopinke and Murtaugh, 2010; Murtaugh et al., 2005). Paraffin sections were cut at 6 μm thickness and collected serially such that sections were spaced approximately 50–80 μm apart, spanning the entire specimen. Frozen sections were cut at 8 μm thickness, and similarly collected such that sections were spaced approximately 60–100 μm apart, spanning the entire specimen.
Primary antibodies used for immunostaining are listed in supplementary material Table S1, and secondary antibodies (raised in donkey) were obtained from Jackson ImmunoResearch. Immunohistochemistry and immunostaining were performed as previously described (Kopinke and Murtaugh, 2010; Murtaugh et al., 2005), with all paraffin sections subjected to high-temperature antigen retrieval. To detect Ki67 and BrdU, sections were subjected to digestion with DNAse I (700 u/μl, in 40 mM Tris-HCl pH 7.4, 10 mM NaCl, 6 mM MgCl2, 10 mM CaCl2) for 15 minutes at room temperature (Ye et al., 2007). Slides stained by immunofluorescence were mounted with Fluoromount-G (Southern Biotech) and imaged on an Olympus IX71 microscope. Photomicrographs were produced with MicroSuite software (Olympus) and processed in Adobe Photoshop, with exposure times and adjustments identical between treatment groups.
TRANSLATIONAL IMPACT.
Clinical issue
Understanding how growth and regeneration of the pancreas are controlled should provide new insights into the etiology and treatment of diseases that affect this organ, including diabetes, pancreatic cancer and pancreatitis. The latter two of these seem to be linked: recent work indicates that pancreatitis can promote acinar cell reprogramming into pancreatic ductal tumors. The Wnt signaling pathway (in which β-catenin is a key mediator) is a crucial regulator of embryonic acinar cell development, but its role in the mature pancreas is more controversial. The numerous proposed roles of β-catenin in both endocrine and exocrine cells of the pancreas are complex and controversial. Some conflicting results have been obtained by manipulating the mouse β-catenin gene with various Cre deleter transgenes, most of which are active in multipotent progenitor cells. In this study, the authors sought to strictly define the genetic requirement for β -catenin in the postnatal pancreas, particularly in establishing, maintaining and regenerating its most abundant constituent, the acinar cell.
Results
The authors developed a quantitative ‘pulse-chase’ knockout and labeling approach to analyze β-catenin function specifically in differentiated acinar cells during their expansion and regeneration. Using this system, they find that β-catenin-deficient acinar cells are dramatically impaired in their ability to proliferate during normal juvenile growth and adult homeostasis, as well as during regeneration from injury caused by experimental pancreatitis. However, β-catenin deletion in acinar cells does not cause cell death or dedifferentiation, nor render the organ more vulnerable to injury. Importantly, the requirement of β-catenin for proliferation seems to be acinar cell specific, because its deletion in islet cells does not detectably impair their expansion or metabolic function.
Implications and future directions
These results clarify the cell-type specificity of β-catenin function in vivo, which can be detected only through a lineage-restricted deletion approach. The Wnt–β-catenin pathway has been implicated in regeneration and tumorigenesis of other tissues, and it is becoming increasingly amenable to pharmacological manipulation as new drugs are developed. For example, small-molecule activators of β-catenin signaling already exist, and this study suggests that they could be useful in accelerating recovery from acute pancreatitis. By contrast, β-catenin inhibitors might prevent the continuous regeneration that sustains chronic pancreatitis. The specificity of β-catenin action implies that such interventions will not affect differentiation state, and therefore should not increase cancer risk. Given that the absence of β-catenin did not disrupt islet cell growth or function, these results also suggest that type-2-diabetes-associated mutations in TCF7L2 (encoding a β-catenin binding partner) might influence diabetes risk via functions other than in the pancreas. Future studies will focus on the molecular mechanisms upstream and downstream of β-catenin in acinar cells, and address the translational potential of β-catenin-focused intervention in disease models.
Image quantification and analysis
To determine R26REYFP labeling indices, we photographed six to ten independent fields (20× or 40× original magnification) per specimen, across multiple sections. Using ImageJ software (NIH), cells co-expressing a given differentiation marker with EYFP were detected by additive image overlay of the DAPI channel with anti-GFP and anti-marker immunofluorescence, and counted using the Analyze Particles function, as described previously (Kopinke and Murtaugh, 2010). Random samples were scored manually in Adobe Photoshop, to confirm overall counting accuracy. Under staining conditions in which additive image overlay was inaccurate, we scored samples manually in Adobe Photoshop CS5, using the Analysis>Count Tool function. Calculations were performed in Microsoft Excel, and all results are reported as mean ± s.e.m. P-values were calculated by two-tailed, unpaired t-test.
Supplementary Material
Acknowledgments
We thank Douglas Melton (Harvard University) for providing Pdx1Cre and ElaCreERT mice, Chris Wright (Vanderbilt University) for providing anti-Pdx1 and -Ptf1a antibodies, and Jan Jensen (Cleveland Clinic) for input on the caerulein-induced pancreatitis model. We thank Brett Baumgartner and Daniel Kopinke for extensive discussions as well as helpful comments on the manuscript.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
L.C.M. and M.D.K. conceived and designed the experiments. M.D.K., H.W. and A.K. performed the experiments. J.-P.D.L.O. and M.A.F. contributed input and experimental support to the caerulein treatment experiments. L.C.M. and M.D.K. analyzed the data and wrote the paper.
FUNDING
This work was supported by a grant from the National Institutes of Health [R01-DK075072 to L.C.M.]; M.D.K. is a predoctoral trainee of the University of Utah Genetics Training Grant [National Institutes of Health T32-GM007464]; and J.-P.D.L.O. was a predoctoral trainee of the University of Utah Developmental Biology Training Grant [National Institutes of Health T32-HD07491].
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.007799/-/DC1
REFERENCES
- Abraham S. C., Wu T. T., Hruban R. H., Lee J. H., Yeo C. J., Conlon K., Brennan M., Cameron J. L., Klimstra D. S. (2002). Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: frequent allelic loss on chromosome 11p and alterations in the APC/beta-catenin pathway. Am. J. Pathol. 160, 953–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaine S. A., Ray K. C., Anunobi R., Gannon M. A., Washington M. K., Means A. L. (2010). Adult pancreatic acinar cells give rise to ducts but not endocrine cells in response to growth factor signaling. Development 137, 2289–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockman D. E. (1997). Morphology of the exocrine pancreas related to pancreatitis. Microsc. Res. Tech. 37, 509–519 [DOI] [PubMed] [Google Scholar]
- Brault V., Moore R., Kutsch S., Ishibashi M., Rowitch D. H., McMahon A. P., Sommer L., Boussadia O., Kemler R. (2001). Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128, 1253–1264 [DOI] [PubMed] [Google Scholar]
- Cano D. A., Sekine S., Hebrok M. (2006). Primary cilia deletion in pancreatic epithelial cells results in cyst formation and pancreatitis. Gastroenterology 131, 1856–1869 [DOI] [PubMed] [Google Scholar]
- Carriere C., Young A. L., Gunn J. R., Longnecker D. S., Korc M. (2009). Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem. Biophys. Res. Commun. 382, 561–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Gu X., Su I. H., Bottino R., Contreras J. L., Tarakhovsky A., Kim S. K. (2009). Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 23, 975–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabernat S., Secrest P., Peuchant E., Moreau-Gaudry F., Dubus P., Sarvetnick N. (2009). Lack of beta-catenin in early life induces abnormal glucose homeostasis in mice. Diabetologia 52, 1608–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La O J. P., Murtaugh L. C. (2009). Notch and Kras in pancreatic cancer: at the crossroads of mutation, differentiation and signaling. Cell Cycle 8, 1860–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La O J. P., Emerson L. L., Goodman J. L., Froebe S. C., Illum B. E., Curtis A. B., Murtaugh L. C. (2008). Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc. Natl. Acad. Sci. USA 105, 18907–18912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leon D. D., Deng S., Madani R., Ahima R. S., Drucker D. J., Stoffers D. A. (2003). Role of endogenous glucagon-like peptide-1 in islet regeneration after partial pancreatectomy. Diabetes 52, 365–371 [DOI] [PubMed] [Google Scholar]
- De Lisle R. C., Logsdon C. D. (1990). Pancreatic acinar cells in culture: expression of acinar and ductal antigens in a growth-related manner. Eur. J. Cell Biol. 51, 64–75 [PubMed] [Google Scholar]
- Desai B. M., Oliver-Krasinski J., De Leon D. D., Farzad C., Hong N., Leach S. D., Stoffers D. A. (2007). Preexisting pancreatic acinar cells contribute to acinar cell, but not islet beta cell, regeneration. J. Clin. Invest. 117, 971–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessimoz J., Bonnard C., Huelsken J., Grapin-Botton A. (2005). Pancreas-specific deletion of beta-catenin reveals Wnt-dependent and Wnt-independent functions during development. Curr. Biol. 15, 1677–1683 [DOI] [PubMed] [Google Scholar]
- Dor Y., Brown J., Martinez O. I., Melton D. A. (2004). Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429, 41–46 [DOI] [PubMed] [Google Scholar]
- Ebert M., Yokoyama M., Ishiwata T., Friess H., Buchler M. W., Malfertheiner P., Korc M. (1999). Alteration of fibroblast growth factor and receptor expression after acute pancreatitis in humans. Pancreas 18, 240–246 [DOI] [PubMed] [Google Scholar]
- Fendrich V., Esni F., Garay M. V., Feldmann G., Habbe N., Jensen J. N., Dor Y., Stoffers D., Jensen J., Leach S. D., et al. (2008). Hedgehog signaling is required for effective regeneration of exocrine pancreas. Gastroenterology 135, 621–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florez J. C. (2007). The new type 2 diabetes gene TCF7L2. Curr. Opin. Clin. Nutr. Metab. Care 10, 391–396 [DOI] [PubMed] [Google Scholar]
- Georgia S., Bhushan A. (2004). Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J. Clin. Invest. 114, 963–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu G., Dubauskaite J., Melton D. A. (2002). Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 129, 2447–2457 [DOI] [PubMed] [Google Scholar]
- Guo X., Ramirez A., Waddell D. S., Li Z., Liu X., Wang X. F. (2008). Axin and GSK3-β control Smad3 protein stability and modulate TGF-β signaling. Genes Dev. 22, 106–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habbe N., Shi G., Meguid R. A., Fendrich V., Esni F., Chen H., Feldmann G., Stoffers D. A., Konieczny S. F., Leach S. D., et al. (2008). Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc. Natl. Acad. Sci. USA 105, 18913–18918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiser P. W., Lau J., Taketo M. M., Herrera P. L., Hebrok M. (2006). Stabilization of {beta}-catenin impacts pancreas growth. Development 133, 2023–2032 [DOI] [PubMed] [Google Scholar]
- Jensen J. N., Cameron E., Garay M. V., Starkey T. W., Gianani R., Jensen J. (2005). Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology 128, 728–741 [DOI] [PubMed] [Google Scholar]
- Ji B., Tsou L., Wang H., Gaiser S., Chang D. Z., Daniluk J., Bi Y., Grote T., Longnecker D. S., Logsdon C. D. (2009). Ras activity levels control the development of pancreatic diseases. Gastroenterology 137, 1072–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y., Cooper B., Gannon M., Ray M., MacDonald R. J., Wright C. V. (2002). The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat. Genet. 32, 128–134 [DOI] [PubMed] [Google Scholar]
- Kopinke D., Murtaugh L. C. (2010). Exocrine-to-endocrine differentiation is detectable only prior to birth in the uninjured mouse pancreas. BMC Dev. Biol. 10, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner J. A., Ciemerych M. A., Sicinska E., Wartschow L. M., Teta M., Long S. Y., Sicinski P., White M. F. (2005). Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol. Cell. Biol. 25, 3752–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bacquer O., Shu L., Marchand M., Neve B., Paroni F., Kerr Conte J., Pattou F., Froguel P., Maedler K. (2011). TCF7L2 splice variants have distinct effects on beta-cell turnover and function. Hum. Mol. Genet. 20, 1906–1915 [DOI] [PubMed] [Google Scholar]
- Lee J. Y., Ristow M., Lin X., White M. F., Magnuson M. A., Hennighausen L. (2006). RIP-Cre revisited, evidence for impairments of pancreatic beta-cell function. J. Biol. Chem. 281, 2649–2653 [DOI] [PubMed] [Google Scholar]
- Lepper C., Conway S. J., Fan C. M. (2009). Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature 460, 627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Habener J. F. (2008). Glucagon-like peptide-1 activation of TCF7L2-dependent Wnt signaling enhances pancreatic beta cell proliferation. J. Biol. Chem. 283, 8723–8735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Habener J. F. (2010). Wnt signaling in pancreatic islets. Adv. Exp. Med. Biol. 654, 391–419 [DOI] [PubMed] [Google Scholar]
- Magami Y., Azuma T., Inokuchi H., Moriyasu F., Kawai K., Hattori T. (2002). Heterogeneous cell renewal of pancreas in mice: [(3)H]-thymidine autoradiographic investigation. Pancreas 24, 153–160 [DOI] [PubMed] [Google Scholar]
- Means A. L., Meszoely I. M., Suzuki K., Miyamoto Y., Rustgi A. K., Coffey R. J., Jr, Wright C. V., Stoffers D. A., Leach S. D. (2005). Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development 132, 3767–3776 [DOI] [PubMed] [Google Scholar]
- Meier J. J., Butler A. E., Saisho Y., Monchamp T., Galasso R., Bhushan A., Rizza R. A., Butler P. C. (2008). Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 57, 1584–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molero X., Adell T., Skoudy A., Padilla M. A., Gomez J. A., Chalaux E., Malagelada J. R., Real F. X. (2007). Pancreas transcription factor 1alpha expression is regulated in pancreatitis. Eur. J. Clin. Invest. 37, 791–801 [DOI] [PubMed] [Google Scholar]
- Morris J. P., IV, Cano D. A., Sekine S., Wang S. C., Hebrok M. (2010). Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J. Clin. Invest. 120, 508–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaugh L. C., Law A. C., Dor Y., Melton D. A. (2005). Beta-catenin is essential for pancreatic acinar but not islet development. Development 132, 4663–4674 [DOI] [PubMed] [Google Scholar]
- Nelson W. J., Nusse R. (2004). Convergence of Wnt, beta-catenin, and cadherin pathways. Science 303, 1483–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien W. T., Klein P. S. (2009). Validating GSK3 as an in vivo target of lithium action. Biochem. Soc. Trans. 37, 1133–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane S. G., Dubus P., Mettus R. V., Galbreath E. J., Boden G., Reddy E. P., Barbacid M. (1999). Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat. Genet. 22, 44–52 [DOI] [PubMed] [Google Scholar]
- Rey J. P., Ellies D. L. (2010). Wnt modulators in the biotech pipeline. Dev. Dyn. 239, 102–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen V. (2009). BMP2 signaling in bone development and repair. Cytokine Growth Factor Rev. 20, 475–480 [DOI] [PubMed] [Google Scholar]
- Rulifson I. C., Karnik S. K., Heiser P. W., ten Berge D., Chen H., Gu X., Taketo M. M., Nusse R., Hebrok M., Kim S. K. (2007). Wnt signaling regulates pancreatic beta cell proliferation. Proc. Natl. Acad. Sci. USA 104, 6247–6252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakorafas G. H., Tsiotou A. G., Peros G. (2007). Mechanisms and natural history of pain in chronic pancreatitis: a surgical perspective. J. Clin. Gastroenterol. 41, 689–699 [DOI] [PubMed] [Google Scholar]
- Schonhoff S. E., Giel-Moloney M., Leiter A. B. (2004). Neurogenin 3-expressing progenitor cells in the gastrointestinal tract differentiate into both endocrine and non-endocrine cell types. Dev. Biol. 270, 443–454 [DOI] [PubMed] [Google Scholar]
- Siveke J. T., Lubeseder-Martellato C., Lee M., Mazur P. K., Nakhai H., Radtke F., Schmid R. M. (2008). Notch signaling is required for exocrine regeneration after acute pancreatitis. Gastroenterology 134, 544–555 [DOI] [PubMed] [Google Scholar]
- Smart N. G., Apelqvist A. A., Gu X., Harmon E. B., Topper J. N., MacDonald R. J., Kim S. K. (2006). Conditional expression of Smad7 in pancreatic beta cells disrupts TGF-beta signaling and induces reversible diabetes mellitus. PLoS Biol. 4, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solar M., Cardalda C., Houbracken I., Martin M., Maestro M. A., De Medts N., Xu X., Grau V., Heimberg H., Bouwens L., et al. (2009). Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev. Cell 17, 849–860 [DOI] [PubMed] [Google Scholar]
- Srinivas S., Watanabe T., Lin C. S., William C. M., Tanabe Y., Jessell T. M., Costantini F. (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanger B. Z., Stiles B., Lauwers G. Y., Bardeesy N., Mendoza M., Wang Y., Greenwood A., Cheng K. H., McLaughlin M., Brown D., et al. (2005). Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell 8, 185–195 [DOI] [PubMed] [Google Scholar]
- Strobel O., Dor Y., Alsina J., Stirman A., Lauwers G., Trainor A., Castillo C. F., Warshaw A. L., Thayer S. P. (2007). In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology 133, 1999–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom A., Bonal C., Ashery-Padan R., Hashimoto N., Campos M. L., Trumpp A., Noda T., Kido Y., Real F. X., Thorel F., et al. (2007). Unique mechanisms of growth regulation and tumor suppression upon Apc inactivation in the pancreas. Development 134, 2719–2725 [DOI] [PubMed] [Google Scholar]
- Tang W., Dodge M., Gundapaneni D., Michnoff C., Roth M., Lum L. (2008). A genome-wide RNAi screen for Wnt/beta-catenin pathway components identifies unexpected roles for TCF transcription factors in cancer. Proc. Natl. Acad. Sci. USA 105, 9697–9702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teta M., Rankin M. M., Long S. Y., Stein G. M., Kushner J. A. (2007). Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev. Cell 12, 817–826 [DOI] [PubMed] [Google Scholar]
- Vacik T., Stubbs J. L., Lemke G. (2011). A novel mechanism for the transcriptional regulation of Wnt signaling in development. Genes Dev. 25, 1783–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells J. M., Esni F., Boivin G. P., Aronow B. J., Stuart W., Combs C., Sklenka A., Leach S. D., Lowy A. M. (2007). Wnt/beta-catenin signaling is required for development of the exocrine pancreas. BMC Dev. Biol. 7, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welters H. J., Kulkarni R. N. (2008). Wnt signaling: relevance to beta-cell biology and diabetes. Trends Endocrinol. Metab. 19, 349–355 [DOI] [PubMed] [Google Scholar]
- Wicksteed B., Brissova M., Yan W., Opland D. M., Plank J. L., Reinert R. B., Dickson L. M., Tamarina N. A., Philipson L. H., Shostak A., et al. (2010). Conditional gene targeting in mouse pancreatic ss-Cells: analysis of ectopic Cre transgene expression in the brain. Diabetes 59, 3090–3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemer S., Elsasser H. P., Adler G. (1992). Hormone-induced pancreatitis. Eur. Surg. Res. 24 Suppl. 1, 29–39 [DOI] [PubMed] [Google Scholar]
- Ye W., Mairet-Coello G., DiCicco-Bloom E. (2007). DNAse I pre-treatment markedly enhances detection of nuclear cyclin-dependent kinase inhibitor p57Kip2 and BrdU double immunostaining in embryonic rat brain. Histochem. Cell Biol. 127, 195–203 [DOI] [PubMed] [Google Scholar]
- Zhang H., Ackermann A. M., Gusarova G. A., Lowe D., Feng X., Kopsombut U. G., Costa R. H., Gannon M. (2006). The FoxM1 transcription factor is required to maintain pancreatic beta-cell mass. Mol. Endocrinol. 20, 1853–1866 [DOI] [PubMed] [Google Scholar]
- Zhou Q., Law A. C., Rajagopal J., Anderson W. J., Gray P. A., Melton D. A. (2007). A multipotent progenitor domain guides pancreatic organogenesis. Dev. Cell 13, 103–114 [DOI] [PubMed] [Google Scholar]
- Zimmermann A., Gloor B., Kappeler A., Uhl W., Friess H., Buchler M. W. (2002). Pancreatic stellate cells contribute to regeneration early after acute necrotising pancreatitis in humans. Gut 51, 574–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.