

Abstract
Objective
To determine which features of incomplete or “nonclassic” forms of cystic fibrosis (CF) are associated with deleterious CF transmembrane conductance regulator gene (CFTR) mutations, and to explore other etiologies for features not associated with deleterious CFTR mutations.
Study design
Clinical features were compared between 57 patients with deleterious mutations in each CFTR and 63 with no deleterious mutations. The Shwachman Bodian Diamond syndrome gene (SBDS) was sequenced to search for mutations in patients with no deleterious CFTR mutations and steatorrhea to determine if any had unrecognized Shwachman-Diamond syndrome (SDS).
Results
The presence of a common CF-causing mutation, absence of the vas deferens, and Pseudomona aeruginosa in the sputum correlated with the presence of two deleterious CFTR mutations, whereas sweat chloride concentration, diagnostic criteria for CF, and steatorrhea did not. However, sweat chloride concentration correlated with CFTR mutation status in patients infected with P aeruginosa. One patient had disease-causing mutations in each SBDS.
Conclusions
Presence of a common CF-causing mutation, absence of the vas deferens and/or P aeruginosa infection in a patient with features of nonclassic CF are predictive of deleterious mutations in each CFTR, whereas steatorrhea in the same context is likely to have etiologies other than CF transmembrane conductance regulator (CFTR) dysfunction.
Persons with classic cystic fibrosis (CF) have clinical manifestations in the pancreas, respiratory tract, male reproductive tract, and sweat gland.1 Approximately 10% of patients with CF have disease manifestations present in only some of the aforementioned organ systems. This incomplete CF phenotype has been termed pancreatic sufficient CF, atypical CF, and variant CF on the basis of organ involvement. Given the redundancy of these definitions, we and others have proposed to group the latter under the single descriptor “nonclassic” CF.2 Loss of function mutations in each copy of the CF transmembrane conductance regulator gene (CFTR) have been shown to cause classic CF, whereas mutations that causes partial loss of function in combination with loss of function mutations or other partial loss of function mutations in the other CFTR have been associated with nonclassic CF. The diagnosis of nonclassic CF can be difficult to make because of unusual presentations and borderline laboratory tests.3–6
Our laboratory has been investigating the role of CF transmembrane conductance regulator (CFTR) in patients with features of nonclassic CF. In a previous study, we found that 41% of the 74 patients studied did not have deleterious mutations identified after exhaustive scanning of the functional regions of CFTR.7 Analysis of clinical data from these patients did not reveal any significant differences in organ system involvement, degree of disease severity or sweat chloride concentration with two, one, or zero CFTR mutations. However, several trends were noted suggesting that analysis of a larger number of patients may reveal significant differences. In this study, we review findings from an expanded collection of 158 patients with features of nonclassic CF. We show that certain clinical manifestations are more likely to be associated with CFTR dysfunction as a result of deleterious CFTR mutations, whereas the presence of other manifestations should prompt a search for other etiologies.
METHODS
Patient Population
Patients were referred by clinicians experienced in the diagnosis of CF and were accepted into this study during a 5-year period from 1998 to 2003. At referral, each patient had one or no mutations after screening for the most common CF-causing mutations. Clinical data were collected for each patient using a form (http://www.hopkinsmedicine.org/cfgenotyping/index.htm) that asked the referring clinician to describe the CF-like clinical manifestations in each of the following organ systems: (1) CF-like respiratory disease defined as one or more of the following: sputum cultures positive for Pseudomonas aeruginosa, chronic productive cough, documented pneumonia, reactive airway disease, abnormal pulmonary function tests (pulmonary function tests were unavailable in 19 children <6 years of age, and 11 adults), abnormal chest x-ray and/or computed tomography (CT) indicating bronchiectasis, infiltrates, or blebs, chronic sinusitis, and/or nasal polyposis; (2) recurrent episodes of pancreatitis or malabsorption manifesting as steatorrhea (fecal fat values >7g/day during a 72-hour period). Fecal fat levels were not available in six patients. In these cases, steatorrhea was confirmed by reduced malabsorption following pancreatic enzyme supplementation (n = 4) or significantly reduced immunoreactive serum trypsinogen levels and clinical evidence of malabsorption (n = 2); and (3) male infertility as a result of absence of the vas deferens by semen analysis, rectal ultrasonography, or physical exam. Patients who had disease manifestations in all three organ systems were excluded. Each patient had two or more measurements of chloride concentration in sweat induced by pilocarpine iontophoresis.
Genotyping of CFTR was performed for each patient using direct DNA sequencing of all 27 exons and bordering intronic regions. Details of this technique have been reported elsewhere.7 A mutation was predicted to be deleterious if: (1) it had been previously reported as a disease-associated mutation (http://www.genet.sickkids.on.ca/cftr/); (2) it created a nonsense codon, induced a frameshift, or altered canonical splice-site sequences; and (3) it created an amino acid substitution in a residue that was conserved in CFTR of many species or was not found in at least 100 normal CFTR. A list of all identified mutations is available from the corresponding author. All studies were approved by The Johns Hopkins Joint Committee on Clinical Investigation, and written consent was obtained for all participants.
Statistical Analysis of Clinical Features
Because the role of CFTR function in patients with only one CFTR mutation is unclear, we performed comparisons of clinical features that were limited to patients with two mutations where CFTR dysfunction was highly likely (n = 57), versus those without any mutations where CFTR function was highly likely to be intact (n = 63). Univariate comparisons were performed for nine variables among patients with two or zero CFTR mutations: sweat chloride concentration, forced expiratory volume in 1 second, forced vital capacity, age of diagnosis, documented steatorrhea, congenital bilateral absence of the vas deferens (CBAVD), abnormal chest x-ray/CT, presence of P aeruginosa in sputum, and nasal polyps. Comparison of P aeruginosa and sweat chloride levels by mutation status was performed by Fisher's exact test. For sweat chloride concentration, forced expiratory volume in 1 second, forced vital capacity, and age at diagnosis we used one-way analysis of variance to compare values between the two groups, and for the remaining (discrete) variables we performed χ2 analysis. Fisher's exact test, analysis of variance, and χ2 analysis were performed using the JMP IN statistical package (version 3.2.6, SAS Institute, Inc., Cary, NC).
Binary logistic regression was performed where the dependent variable was the presence of two CFTR mutations, or not. The seven independent variables included: average sweat chloride concentration, age at diagnosis, steatorrhea, CBAVD, abnormal chest x-ray/CT, nasal polyps, and positive cultures for P aeruginosa. Measurements of pulmonary function were not reported for 36 patients, and were therefore not included in the logistic regression analysis. The logistic regression was first performed by including all variables in the model, as well as by using forward stepwise analysis. Logistic regression was performed using the Statistical Package for the Social Sciences for Windows (11.5.0, SPSS Inc., Chicago, Ill).
Genetic Analysis of SBDS
Steatorrhea as a result of pancreatic insufficiency is a common feature of patients with Shwachman-Diamond Syndrome (SDS; online mendelian inheritance in man [OMIM] 260400), which has recently been shown to be caused by mutations in the Shwachman-Bodian-Diamond syndrome gene (SBDS).8SBDS was analyzed in 18 patients who did not have two CFTR mutations, and who had steatorrhea. To find mutations in SBDS, exons and bordering intronic regions were amplified from genomic DNA using polymerase chain reactions (PCR). SBDS is approximately 5.8Mb from a pseudogene (ΨSBDS) that shares 97% nucleotide identity. Thus, PCR primers were designed to discriminate between SBDS and its highly conserved pseudogene.8 PCR products were purified using Qiaquick purification kits (Qiagen, Valencia, Calif ), sequenced using ABI dye-terminator sequencing reactions (Applied Biosystems, Foster City, Calif), and analyzed using the Sequencher analysis program (Gene Codes, Ann Arbor, Mich).
RESULTS
A total of 158 patients with manifestations compatible with nonclassic CF were enrolled in the study (Table I), 74 of whom were described in a previously published paper.7 Seventy-one patients were referred with a single mutation identified by standard DNA-based tests that typically identify approximately 85% of known CF-causing alleles, whereas 87 had no common CF-causing mutations. We identified two known deleterious or putative deleterious CFTR mutations in 57 patients, a single deleterious mutation in 38 patients, and no deleterious CFTR mutations in 63 patients. At referral, 78 patients met the diagnostic criteria for CF whereas 80 did not.9 We then examined whether mutation status at referral or diagnostic criteria at referral predicted which patients had CFTR dysfunction (ie, two deleterious CFTR mutations). Of the 57 patients with two deleterious CFTR mutations, 46 patients presented with a common CF-causing mutation (81% sensitivity), but only 31 met diagnostic criteria for CF (54% sensitivity). Of the 101 patients who did not have two deleterious CFTR mutations, 76 patients presented without a common CF-causing mutation (75% specificity), whereas 54 patients did not meet diagnostic criteria (53% specificity). Thus, presentation with a common CF-causing mutation (P <.000001), and not the CF diagnostic criteria (P = .41), was more predictive of CFTR dysfunction in a patient with clinical features of nonclassic CF.
Table I.
Clinical features of 158 patients enrolled in this study
| Respiratory | Gastrointestinal | Male reproductive | Two organ systems | |
|---|---|---|---|---|
| Sweat [Cl-] (mmol/L) | ||||
| >60 | 45 | 9 | 1 | 13 |
| 40–60 | 34 | 3 | 2 | 20 |
| <40 | 15 | 2 | 0 | 14 |
| Total | 94 | 14 | 3 | 47 |
To identify any major differences in clinical presentation between patients with CFTR dysfunction versus other etiologies, we compared clinical features between patients with two or zero deleterious CFTR mutations. Univariate analysis of key clinical features revealed that the mean sweat chloride concentration was near the diagnostic cutoff level for the two groups, and was neither sensitive nor specific for the presence of CFTR mutations (Figure 1). The extent of airways disease as reflected by pulmonary function tests, abnormal chest x-rays, and the frequency of nasal polyps were equally variable among the two genotype groups (Figures 1 and 2). Patients without CFTR mutations were diagnosed at an earlier age and had a higher frequency of steatorrhea than patients with 2 CFTR mutations (Figures 1 and 2). On the other hand, the frequency of congenital absence of the vas deferens and presence of P aeruginosa in the sputum on at least one occasion was significantly more common in patients with CFTR mutations (Figure 2).
Figure 1.
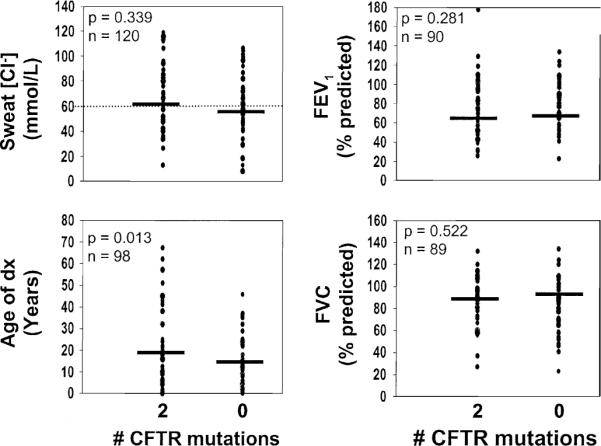
Comparison of quantitative features among patients with two or zero CFTR mutations. Each data point represents a value from a single patient, and horizontal bars indicate mean values. The diagnostic cutoff for sweat chloride concentration is shown with the horizontal dotted line.
Figure 2.
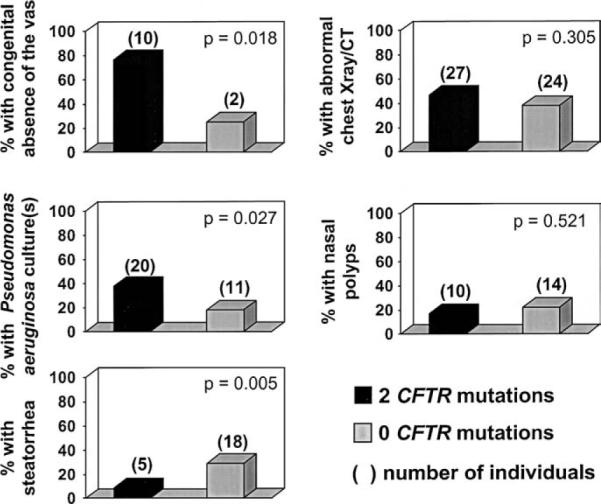
Comparison of the frequency of clinical features among patients with two or zero CFTR mutations. Gray bars represent the frequency of each feature in each genotype group, and numbers above the bars are the actual number of patients with the clinical feature in each group.
We also investigated whether particular combinations of clinical features correlated with the presence of deleterious CFTR mutations. Intriguingly, patients with P aeruginosa infections and sweat chloride concentrations <40 mmol/L were more likely to have zero CFTR mutations, whereas those with P aeruginosa infection and sweat chloride concentrations greater than >mmol/L were more likely to have two CFTR mutations (P = .003) (Figure 3). Binary logistic regression analysis using seven presenting features as independent variables resulted in a model that predicted 71.0% of the correct outcome for the dependent variable (presence of two or zero mutations). However, an alternative model using only three of the seven variables correctly predicted 66.8% of the outcome (Table II). Thus, the majority of the variation in the total regression model could be explained by the combination of absence of the vas deferens, positive P aeruginosa culture, and absence of steatorrhea.
Figure 3.
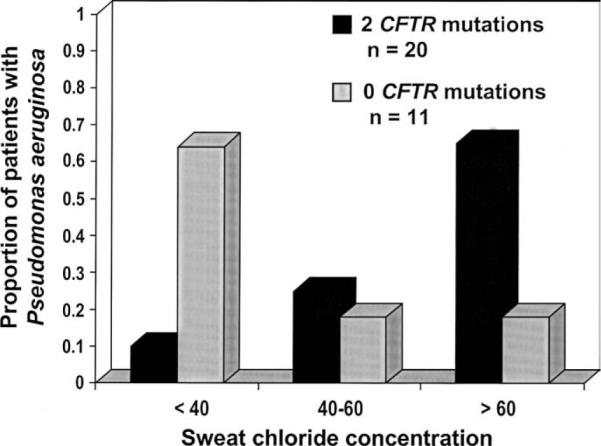
The proportion of patients with P aeruginosa infection and normal (<40 mmol/L), borderline (40–60 mmol/L), or elevated (>60 mmol/L) sweat chloride concentration between patients with either two or zero CFTR mutations. A higher proportion of patients with P aeruginosa infection and two CFTR mutations had elevated sweat chloride concentration, whereas patients with P aeruginosa infection but without CFTR mutations were more likely to have normal sweat chloride concentration.
Table II.
Logistic regression analysis using clinical features as independent variables and the presence of two CFTR mutations versus zero CFTR mutations as the dependent variable
| All entered |
Alternate model: three entered |
|||
|---|---|---|---|---|
| Independent variable | β | P value | β | P value |
| Absence of the vas deferens | 2.94 | 0.01 | 2.85 | .01 |
| Steatorrhea | −1.62 | 0.03 | −1.64 | .02 |
| P aeruginosa positive culture | 0.97 | 0.11 | 1.11 | .04 |
| Age at diagnosis | 0.02 | 0.44 | ||
| Average sweat Cl− | 0.01 | 0.61 | ||
| Nasal polyps | −0.92 | 0.13 | ||
| Abnormal chest x-ray/CT | 0.35 | 0.51 | ||
| Model χ2 | 25.9 | 22.60 | ||
| P value | P = .001 | P <.001 | ||
| R2 | 0.243 | 0.216 | ||
| % Correct prediction | 71.0 | 68.8 | ||
Because steatorrhea as a presenting feature in nonclassic patients was not correlated with CFTR dysfunction (ie, two deleterious CFTR mutations), we searched for another molecular etiology in this group of patients. Patients with SDS have steatorrhea because of pancreatic exocrine insufficiency as a result of hyposecretion of enzymes from pancreatic acinar cells, rather than obstruction of the pancreatic duct as seen in CF. Because patients with nonclassic CF or SDS can have steatorrhea, we examined the gene responsible for SDS (SBDS) in the 18 patients without CFTR mutations who had steatorrhea. Sequence analysis revealed that one of the 18 patients had the most common mutation known to cause SDS (258+2T−>C) and a single base pair deletion mutation in exon 1 (123delC). Analysis of the SBDS in each parent confirmed that these mutations did not occur in the same gene, but were inherited independently from each parent. The patient had steatorrhea, low serum trypsinogen, chronic sinusitis, asthma, recurrent pulmonary infections with Staphylococcus aureus positive cultures, and mild digital clubbing. Sweat chloride concentrations were 44 and 49 mmol/L on 70 and 60 mg of sweat, respectively. The patient exhibited several behavioral problems and was diagnosed with attention deficit disorder and Tourette's syndrome. Absolute neutrophil counts (ANC) over a 1-year period revealed mild, variable neutropenia (ANC 600–1700/mL), with normal platelets (158,000–190,000/mL), and hemoglobin (12.1–13.7 g/dL). There were no long bone or rib abnormalities evident upon examination of x-rays.
DISCUSSION
The diagnosis of CF is based primarily on clinical manifestations accompanied by evidence of CFTR dysfunction such as elevated sweat chloride concentration, a characteristic nasal potential difference profile, or two deleterious CFTR mutations.9 Although the diagnostic criteria are sensitive and specific for classic CF as a result of CFTR dysfunction,1 the criteria were not able to accurately identify those with CFTR involvement (as defined by the presence of known deleterious or predicted deleterious mutations in each CFTR) in these patients with features of nonclassic CF. Furthermore, an ion transport defect documented by elevated sweat chloride concentration did not differentiate patients with two or zero CFTR mutations. This finding demonstrates that the “gold standard” test for classic CF was neither sensitive nor specific for CFTR dysfunction in these patients. Nasal potential difference measurement may provide a more sensitive assay for ion transport defects that could differentiate etiology in patients with nonclassic CF.10–12 Unfortunately, this test was performed on too few patients to incorporate into our analyses. However, the presence of one common CF-causing mutation was a useful predictor of those with CFTR dysfunction as the etiology of their nonclassic CF phenotype.
In addition to the presence of a common CF mutation, two clinical features, absence of the vas deferens and infection with P aeruginosa, were highly associated with the presence of two deleterious CFTR mutations in patients presenting with nonclassic CF. Although absence of the vas deferens in men with nonclassic CF is a sensitive clinical indicator of CFTR involvement, only 36% of males in this study were examined for CBAVD by either semen analysis, palpation of the vas, or ultrasonography. This is likely to be a result of the fact that many males are not screened for CBAVD until it presents as infertility in adulthood. The results of this study suggest that an evaluation for reproductive tract abnormalities should be considered for every male who presents with clinical features of nonclassic CF.
Infection with P aeruginosa is a common feature of classic CF but a highly variable feature in patients with nonclassic CF. However, two observations demonstrate a close relationship between infection with this organism, CFTR dysfunction, and abnormal ion transport in patients with nonclassic CF. First, patients with two deleterious CFTR mutations were significantly more likely to have a sputum culture positive for P aeruginosa. Second, in this study patients with two CFTR mutations and P aeruginosa were more likely to have elevated sweat chloride concentration, whereas patients with zero CFTR mutations and P aeruginosa were more likely to have a normal sweat chloride concentration. These findings are somewhat unexpected because sweat chloride concentration does not differ between genotype groups as a whole, but differs only when considering patients with P aeruginosa infection. The positive correlation between P aeruginosa infection and sweat chloride concentration in patients with CFTR defects suggests that susceptibility to infection is specifically correlated with CFTR dysfunction and an electrolyte transport defect.
Pancreatic insufficiency seen in patients with CF is typically associated with complete loss of CFTR function that also results in the development of severe lung disease and highly elevated sweat electrolytes.13 Most mutations that cause severe loss of CFTR function occur in the coding regions of CFTR and should be readily identified by the genetic analysis performed here.1 Thus, the large number of patients with nonclassic CF referred with steatorrhea in the absence of CFTR mutations was unexpected. However, other studies have reported patients with atypical or mild presentations of CF with pancreatic insufficiency in the absence of two CFTR mutations.3,14,15 The majority of patients in our study had steatorrhea documented by elevated fecal fat levels. This method does not discriminate between fat malabsorption as a result of pancreatic exocrine insufficiency versus primary gastrointestinal absorptive defects, indicating that other causes of steatorrhea were possible.
The latter concept was evaluated by sequencing the SBDS in each of the patients with nonclassic CF with steatorrhea who had no deleterious CFTR mutations. One patient was found to have a loss of function mutation in each SBDS that would predict a classic presentation of SDS. However, the borderline elevated sweat chloride concentration contributed to a diagnosis of nonclassic CF in this patient. In retrospect, the sweat chloride concentrations were determined from low volumes of sweat, which can in some cases give erroneous quantification of electrolytes.16 Furthermore, the patient exhibited transient neutropenia that was not recognized as being consistent with SDS.17 This patient appears to be an atypical form of SDS rather than CF. Given that only 1 of 18 patients with steatorrhea had SDS, unrecognized SDS is not likely to account for a large fraction of nonclassic CF cases with steatorrhea.
Ascertaining the etiology of atypical presentations of single gene disorders can be difficult, and often cannot be based on clinical or genetic data alone. Recognizing that molecular abnormalities other than CFTR dysfunction can masquerade as nonclassic CF should trigger re-evaluation of the etiology in certain nonclassic cases. In such cases, integration of clinical, genetic and nongenetic information may help in classifying the phenotypic variability of nonclassic CF into distinct etiologic subgroups.18 This may be of particular importance for patients with features of nonclassic CF, as prognosis and treatment is likely to depend on the etiology of their disease.
Acknowledgments
We are indebted to the DNA Diagnostic Laboratory at Johns Hopkins and the clinicians and staff of the CF Centers who referred patients. Most of all, we would like to thank the patients and families for their willingness to participate in this study.
This study was funded by grants from the NIH and the North American CF Foundation to G.R.C.
Glossary
- CF
Cystic fibrosis
- CFTR
Cystic fibrosis transmembrane conductance regulator
- CFTR
Cystic fibrosis transmembrane conductance regulator gene
- CT
Computed tomography
- CBAVD
Congenital bilateral absence of the vas deferens
- PCR
Polymerase chain reactions
- SDS
Shwachman-Diamond syndrome
- SDBS
Shwachman-Bodian-Diamond syndrome
REFERENCES
- 1.Welsh MJ, Ramsey BW, Accurso FJ, Cutting GR. Cystic fibrosis. In: Scriver CR, Beaudet AL, Valle D, Sly WS, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill, Inc.; New York: 2001. pp. 5121–88. [Google Scholar]
- 2.Knowles MR, Durie PR. What is cystic fibrosis? N Engl J Med. 2002;347:439–42. doi: 10.1056/NEJMe020070. [DOI] [PubMed] [Google Scholar]
- 3.Wallis C. Atypical cystic fibrosis: diagnostic and management dilemmas. J R Soc Med. 2003;96(suppl 43):2–10. [PMC free article] [PubMed] [Google Scholar]
- 4.Boyle MP. Nonclassic cystic fibrosis and CFTR-related diseases. Curr Opin Pulm Med. 2003;9:498–503. doi: 10.1097/00063198-200311000-00009. [DOI] [PubMed] [Google Scholar]
- 5.Bush A, Wallis C. Time to think again: cystic fibrosis is not an “all or none” disease. Pediatr Pulmonol. 2000;30:139–44. doi: 10.1002/1099-0496(200008)30:2<139::aid-ppul9>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 6.Rosenstein BJ. Nonclassic cystic fibrosis: a clinical conundrum. Pediatr Pulmonol. 2003;36:10–2. doi: 10.1002/ppul.10286. [DOI] [PubMed] [Google Scholar]
- 7.Groman JD, Meyer ME, Wilmott RW, Zeitlin PL, Cutting GR. Variant cystic fibrosis phenotypes in the absence of CFTR mutations. N Engl J Med. 2002;347:401–7. doi: 10.1056/NEJMoa011899. [DOI] [PubMed] [Google Scholar]
- 8.Boocock GR, Morrison JA, Popovic M, Richards N, Ellis L, Durie PR, et al. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 2003;33:97–101. doi: 10.1038/ng1062. [DOI] [PubMed] [Google Scholar]
- 9.Rosenstein BJ, Cutting GR. The diagnosis of cystic fibrosis: a consensus statement. J Pediatr. 1998;132:589–95. doi: 10.1016/s0022-3476(98)70344-0. [DOI] [PubMed] [Google Scholar]
- 10.Delmarco A, Pradal U, Cabrini G, Bonizzato A, Mastella G. Nasal potential difference in cystic fibrosis patients presenting borderline sweat test. Eur Respir J. 1997;10:1145–9. doi: 10.1183/09031936.97.10051145. [DOI] [PubMed] [Google Scholar]
- 11.Wilson DC, Ellis LE, Zielenski J, Corey M, Ip WF, Tsui LC, et al. Uncertainty in the diagnosis of cystic fibrosis: possible role of in vivo nasal potential difference measurements. J Pediatr. 1998;132:596–9. doi: 10.1016/s0022-3476(98)70345-2. [DOI] [PubMed] [Google Scholar]
- 12.Wilschanski M, Famini H, Strauss-Liviatan N, Rivlin J, Blau H, Bibi H, et al. Nasal potential difference measurements in patients with atypical cystic fibrosis. Eur Respir J. 2001;17:1208–15. doi: 10.1183/09031936.01.00092501. [DOI] [PubMed] [Google Scholar]
- 13.Kerem E, Corey M, Kerem B-S, Rommens J, Markiewicz D, Levison H, et al. The relation between genotype and phenotype in cystic fibrosis: analysis of the most common mutation (deltaF508) N Engl J Med. 1990;323:1517–22. doi: 10.1056/NEJM199011293232203. [DOI] [PubMed] [Google Scholar]
- 14.Wine JJ, Kuo E, Hurlock G, Moss RB. Comprehensive mutation screening in a cystic fibrosis center. Pediatrics. 2001;107:280–6. doi: 10.1542/peds.107.2.280. [DOI] [PubMed] [Google Scholar]
- 15.Hughes D, Dork T, Stuhrmann M, Graham C. Mutation and haplotype analysis of the CFTR gene in atypically mild cystic fibrosis patients from Northern Ireland. J Med Genet. 2001;38:136–9. doi: 10.1136/jmg.38.2.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LeGrys VA, Burritt MF, Gibson LE, Hammond KB, Kraft K, Rosenstein BJ. Sweat Testing: Sample Collection and Quantitative Analysis; Approved Guideline. NCCLS; Villlanova, Pa: 1994. [Google Scholar]
- 17.Ginzberg H, Shin J, Ellis L, Morrison J, Ip W, Dror Y, et al. Shwachman syndrome: phenotypic manifestations of sibling sets and isolated cases in a large patient cohort are similar. J Pediatr. 1999;135:81–8. doi: 10.1016/s0022-3476(99)70332-x. [DOI] [PubMed] [Google Scholar]
- 18.Robin NH, Biesecker LG. Considerations for a multiaxis nomenclature system for medical genetics. Genet Med. 2001;3:290–3. doi: 10.1097/00125817-200107000-00004. [DOI] [PubMed] [Google Scholar]