

SUMMARY
Cardio-facio-cutaneous (CFC) syndrome is caused by germline mutations in KRAS, BRAF and MEK1/2. The highly selective and potent MEK inhibitors that have been developed as anti-cancer agents hold potential as therapeutics for CFC syndrome. We have previously shown that the effects of CFC mutations on zebrafish gastrulation can be prevented by a 1-hour treatment with MEK inhibitors within a specific developmental time-window. However, MEK activity is essential for normal development and PD0325901 treatment outside this treatment window leads to additional developmental defects in MEK-dependent tissues. We now test ten different doses of PD0325901 at six developmental time points and assess the effects on body axis length, heart development and craniofacial structures in zebrafish embryos. Notably, we find that a continuous low-level dose of PD0325901 that has only minor inhibition of MEK activity can prevent the action of both the common CFC BRAFQ257R kinase-active allele and the BRAFG596V kinase-impaired mutant allele through the first 5 days of development. These results provide a detailed study of the effects of PD0325901 in development and show that, unlike in cancer, which requires robust inhibition of MAPK signalling, a partial reduction in phospho-ERK1/2 activity is sufficient to moderate the developmental effects of BRAFCFC mutations.
INTRODUCTION
Animal models of disease provide an important opportunity to test the action of existing drugs in new disease contexts. Germline mutations in the RAS-MAPK signalling cascade are found in a spectrum of overlapping developmental syndromes, collectively referred to as the ‘RASopathies’ (Tidyman and Rauen, 2009). Rare genetic developmental disorders such as RASopathies are not a focus of drug development. However, drugs with high specificity and efficacy for the RAS-MAPK pathway, although designed as anti-cancer therapies, are obvious potential therapies for RASopathies (Sebolt-Leopold, 2008; Rauen et al., 2011; Pratilas and Solit, 2010). PD0325901 is a highly selective small-molecule inhibitor of MEK1 and MEK2 in vitro and in vivo (Sebolt-Leopold, 2008). In clinical trials, PD0325901 has shown effective inhibition of MEK activity for individuals with MAPK-activated solid tumours (Haura et al., 2010; LoRusso et al., 2010). Thus, although designed as anti-cancer drugs, MEK inhibitors hold potential for use in additional clinical settings.
The RASopathies spectrum includes cardio-facio-cutaneous syndrome (CFC), Costello syndrome (CS), Noonan syndrome (NS), LEOPARD syndrome (LS), neurofibromatosis type 1 (NF1) and Legius syndrome. Clinical features of CFC syndrome include heart malformations, prominent facial features, sparse eyebrows, curly hair, increased number of nevi and neurocognitive delay (Roberts et al., 2006). In vitro analysis of the mutations in BRAF and MEK that are identified in individuals with CFC syndrome has shown some to be kinase-activating and some kinase-impaired (Rodriguez-Viciana et al., 2006). However, we have shown that all tested CFC mutations have gain-of-function activity in vivo (Anastasaki et al., 2009). Expression of BRAFCFC and MEKCFC mutant alleles in zebrafish embryos causes cell movement defects during early development, indicative of activated FGF-MAPK signalling in gastrulation cell movements (convergence-extension) (Krens et al., 2008). In vitro, kinase-activating MEKCFC mutations are responsive to inhibition of MEK and RAF (Senawong et al., 2008). In vivo, FGFR and MEK inhibitors can prevent cell movement defects in BRAFCFC- and MEKCFC-expressing zebrafish embryos (Anastasaki et al., 2009). Notably, in our early embryonic CFC model, a 1-hour treatment window at 4 hours postfertilisation (hpf) was sufficient to restore normal gastrulation and embryonic formation up to 24 hpf (Anastasaki et al., 2009).
However, identification of an early development treatment window is not easily extrapolated to a timed treatment of embryos in utero. Moreover, embryos that had undergone longer treatments often had serious additional developmental defects associated with strong inhibition of MEK activity. Here, we use the zebrafish system as a model for continuous low-level MEK inhibition in developing vertebrates. First, we perform a detailed analysis of the effects of ten different doses of PD0325901 and assess the impact during the first 5 days of development. Notably, a very low treatment dose with only minimal MEK inhibition is sufficient to prevent the deleterious effects of BRAFCFC in developing zebrafish. We suggest that continuous minimal inhibition of MAPK signalling might be sufficient to prevent some of the developmental features caused by CFC mutations in developing children. This study provides a proof-of-principle that low treatment doses of kinase inhibitors can be effective in ameliorating the effects of CFC alleles in development, and could be crucial for designing future CFC syndrome clinical trials (Rauen et al., 2010; Rauen et al., 2011).
RESULTS
Effective MEK inhibition by PD0325901 in zebrafish development
MEK signalling is crucial for normal embryonic development. We have previously found that the MEK inhibitor PD0325901 (Fig. 1A) effectively inhibits phospho-ERK1/2 activity in zebrafish and is able to counter the effects of mutant BRAF and MEK signalling in gastrulation (Anastasaki et al., 2009). However, this dose has deleterious effects on other developing organs (Anastasaki et al., 2009; Grzmil et al., 2007). To determine whether low-level MEK inhibition can be tolerated during development, we tested different concentrations of PD0325901 during the first 4 days of development. First, we tested a range of PD0325901 doses to determine suboptimal concentrations as measured by levels of phospho-ERK1/2 in the whole embryo. Zebrafish embryos (4 hpf) were treated with increasing concentrations of PD0325901 for 6 hours, and protein extracts from the whole animal were measured for phospho-ERK1/2 activity (Fig. 1B). Although all embryos expressed approximately equal levels of total ERK1/2 protein, almost all phospho-ERK1/2 isoforms were lost after 0.7 μM PD0325901 treatment and strong reductions in phospho-ERK1/2 signals were detected with 0.5 and 0.6 μM PD0325901. At 0.1 μM, at least half of the phospho-ERK1/2 isoforms remained detectable. Thus, PD0325901 treatment can be tightly calibrated to generate a range of phospho-ERK1/2 levels in the developing embryo.
Fig. 1.
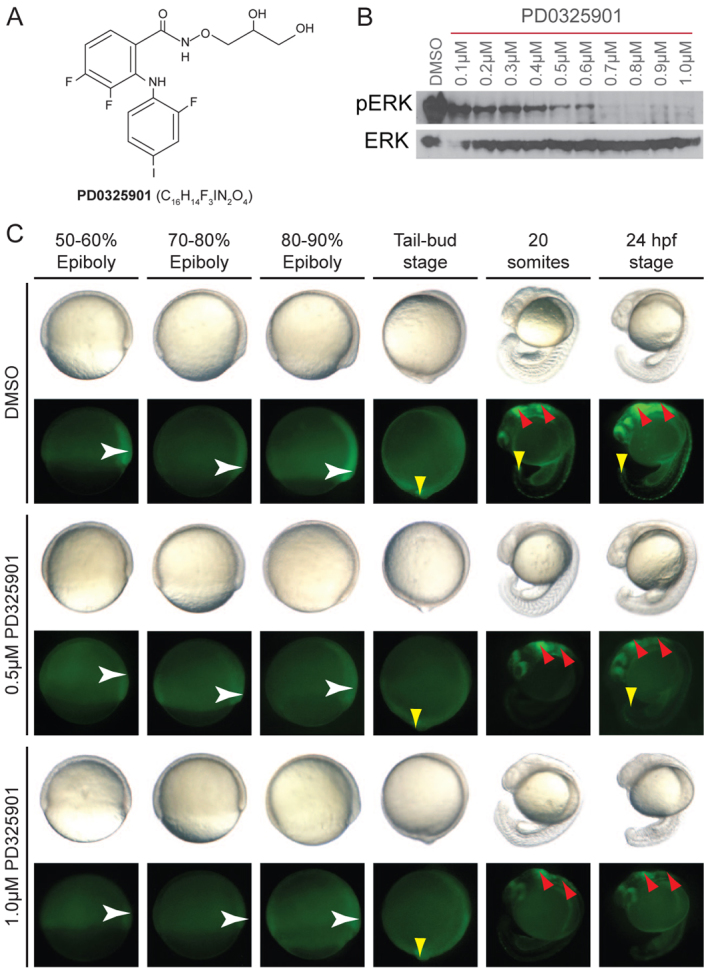
PD0325901 is an effective MEK inhibitor in zebrafish development. (A) Chemical structure of the selective MEK inhibitor PD0325901. (B) Western blot of 10-hpf zebrafish embryos treated with increasing concentrations of PD0325901 from 4 hpf, and immunostained with anti-phospho-ERK1/2 and anti-ERK1/2 antibodies. (C) Images of developing zebrafish treated with DMSO, or 0.5 μM or 1.0 μM PD0325901. Zebrafish were imaged under brightfield lighting conditions (top row), or using fluorescence microscopy to visualise GFP expressed from the dusp6 promoter (dusp6-GFP; bottom row). GFP signal was first detected in gastrulating embryos at 50–60% epiboly (white arrowhead). The expression area expanded towards the vegetal pole during epiboly (white arrowhead) and at the tail-bud stage the expression was more diffused with higher expression levels at the tail bud (yellow arrowhead). By 24 hpf, GFP expression was detected in the brain (red arrowhead), along the spinal column and in the tail bud (yellow arrowhead). n=30 embryos per treatment condition.
Western blots provide an indication of the overall level of MAPK signalling; however, the MAPK signalling pathway is activated in a highly specific temporal and spatial manner during development. We wanted to visualize how PD0325901 affected MAPK signalling in developing tissues. Dusp6 is a protein phosphatase that is upregulated by FGF-MAPK signalling as part of a regulatory negative-feedback loop. The transgenic zebrafish line expressing GFP from the dusp6 promoter (dusp6-GFP) (Molina et al., 2007) allows for visualization of activated MAPK signalling in living tissues, including the developing tail bud, the brain and pharyngeal arches (Fig. 1C). Treatment with 0.5 μM PD0325901 from 4 hpf to 24 hpf caused a reduction in GFP levels in all tissues, with reduced levels remaining in the developing tail bud and brain (red arrows, Fig. 1C). Treatment with 1.0 μM conferred even greater losses of GFP signal, such that almost no signal was detectable during epiboly, and very minor levels were detected in the brain coupled with a complete loss of signal detectable in the tail bud (Fig. 1C). These data show that PD0325901 treatment can effectively target the MAPK pathway within multiple tissues in the developing zebrafish.
Systematic analysis of the MEK inhibitor PD0325901 in zebrafish development
We next wanted to determine whether there was an effective MEK inhibitor treatment dose that could be tolerated by wild-type developing zebrafish. Wild-type zebrafish embryos were collected at 4, 10, 24, 30, 48 and 72 hpf, and 40–60 embryos at each stage were treated with ten different doses of PD0325901 (ranging from 0.1–1.0 μM) (Fig. 2A). The embryos were imaged daily and assessed for their overall length, craniofacial development (by Alcian Blue staining) and heart development (Fig. 2A; supplementary material Figs S1–S3) (Grzmil et al., 2007). Phenotypes were scored from none (0) to mild (+), moderate (++) and severe (+++). Scores were then translated to a colour (blue, yellow, orange and red, respectively) and arranged as a heat map. In this way, a visual overview of over 500 data points could be rapidly assessed (Fig. 2B).
Fig. 2.

Wild-type embryo sensitivity to PD0325901 treatments. (A) Schematic overview of the design of the PD0325901 treatment regimes. Wild-type embryos were treated with ten increasing concentrations of PD0325901 (0.1–1.0 μM) to identify the most affected tissues at different stages during development. Zebrafish embryos were treated at six different developmental stages (4, 10, 24, 30, 48 and 72 hpf) until 96 hpf. The embryos were monitored daily and were assessed for body axis, cardiac morphology and craniofacial structure phenotypes at 96 hpf. (B) Heat-map of tissue sensitivity to PD0325901 treatment. Each panel shows tissue sensitivity after the addition of increasing concentrations of PD0325901 (0.1–1.0 μM) at a different time in development. Phenotypes were scored for sensitivity to PD0325901, and these data were translated to a colour score in the heat map: 0, no phenotype, blue; +, mild phenotype, yellow; ++, moderate phenotype, orange; +++, severe phenotype, red. PA, pharyngeal arches; MC, Meckel’s cartilage; CH, ceratohyal cartilage; BA, branchial arches. Embryos treated with 0.2 μM PD0325901 from 4 hpf had a normal A-P axis but showed mild malformation of the tail tip (*). The heart valve of embryos treated with 0.1 μM PD0325901 from 48 hpf and with all ten concentrations of PD0325901 from 72 hpf was blocked at 5 dpf (**).
FGF-MAPK signalling is required for gastrulation cell movements that determine the development of the posterior body length. We found that embryos were highly sensitive to PD0325901 when treated before 4 hpf (data not shown). Embryos treated at 4 hpf had a shorter anteroposterior (A-P) axis, which correlated with dose such that concentrations above 0.6 μM PD0325901 led to a severely shortened embryo whereas 0.2–0.4 μM of the drug caused only a mild effect on embryo length, and 0.1 μM had no effect on the length of the A-P axis (supplementary material Fig. S1). These results were consistent with our observations in the dusp6-GFP line (Fig. 1C), in which MAPK activity in the gastrulating embryo was reduced by treatment with 0.5 μM PD0325901 and almost undetectable with 1.0 μM PD0325901 treatment conditions. Drug administration at time points after 4 hpf had no effect on the A-P body axis, consistent with an early role for FGF-MAPK in establishing body length. Thus, MEK activity is essential before 10 hpf but A-P body axis development can tolerate a partial reduction in MAPK signalling below a specific dose threshold.
The vertebrate jaw derives from neural crest cells and activated MAPK signalling is required for proper specification of craniofacial components (Walshe and Mason, 2003; Crump et al., 2004; Wilson et al., 2004; Komisarczuk et al., 2008). Pharyngeal arch development was highly sensitive to PD0325901 treatment, with the most severe phenotype being associated with early and high (0.8–1.0 μM PD0325901) treatment conditions. The first and second branchial arches (BAs) were the least sensitive to the drug, and treatments after 24 hpf had no effect on the first BA and minimal effects on the second BA. Arches 3, 4 and 5 were highly sensitive to MEK inhibition at 4 hpf and 10 hpf (0.7–1.0 μM), with embryos becoming progressively less sensitive as they developed passed 24 hpf. Formation of the Meckel’s cartilage (MC) and ceratohyal cartilage (CH) were highly affected by MEK inhibition; concentrations as low as 0.5 μM PD0325901 at 4 hpf and 0.8 μM PD0325901 at 48 hpf caused moderate-to-severe anomalies (supplementary material Fig. S2). MEK inhibition in embryos older than 3 days postfertilisation (dpf) did not promote an overt craniofacial abnormal phenotype (data not shown), probably because all visible structures were already formed. These findings indicate that zebrafish jaw development is highly sensitive to PD0325901 treatment and suggests that MEK signalling is required at multiple stages for normal jaw development.
Administration of all PD0325901 concentrations at 4, 10, 24, 30 and 48 hpf led to heart anomalies in most embryos by 4 dpf (supplementary material Fig. S3). The phenotype was reminiscent of that promoted by a previous generation MEK inhibitor, CI-1040 (Grzmil et al., 2007). The embryos developed cardiac oedemas and blockage of the bulbus arteriosus, causing restricted blood flow and exit from the heart chambers. The cardiovascular system of embryos incubated at 72 hpf seemed to be unaffected 24 hours later, but the embryos displayed the described aberrant cardiac phenotype after a 48-hour incubation (data not shown). Notably, the PD0325901-induced heart defect was always observed 2 days after treatment, regardless of the developmental stage (Fig. 2B). Thus, although MAPK signalling might be important for normal heart development, these results suggest that either active MEK activity is required to maintain heart integrity, or cardiac oedemas and valve blockage are off-target, toxic side effects of PD0325901 treatment.
Small-molecule prevention of BRAFCFC phenotypes
Given the sensitivity of the developing zebrafish to inhibition of MEK activity, we investigated whether low doses of PD0325901 (0.1–0.2 μM) were sufficient to suppress activity of the BRAFCFC allele without the unwanted effects caused by the inhibitor. First, we established our model for CFC mutant allele expression. Single-cell embryos were injected with 35 pg of mRNA expressing either human BRAFWT, the kinase-activating and most common CFC mutation BRAFQ257R, or the kinase-inactivating BRAFG596V mutation. We have previously shown that overexpression of BRAFWT has no effect on zebrafish, suggesting that developing embryos can compensate for the increased wild-type BRAF activity (Fig. 3A). This is consistent with the inability of transgenic BRAFWT to induce nevi and/or melanoma in zebrafish. By contrast, overexpression of either BRAFQ257R or BRAFG596V promotes cell movement defects during gastrulation, consistent with enhanced activation of the MAPK signalling pathway in vivo (Fig. 3A). These embryos continue to develop, but with a shortened body axis (Fig. 3A). Increased nevi count is a feature of the CFC syndrome and, likewise, we found that transgenic BRAFG596V is also able to promote nevi development when expressed under the melanocyte-specific mitfa promoter (Fig. 3B). Thus, although BRAF mutations are not expressed from the endogenous locus in our model, BRAFCFC RNA overexpression or expression of transgenic BRAFCFC can serve as a model for CFC mutant allele expression in animals.
Fig. 3.

Continuous suboptimal PD0325901 treatment prevents zebrafish BRAFCFC phenotypes. (A) Wild-type embryos were injected with human BRAFWT, the kinase-activating and most common CFC allele BRAFQ257R, or the kinase-inactivating BRAFG596V, and imaged at the indicated developmental time post-fertilisation. (B) Images of untreated control zebrafish (top panel) and a zebrafish expressing the kinase-inactivating BRAFG596V allele under the melanocyte-specific mitfa promoter (bottom panel). Insert image depicts a high-magnification image of ectopic melanisation and nevi formation. (C) Continuous treatment of BRAFWT- and BRAFCFC-injected embryos with a suboptimal dose (0.2 μM) of PD0325901 from 4 hpf through 4 dpf. (D) Western blot of 10-hpf wild-type [uninjected (Un)] or BRAFCFC embryos treated with 0.2 μM of PD0325901 from 4 hpf, and immunostained with anti-phospho-ERK1/2 and anti-ERK1/2 antibodies.
Next, we tested the sensitivity of BRAFCFC alleles to MEK inhibition in zebrafish embryos. We have previously shown that BRAFCFC and MEKCFC mutations are responsive to an optimal short treatment dose of two MEK inhibitors. We wanted to identify a continuous and low-dose MEK inhibitor treatment that could prevent the action of the BRAFCFC mutation while not causing additional developmental abnormalities as a result of MEK inhibition. Our analysis indicated that embryos had to be treated early enough to prevent BRAFCFC gastrulation defects and with less than 0.3 μM PD0325901 to prevent drug-induced developmental defects. Single-cell embryos were injected with 35 pg of human BRAFWT, BRAFQ257R or BRAFG596V mRNA and these embryos were treated from 4 hpf to 4 dpf with a continuous dose of 0.1 or 0.2 μM PD0325901 (Fig. 3C). BRAFWT embryos treated with a continuous 0.1 μM PD0325901 dose were grossly normal. However, this treatment dose was insufficient to prevent the action of BRAFQ257R and BRAFG596V activity: CFC embryos were elongated during gastrulation and severely shortened in the A-P axis (supplementary material Fig. S4). By comparison, BRAFQ257R and BRAFG596V embryos treated with a continuous 0.2 μM PD0325901 dose were only mildly elongated during gastrulation and were slightly delayed in development, but were able to develop a normal body axis by 24 hpf (Fig. 3C). Interestingly, this treatment dose caused a minor tail abnormality in BRAFWT embryos, suggesting that 0.2 μM PD0325901 treatments are at the limit of tolerance for BRAFWT-injected embryos. Despite the treatment conditions allowing axial development, all 0.2 μM PD0325901-treated embryos nevertheless developed a cardiac valve closure. Western blotting confirmed that phospho-ERK1/2 was present in 0.2 μM PD0325901-treated BRAFQ257R and BRAFG596V embryos (Fig. 3D). Thus, in our zebrafish model, we can identify a low-dose and permissive MEK inhibition treatment for CFC embryos.
DISCUSSION
The importance of the RAS-MAPK signalling pathway in cancer has led to the development of highly effective and specific inhibitors of RAS, BRAF and MEK that hold potential for the reduction of the severity and progression of the RASopathies (Tidyman and Rauen, 2009). Some inhibitors have already been tested in preclinical mouse models: MEK inhibitors can effectively limit some of the features of activated MAPK signalling in an FGFR model of Apert syndrome, as well as in a RAF model of Noonan syndrome (Chen et al., 2010; Wu et al., 2011). In zebrafish, expression of RAS, BRAF and MEK Noonan and CFC syndrome mutations cause cell movement defects that can be prevented with the MEK inhibitors PD0325901 and/or the closely related CI-1040 (Anastasaki et al., 2009; Runtuwene et al., 2011). Altering the MAPK output can also restore impaired learning in fruit flies expressing SHP2 Noonan syndrome mutations (Pagani et al., 2009). Thus, animal models of developmental disease in a wide range of species can respond to treatments designed to target altered signalling in human cancer.
Short treatments with MEK inhibitors at key developmental stages could thus prevent the serious consequences of CFC mutations in development. This strategy is possible in zebrafish because of the ex utero fertilization, the ability to clearly distinguish each developmental stage under the light microscope, and the rapid uptake of the compound from the fish water. This approach would be much more difficult to apply to in utero mammalian models, or even people. To address this, we tested continual MEK inhibitor treatment in the context of a zebrafish model of CFC syndrome. We identify three key findings relevant to MEK inhibitor treatments in animals. First, through systematic analysis of drug-development combinations, we identify that a 0.1–0.2 μM treatment dose of PD0325901 has little effect on normal development. These doses have only a partial effect on MEK activity as assessed by phospho-ERK1/2 activity and dusp6-GFP expression. Second, continuous treatment with a 0.2 μM dose of MEK inhibitor is sufficient to prevent the effects of both kinase-active and kinase-impaired BRAFCFC alleles. Thus, unlike cancer treatment strategies, which aim to fully inhibit MEK activity (e.g. Solit et al., 2006; Sharma and Settleman, 2007), MAPK signalling developmental disorders might benefit from only a partial inhibition of the pathway. This concept was also illustrated by the ability of upstream FGFR inhibitors to rescue downstream BRAFCFC mutations in zebrafish, presumably because reducing endogenous FGF signalling helped reduce the total MAPK signalling output (Anastasaki et al., 2009). Interestingly, in treatment conditions optimal for BRAFCFC embryos, the embryos still showed some of the effects of increased MAPK signalling and slight developmental delay at 10 hpf, but once passed this stage developed normally. This might reflect the potential for the developing animal to tolerate some variations in signalling provided that it is below a critical threshold. Finally, we find that PD0325901 causes zebrafish embryonic heart defects. This effect appeared at all treatment doses, suggesting that either MEK activity is required to maintain heart integrity and/or that this defect reflects a toxic side effect. By contrast, adult zebrafish can tolerate 0.01–1.0 μM PD0325901 in fish water for at least 7 days (Jennifer Richardson, personal communication), suggesting that the effects might be specific to the embryonic zebrafish heart. Thus, in our model, treatment does not necessarily require a highly effective MEK inhibitor; however, the inhibitor needs to be highly specific in vivo to permit normal development. The importance of small-molecule specificity is underscored by the finding that the MEK inhibitor U0126 also targets copper metabolism pathways in vivo (Ishizaki et al., 2010).
Our approach provides a starting point for the potential to use MEK inhibitors in the treatment of CFC syndrome. Other inhibitors of the RAS-MAPK signalling pathway could also be valuable (Wilkie, 2007; Tidyman and Rauen, 2009). For example, new BRAF inhibitors have been highly effective in BRAF mutant melanoma treatment (Yang et al., 2010; Chapman et al., 2011), but side effects include the development of squamous-cell carcinomas in tissues that lack the somatic BRAF mutation; it is unknown how BRAF inhibitors might work in the context of germline BRAF mutations. A recent BRAFCFC mouse model might assist in addressing this question (Urosevic et al., 2011). The clinical features of CFC syndrome continue to progress after birth, and MEK inhibitors might be useful in managing and reducing the severity of clinical features during childhood. Importantly, in the preclinical mouse model of Noonan syndrome, postnatal administration of PD0325901 reduced lethality and the severity of the Noonan pathology (Chen et al., 2010; Wu et al., 2011). Although toxicity will always be a concern, we are encouraged that very low partial MEK inhibition treatments are effective in our zebrafish CFC syndrome model. We suggest that animal disease models such as this provide an important opportunity to test the action of available drugs in disease contexts that would otherwise remain unexplored in the drug development industry.
METHODS
Zebrafish husbandry
Adult and embryonic zebrafish were maintained and raised at 28.5°C. Embryos were collected from pair matings of wild-type AB adults. All embryos were maintained in six-well plates in 6 ml E3 embryo medium alone, with added DMSO (control) or PD0325901. A maximum of ten larvae were placed in each well and the medium was refreshed daily.
Capped mRNA microinjections
Capped human mRNA was in vitro transcribed using mMESSAGE mMACHINE (Ambion) as previously described (Anastasaki et al., 2009). Capped BRAFQ257R mRNA (1 nl) was injected at a final concentration of 35 ng/μl directly into the cytoplasm of one-cell-stage wild-type embryos. Microinjections were performed using a nitrogen-powered Picospritzer III injector (Intracel) and Nikon SMZ 1000 dissection microscope.
Pharmacological treatment
For the systematic characterisation of the effects of PD0325901, wild-type embryos were incubated in E3 embryo medium enriched with PD0325901 (University of Dundee) at final concentrations ranging from 0.1 to 1.0 μM. The medium was refreshed daily to maintain the drug activity. For the rescue of the BRAFCFC-expressing embryos, injected embryos were incubated in 0.1, 0.2 and 1.0 μM PD0325901 at 4 hpf. Control-treated embryos were incubated in E3 with DMSO at the same final concentration as PD0325901.
Immunoblotting
Immunoblotting was performed with whole-embryo lysates from 4-hpf individuals. The embryos were ribolysed for 1 minute in protein extraction buffer [2 M Tris, pH 7.5, 5 M NaCl, 1% NP40, Na deoxycholate, 10% SDS, 0.5 M NaF, 1 M β-glycosyl phosphate, protease inhibitor cocktail tablet (Roche)]. The protein content was measured with a spectrophotometer and the samples were normalized, accordingly. Total protein extracts were probed with anti-p44/4-MAPK (1:2000; #9102) and anti-phospho-p44/42-MAPK (E10; 1:2000; #9106) antibodies (Cell Signaling Technology).
Alcian Blue cartilage staining
4-dpf embryos were fixed in freshly prepared 4% PFA (in PBS) at room temperature for 7 hours. The embryos were subsequently thoroughly washed in 0.1% PBS-Tw and stained in freshly prepared syringe-filtered 0.1% Alcian Blue solution (in acetic acid) at room temperature for approximately 7 hours. Following complete staining, the samples were rinsed in 100% ethanol, serially rehydrated in PBS-Tw and stored at 4°C. The tissue was cleared in 0.05% trypsin (Invitrogen) for a minimum of 2 hours at 30°C until the brain tissue was translucent, followed by short room-temperature bleaching in 1% KOH in 3% hydrogen peroxide solution to remove melanocyte pigmentation. The samples were then thoroughly washed in PBS-Tw and mounted in 70% glycerol to be stored and imaged.
TRANSLATIONAL IMPACT.
Clinical issue
The RASopathies, including cardio-facio-cutaneous syndrome (CFC), Noonan syndrome and others, are a series of rare genetic syndromes caused by germline mutations in the RAS-MAPK signalling pathway. Treatment of these syndromes might benefit from the products in the intensive drug development pipeline targeted at inhibiting the activated RAS-MAPK signalling that underlies some cancers. The clinical features of the RASopathies continue to progress after birth and, thus, there might be potential for both pre- and post-natal treatment with such drugs. However, how these drugs could be used in the context of the RASopathies, which are developmental diseases, rather than in the context of anti-cancer therapy is unknown.
Results
In this paper, the authors use zebrafish embryos as a tool to explore different timing and treatment doses of a clinically active inhibitor of the RAS-MAPK pathway – the MEK inhibitor PD0325901 – during development. Previously, this group showed in zebrafish that mutated forms of BRAF and MEK found in humans with CFC are sensitive to MEK inhibitors, but that a short treatment time is necessary to prevent additional unwanted effects of MEK inhibition. Here, they explore the potential for continuous MEK inhibitor treatment to alleviate the defects caused by CFC-associated BRAF mutations. They find that continuous and partial MEK inhibition can be tolerated during zebrafish development, and can prevent the action of CFC-associated BRAF mutations at multiple developmental stages. Thus, MEK inhibitors might have potential in the management of CFC syndrome in very low doses that only partially inhibit MEK activity.
Implications and future directions
Animal models of disease are important preclinical tools for exploring how drugs can be used for indications other than the diseases they were initially designed to treat. This study suggests that very low doses of MEK inhibitors can prevent the developmental effects of CFC-associated BRAF mutations while not affecting the development of normal tissues that require MEK signalling. Future directions include testing similar approaches in pre- and post-natal preclinical mammalian models, and chemical optimisation of MEK inhibitors to reduce toxic side effects.
Supplementary Material
Acknowledgments
We are grateful to the families of CFC International for enabling this research; Ian Jackson and David Fitzpatrick for helpful comments; Michael Tsang for the dusp6-GFP transgenic line; and Karthika Paranthaman for zebrafish husbandry.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
C.A. performed all experiments; K.A.R. contributed reagents, assisted with the experimental approach and commented on the manuscript; C.A. and E.E.P. designed experiments, analysed the data and wrote the manuscript; E.E.P. acquired funding.
FUNDING
This work was funded by the Wellcome Trust; and the Medical Research Council.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.008672/-/DC1
REFERENCES
- Anastasaki C., Estep A. L., Marais R., Rauen K. A., Patton E. E. (2009). Kinase-activating and kinase-impaired cardio-facio-cutaneous syndrome alleles have activity during zebrafish development and are sensitive to small molecule inhibitors. Hum. Mol. Genet. 18, 2543–2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman P. B., Hauschild A., Robert C., Haanen J. B., Ascierto P., Larkin J., Dummer R., Garbe C., Testori A., Maio M., et al. (2011). Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 364, 2507–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P. C., Wakimoto H., Conner D., Araki T., Yuan T., Roberts A., Seidman C. E., Bronson R., Neel B. G., Seidman J. G., et al. (2010). Activation of multiple signaling pathways causes developmental defects in mice with a Noonan syndrome-associated Sos1 mutation. J. Clin. Invest. 120, 4353–4365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump J. G., Maves L., Lawson N. D., Weinstein B. M., Kimmel C. B. (2004). An essential role for Fgfs in endodermal pouch formation influences later craniofacial skeletal patterning. Development 131, 5703–5716 [DOI] [PubMed] [Google Scholar]
- Grzmil M., Whiting D., Maule J., Anastasaki C., Amatruda J. F., Kelsh R. N., Norbury C. J., Patton E. E. (2007). The INT6 cancer gene and MEK signaling pathways converge during zebrafish development. PLoS ONE 2, e959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haura E. B., Ricart A. D., Larson T. G., Stella P. J., Bazhenova L., Miller V. A., Cohen R. B., Eisenberg P. D., Selaru P., Wilner K. D., et al. (2010). A phase II study of PD-0325901, an oral MEK inhibitor, in previously treated patients with advanced non-small cell lung cancer. Clin. Cancer Res. 16, 2450–2457 [DOI] [PubMed] [Google Scholar]
- Ishizaki H., Spitzer M., Wildenhain J., Anastasaki C., Zeng Z., Dolma S., Shaw M., Madsen E., Gitlin J., Marais R., et al. (2010). Combined zebrafish-yeast chemical-genetic screens reveal gene-copper-nutrition interactions that modulate melanocyte pigmentation. Dis. Model. Mech. 3, 639–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komisarczuk A. Z., Topp S., Stigloher C., Kapsimali M., Bally-Cuif L., Becker T. S. (2008). Enhancer detection and developmental expression of zebrafish sprouty1, a member of the fgf8 synexpression group. Dev. Dyn. 237, 2594–2603 [DOI] [PubMed] [Google Scholar]
- Krens S. F., He S., Lamers G. E., Meijer A. H., Bakkers J., Schmidt T., Spaink H. P., Snaar-Jagalska B. E. (2008). Distinct functions for ERK1 and ERK2 in cell migration processes during zebrafish gastrulation. Dev. Biol. 319, 370–383 [DOI] [PubMed] [Google Scholar]
- LoRusso P. M., Krishnamurthi S. S., Rinehart J. J., Nabell L. M., Malburg L., Chapman P. B., DePrimo S. E., Bentivegna S., Wilner K. D., Tan W., et al. (2010). Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin. Cancer Res. 16, 1924–1937 [DOI] [PubMed] [Google Scholar]
- Molina G. A., Watkins S. C., Tsang M. (2007). Generation of FGF reporter transgenic zebrafish and their utility in chemical screens. BMC Dev. Biol. 7, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani M. R., Oishi K., Gelb B. D., Zhong Y. (2009). The phosphatase SHP2 regulates the spacing effect for long-term memory induction. Cell 139, 186–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratilas C. A., Solit D. B. (2010). Targeting the mitogen-activated protein kinase pathway: physiological feedback and drug response. Clin. Cancer Res. 16, 3329–3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauen K. A., Schoyer L., McCormick F., Lin A. E., Allanson J. E., Stevenson D. A., Gripp K. W., Neri G., Carey J. C., Legius E., et al. (2010). Proceedings from the 2009 genetic syndromes of the Ras/MAPK pathway: From bedside to bench and back. Am. J. Med. Genet. A 152, 4–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauen K. A., Banerjee A., Bishop W. R., Lauchle J. O., McCormick F., McMahon M., Melese T., Munster P. N., Nadaf S., Packer R. J., et al. (2011). Costello and cardio-facio-cutaneous syndromes: Moving toward clinical trials in RASopathies. Am. J. Med. Genet. C 157, 136–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A., Allanson J., Jadico S. K., Kavamura M. I., Noonan J., Opitz J. M., Young T., Neri G. (2006). The cardiofaciocutaneous syndrome. J. Med. Genet. 43, 833–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P., Tetsu O., Tidyman W. E., Estep A. L., Conger B. A., Cruz M. S., McCormick F., Rauen K. A. (2006). Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science 311, 1287–1290 [DOI] [PubMed] [Google Scholar]
- Runtuwene V., van Eekelen M., Overvoorde J., Rehmann H., Yntema H. G., Nillesen W. M., van Haeringen A., van der Burgt I., Burgering B, den Hertog J. (2011). Noonan syndrome gain-of-function mutations in NRAS cause zebrafish gastrulation defects. Dis. Model. Mech. 4, 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebolt-Leopold J. S. (2008). Advances in the development of cancer therapeutics directed against the RAS-mitogen-activated protein kinase pathway. Clin. Cancer Res. 14, 3651–3656 [DOI] [PubMed] [Google Scholar]
- Senawong T., Phuchareon J., Ohara O., McCormick F., Rauen K. A., Tetsu O. (2008). Germline mutations of MEK in cardio-facio-cutaneous syndrome are sensitive to MEK and RAF inhibition: implications for therapeutic options. Hum. Mol. Genet. 17, 419–430 [DOI] [PubMed] [Google Scholar]
- Sharma S. V., Settleman J. (2007). Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev. 21, 3214–3231 [DOI] [PubMed] [Google Scholar]
- Solit D. B., Garraway L. A., Pratilas C. A., Sawai A., Getz G., Basso A., Ye Q., Lobo J. M., She Y., Osman I., et al. (2006). BRAF mutation predicts sensitivity to MEK inhibition. Nature 439, 358–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidyman W. E., Rauen K. A. (2009). The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 19, 230–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urosevic J., Sauzeau V., Soto-Montenegro M. L., Reig S., Desco M., Wright E. M., Cañamero M., Mulero F., Ortega S., Bustelo X. R., et al. (2011). Constitutive activation of B-Raf in the mouse germ line provides a model for human cardio-facio-cutaneous syndrome. Proc. Natl. Acad. Sci. USA 12, 5015–5020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walshe J., Mason I. (2003). Fgf signalling is required for formation of cartilage in the head. Dev. Biol. 264, 522–536 [DOI] [PubMed] [Google Scholar]
- Wilkie A. O. (2007). Cancer drugs to treat birth defects. Nat. Genet. 39, 1057–1059 [DOI] [PubMed] [Google Scholar]
- Wilson Y. M., Richards K. L., Ford-Perriss M. L., Panthier J. J., Murphy M. (2004). Neural crest cell lineage segregation in the mouse neural tube. Development 131, 6153–6162 [DOI] [PubMed] [Google Scholar]
- Wu X., Simpson J., Hong J. H., Kim K. H., Thavarajah N. K., Backx P. H., Neel B. G., Araki T. (2011). MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J. Clin. Invest. 121, 1009–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Higgins B., Kolinsky K., Packman K., Go Z., Iyer R., Kolis S., Zhao S., Lee R., Grippo J. F., et al. (2010). RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res. 70, 5518–5527 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.