

Abstract
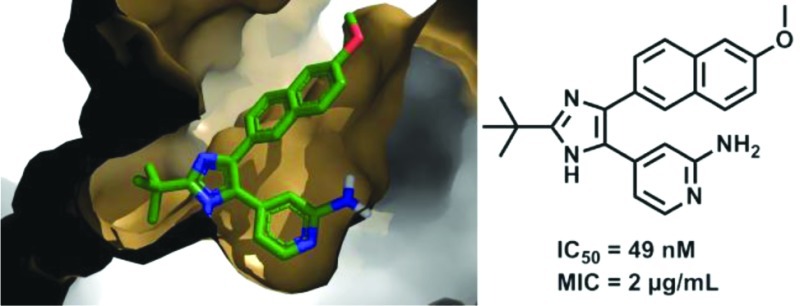
Mycobacterium tuberculosis glutamine synthetase (MtGS) is a promising target for antituberculosis drug discovery. In a recent high-throughput screening study we identified several classes of MtGS inhibitors targeting the ATP-binding site. We now explore one of these classes, the 2-tert-butyl-4,5-diarylimidazoles, and present the design, synthesis, and X-ray crystallographic studies leading to the identification of MtGS inhibitors with submicromolar IC50 values and promising antituberculosis MIC values.
Introduction
Mycobacterium tuberculosis, the causative agent of tuberculosis, is one of the world’s most deadly pathogens, leading to almost 1.7 million deaths annually despite more than 100 years of research.1 Rapidly emerging drug-resistant and multidrug-resistant strains have provided additional impetus to the search for new therapeutic agents to combat the disease.2,3M. tuberculosis glutamine synthetase (MtGS, EC 6.3.1.2) catalyzes the conversion of glutamate, ammonia, and ATP to glutamine, phosphate, and ADP.4MtGS plays a key role in controlling the ammonia levels within infected host cells and so contributes to the pathogen’s capacity to inhibit phagosome acidification and phagosome–lysosome fusion.5,6 Furthermore, MtGS is believed to be involved in cell wall biosynthesis; it is found extracellularly in large quantities, which is related to a role in the production of the poly-l-glutamate–glutamine that is a major component of the cell wall in pathogenic mycobacteria.7 Treatment of M. tuberculosis with antisense oligonucleotides to GS mRNA8 or with the GS inhibitor l-methionine-S-sulfoximine (MSO, 1, Figure 1a)9 inhibits biosynthesis of poly-l-glutamate–glutamine and the bacterial growth.8,9 Compound 1 also shows in vivo efficacy in a guinea pig model, suggesting GS as a promising and druggable target in the treatment of M. tuberculosis.10 The structure of MtGS incubated with 1, which is phosphorylated in situ to form the transition-state analogue l-methionine-S-sulfoximine phosphate (MSO-P, 2, Figure 1a), has been reported showing how the inhibitor interacts in the amino acid binding site.11
Figure 1.

(a) Reference GS inhibitor 1 and phosphorylated transition-state analogue 2. These two compounds bind to the amino acid-binding site of MtGS. (b) Common scaffold of the HTS hits of cluster 2. (c, d) Two most potent compounds from cluster 2 in the HTS study. Compounds 3 and 7a interact with the ATP-binding site of MtGS.12
In the present study we explore a new class of MtGS inhibitors derived from a recent high throughput screening (HTS) study.12,13 This screen targeted the ATP-binding site of MtGS with the goal of moving away from the amino acid binding site,14 which is largely conserved in human GS. The 2-tert-butyl-4-aryl-5-pyridylimidazole motif was the common structure in the hits assigned to cluster 2, with the 4-position substituted with various aryl groups (Figure 1b). From more than 20 compounds in this class, the two best were chosen for further SAR studies (3 and 7a in Figure 1c,d).
Results and Discussion
We first sought a synthetic route to the two most active HTS hits in order to resynthesize them, confirm their activity, and initiate X-ray crystallographic studies. This proved to be problematic; despite exploration of various synthetic routes, we were never able to resynthesize 3. However, the trisubstituted imidazole 7a could be readily synthesized and was therefore used in the hit expansion of this cluster. The synthesis started from 2-bromo-6-methoxynaphthalene (4) with two consecutive Sonogashira couplings followed by an oxidation and cyclization to form the imidazole ring (see Scheme 1). The ethyne was introduced by a fast microwave assisted15 Sonogashira method16,17 using ethynyltrimethylsilane, dichlorobis(triphenylphosphine)palladium, copper iodide, and acetonitrile/diethylamine (1:1) as solvent. In situ deprotection with saturated K2CO3 in methanol gave the 2-ethynyl-6-methoxynapthalene (5) in 85% yield. By use of the same protocol, the 4-pyridyl moiety was incorporated in moderate yield (6a, 47%). Oxidation of the ethyne 6a to the diketone intermediate with potassium permanganate18 in aqueous acetone proved to be a very sensitive reaction and often led to oxidative cleavage of the triple bond. Changing the standard buffer system NaHCO3/MgSO4 to NaH2PO4/Na2HPO4, and thus lowering the pH from 7.5 to 5, provided a more robust protocol to furnish the diketone. In our attempts to prepare 3, the identical oxidation conditions with the corresponding quinoline intermediate only gave overoxidation, and no conditions were identified that suppressed oxidative cleavage. Finally the imidazole ring was synthesized by in situ trapping of the diketone with various aldehydes and ammonium acetate allowing straightforward variation of the tert-butyl group position (R2). The resynthesized HTS hit 7a had IC50 = 3.1 μM, which is 30-fold higher than the IC50 obtained in the HTS study (0.1 μM), due to our more stringent assay conditions.12 Unfortunately, neither 3 (from the AstraZeneca library) nor the resynthesized 7a was active in the mycobacterial growth assay (MIC > 32 μg/mL).
Scheme 1. Synthetic Route to Compounds 7a–m.

Reagents and conditions: (a) ethynyltrimethylsilane, Pd(PPh3)2Cl2, CuI, MeCN, diethylamine, microwave 120 °C, 15 min, then K2CO3, MeOH, rt, 2 h, 85%; (b) bromoaryl/heteroaryl, Pd(PPh3)2Cl2, CuI, MeCN, diethylamine, microwave 80–120 °C, 15 min, 22–63%; (c) KMnO4, phosphate buffer; (d) aldehyde, ammonium acetate, n-butanol, 50–65 °C, 0.5–5 h, 10–63%.
Since numerous 4-substituted arylimidazoles of 7a (Figure 1) had already been identified and evaluated in the HTS study, we focused instead on optimizing positions 2 (the tert-butyl group) and 5 (the 4-pyridyl group). The importance of the position of the nitrogen in the 4-pyridyl ring was first examined by synthesizing the phenyl and pyrimidine analogues (Table 1, 7b–d). The same synthetic strategy as described above was applied, replacing the 4-pyridyl with aryl/heteroaryl groups introduced in the second Sonogashira reaction. Cyclization with pivalaldehyde gave the targets 7b–d in which the nitrogen was either removed (7b) or its position altered (7c–d). The results clearly indicate the importance of having a nitrogen in the 4-position of the pyridine ring; all compounds lacking this feature were inactive (IC50 > 25 μM).
Table 1. Activities of Trisubstituted Imidazoles.

To investigate the importance of steric bulk in position 2 of the imidazole ring, compounds with smaller alkyl chains were synthesized. Cyclizing the diketone with propionaldehyde, acetaldehyde, and formaldehyde gave ethyl, methyl, and hydrogen, respectively, at position 2 of the imidazole ring (7e–g in Table 1). Removal of two methyl groups generated a slightly more active compound (7e, 2.2 μM vs 7a, 3.1 μM). Removal of yet another methyl group yielded 7f, which was inactive. Likewise, complete removal of the tert-butyl group gave the inactive 7g. Clearly, there is a preference for relatively bulky substituents in this position.
Parallel studies at this point produced the structure of MtGS in complex with 2, 3 (obtained from AstraZeneca’s in-house compound library), phosphate, and magnesium at 2.15 Å resolution (crystallographic R-factor 22.5%, Figure 2a). An overlay of this structure on that of the complex with 2, ADP, and magnesium (PDB entry 2BVC)11 is shown in Figure 2b; an rmsd of 0.2 Å is obtained when the Cα's of residues 4–478 in the A chains are compared. Compound 2 occupies exactly the same position in the amino acid binding site in each case; all three of the metal ions are also in equivalent locations. The 4-pyridyl moiety of 3 is at essentially the same place as the six-membered pyrimidine ring of ADP, with the inhibitor’s ring nitrogen providing the only hydrogen bond to protein (via the side chain of Ser280) in an interaction equivalent to that of N1 of ADP’s adenine ring. The interaction with this serine explains why a nitrogen at the 4-position of the pyridine ring is so important for binding affinity (vide supra). The imidazole ring and tert-butyl moieties of 3 occupy roughly the same space as the ribose of the nucleotide. Thus, the R2 group is found at the end of the ribose proximal to its linkage with the phosphates. A phosphate ion assumes the position of the β-phosphate of ADP. The quinoline moiety extends out toward the solvent, making van der Waals interactions with the protein only near Ala362 (not shown). Correspondingly, the electron density of this group is poorer than that of the rest of the ligand, although density in the maps averaged over all six subunits is clear (see Supporting Information). Efforts to cocrystallize 7a with MtGS were unsuccessful. However, we hypothesize that the binding modes of 3 and 7a are very similar, which is supported by our docking studies. The comparisons in Figure 2b led us to make 7i; docking studies indicated that the hydroxyl group of this compound could occupy the same position as O3′ of ADP and form an equivalent hydrogen bond to the backbone carbonyl of Tyr230 (Figure 2b). In the original synthetic route to 7i, the hydroxyl was benzyl protected (7h) to ease the synthesis. However, all attempts to deprotect the corresponding alcohol by hydrogenation or oxidative protocols were unsuccessful. Compound 7h was evaluated in the MtGS assay but did not show any inhibitory activity at 25 μM. An alternative protective group strategy was then employed in which 2-(tert-butyldimethylsilyloxy)acetaldehyde was instead used in the imidazole ring cyclization. The silyloxy group allowed smooth deprotection with tetrabutylammonium fluoride to yield 7i. Disappointingly, 7i was also inactive in the enzymatic assay. The remaining compounds (7j–m) in Table 1 were similarly suggested to reach into the sugar-binding site of the ATP pocket. In the final step of the synthesis leading up to these compounds, aldehydes are used as reagents (see Scheme 1). We therefore created a set of virtual compounds by merging aldehydes found in-house and in the ACD database, using the Legion software.19 The compounds were then docked to the nucleotide-binding site using Glide, and the fit was evaluated based on the Glide score and visual inspection. Despite their promising docking poses, 7j–m all lacked inhibitory activity at 25 μM.
Figure 2.
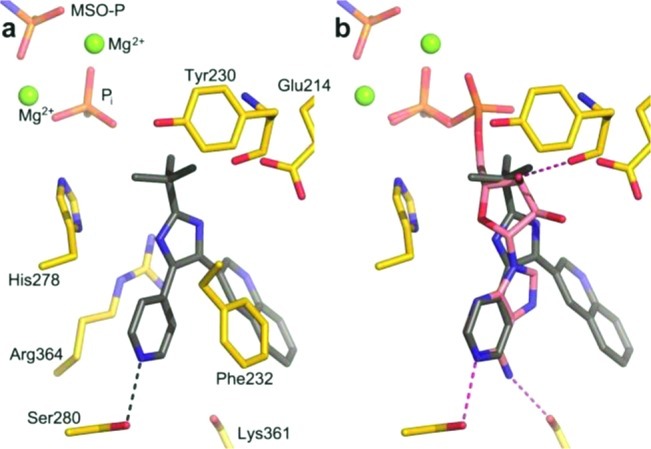
(a) MtGS (yellow carbons) in complex with 2 (MSO-P), 3 (gray carbons), phosphate, and magnesium. Hydrogen bond between 3 and Ser280 is shown as a black dashed line. Nearby magnesium ions are green spheres. (b) Superposition of the MtGS structure with 3 on that with bound ADP (PDB entry 2BVC) allows comparison to nucleotide (pink carbons) binding. Protein/ADP hydrogen bonds are pink dashed lines.
Going back to the superimposed X-ray structures, we decided to substitute the 4-pyridyl group of 7a with a 2-aminopyridine-4-yl, which would possibly allow the formation of a hydrogen bond to the backbone carbonyl oxygen of Lys361, similar to the interaction seen for N6 of ADP (Figure 2b). Another synthetic approach was employed in the synthesis of the aminopyridines 11a–d, avoiding the sensitive oxidation of the triple bond (Scheme 2). Ethyl 6-methoxy-2-naphthoate (8) was synthesized in 93% yield by a palladium-catalyzed carbonylation of 2-bromo-6-methoxynaphthalene (4) utilizing ethanol as the nucleophile.20 In the next step, the fluorine of 2-fluoro-4-methylpyridine was incorporated as a handle to allow nucleophilic aromatic substitutions in later steps. Compound 9 was produced in acceptable yield (64%) by deprotonation of 2-fluoro-4-methylpyridine with NaHMDS via nucleophilic substitution of 8. As an alternative to creating the imidazole ring from the diketone, an α-bromination was performed on 9 followed by cyclization with ammonium acetate and pivalaldehyde. Attempts to resynthesize the HTS hit 3 by this route again failed because we were never able to substitute the ethoxy of ethyl quinoline-3-carboxylate with 4-methylpyridine. At this point, it was encouraging to see that the fluorine in 10 contributed to an approximate 2.5-fold increase of affinity compared to the hit 7a (IC50 of 1.2 μM vs 3.1 μM, Table 1). The aminopyridine 11a was synthesized via microwave-assisted nucleophilic substitution of the fluorine of 10 with diphenylmethanamine followed by deprotection through catalytic hydrogenation. Remarkably, 11a was over 60 times better than the reference 7a (0.049 μM vs 3.1 μM, Table 1), suggesting an important new interaction with the enzyme. For comparison, the methylamino and the dimethylamino groups were incorporated in a similar way. Compound 11b, having one methyl group, showed a 100-fold loss in activity compared to 11a, making it even less potent than 7a. Substituting the fluorine with a dimethylamino group resulted in 11c, which was inactive. This suggested that having a hydrogen bond donating group in the 2-position of the pyridine ring was advantageous. As an alternative donating group, we introduced the 2-pyridone ring (11d), which we believed would bind in its 2-hydroxy tautomer. However, on the basis of the lack of activity of this compound, one can speculate that it predominantly exists in the pyridone form.
Scheme 2. Synthetic Route to Compounds 11a–d.

Reagents and conditions: (a) EtOH, Pd(OAc)2, Xantphos, DBU, Mo(CO)6, microwave 120 °C, 30 min, 93%; (b) 2-fluoro-4-methylpyridine, NaHMDS, THF, 0 °C, 2 h, 64%; (c) HBr, DMSO, 70 °C, 2 h, then pivalaldehyde, ammonium acetate, n-butanol, 50 °C, 2 h, 71%. (d) 11a: (i) diphenylmethanamine, dioxane, microwave 200 °C, 10 h, (ii) Pd/C, NH4OAc, MeOH, microwave 120–140 °C, 2 h, 28%. 11b: methylamine (2.0 M in THF), microwave 150 °C, 17 h, 18%. 11c: ammonium hydroxide, DMF, microwave 150 °C, 12 h, 85%. 11d: AcOH, H2O, microwave 190 °C, 2 h, 91%.
At this time, we succeeded in obtaining a crystal structure of 11a bound to MtGS together with 2, phosphate, and magnesium at 2.26 Å resolution (R-factor 19.5%, Figure 3a). The complexes with 3 and 11a are overlaid in Figure 3b; again, the protein structures are highly similar, with an rmsd of only 0.1 Å when the Cα's of residues 4–478 are compared. Ser280 of MtGS was seen to interact with both nitrogens of the 2-aminopyridine-4-yl moiety of 11a (only the hydrogen bond with the ring nitrogen was present in the complex with 3), which shifts the ligand ∼0.5 Å deeper into the binding site. Inspection of the 11a complex indicates that the distance is too great (∼3.5 Å) to support a hydrogen bond between the 2-amino group and the backbone carbonyl oxygen of Lys361. Apart from very small changes at the side chain of Phe232 and the main chain near residue Ala362, the protein structures are essentially identical. The compounds that inhibited MtGS were also evaluated for their activity against the M. tuberculosis strain H37Rv. The hit 7a and 7e lacked antibacterial activity (MIC > 32 μg/mL), while the 2-pyridyl substituted compounds had promising MIC values; the most potent compound as measured by the enzymatic inhibition, 11a, also gave the best MIC (2 μg/mL; see Table 1). To assess the potential cytotoxicity of 11a, we performed a standard mammalian cell proliferation assay22 using A549 cells; 11a had an IC50 of 24.7 μm in this test, which is almost 5-fold greater than the MIC of 5.4 μM (2 μg/mL). This moderate level of cytotoxicity is not unusual among compounds in the early stages of drug development, and the situation is expected to improve as more effective enzyme inhibition and antibacterial activity are attained.
Figure 3.
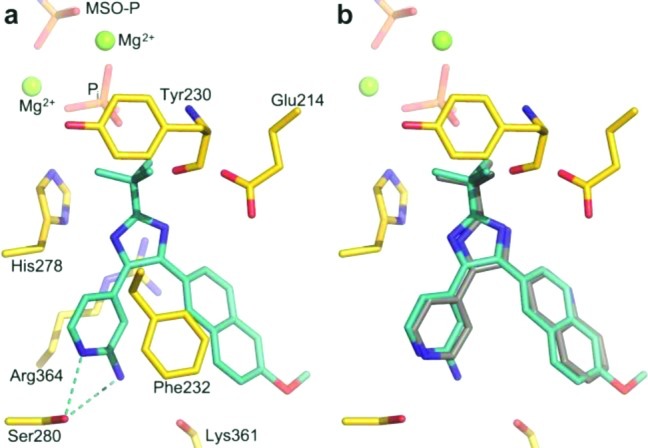
(a) MtGS (yellow carbons) in complex with 2, 11a (cyan carbons), phosphate, and magnesium. Hydrogen bonds are shown as cyan dashed lines. (b) Superposition of the protein allows comparison of binding of 3 (gray carbons) and 11a.
Conclusions
A recent HTS study targeting the ATP-binding site of MtGS produced several active classes of compounds. One of these, the 2-tert-butyl-4,5-diarylimidazoles, contained several hits, and a complex with one of the inhibitors with MtGS could be obtained by X-ray crystallography. On the basis of this structure, our initial SAR explorations could be rationalized as well as the importance of having a nitrogen, acting as a hydrogen bond acceptor, in the 4-position of the pyridine ring. Building out from the tert-butyl group, a series of compounds was designed to reach into the ribose-binding site of the ATP pocket. However, these compounds lacked inhibitory activity at 25 μM. An alternative synthetic strategy in which a 2-amino group was introduced into the 4-pyridyl ring was then pursued. This gave us our best inhibitor (11a) with IC50 = 0.049 μM on MtGS and an MIC = 2 μg/mL against M. tuberculosis. We were also able to obtain an X-ray structure of this compound bound to MtGS, which showed that instead of forming a hydrogen bond to the backbone carbonyl oxygen of Lys361 as predicted from docking studies, the 2-amino group formed an additional interaction with the hydroxyl oxygen of Ser280 in its primary binding mode.
Experimental Procedures
General Methods
Microwave-assisted reactions were performed in sealed vials dedicated for microwave processing, using a Smith synthesizer. NMR spectra were recorded on a Varian Mercury Plus for 1H at 399.9 MHz and for 13C NMR at 100.5 MHz. Analytical HPLC–UV/MS analysis of pure products were performed on a Gilson HPLC system with a Chromolith SpeedROD RP-18e column (50 mm × 4.6 mm) equipped with a Finnigan AQA quadrupole mass spectrometer using a 4 mL/min CH3CN/H2O gradient (0.05% HCOOH) and detection by UV (DAD) and MS (ESI+). All compounds were determined to be >95% pure by HPLC–UV at 254 nm.
4-(2-tert-Butyl-4-(6-methoxynaphthalen-2-yl)-1H-imidazol-5-yl)pyridin-2-amine (11a)
To a microwave vial (2–5 mL) were added 10 (50 mg, 0.13 mmol), diphenylmethanamine (1.5 mL), and dioxane (1.5 mL). The vial was then sealed under air and heated at 200 °C by microwave irradiation for 10 h. After cooling, the mixture was filtered through a plug of silica, eluted with EtOAc/isohexane (1:1) and the filtrate concentrated in vacuo. The crude mixture was then taken up in MeOH (2.0 mL) and transferred to a microwave vial (2–5 mL) loaded with Pd/C (10%, 5 mg) and ammonium acetate (100 mg, 1.3 mmol). The vial was then sealed under air and heated at 120 °C by microwave irradiation for 2 h. After the mixture was cooled, Pd/C (10%, 10 mg) and ammonium acetate (200 mg, 2.6 mmol) were added, and the mixture was heated by microwave irradiation for a further 20 min at 140 °C. After cooling, the mixture was diluted with EtOAc and saturated NaHCO3 (10 mL each) and the two layers were separated. The aqueous layer was washed twice with EtOAc (10 mL), and the combined organic phases were concentrated in vacuo. The crude product was thereafter purified by silica column flash chromatography, eluting with EtOAc/methanol/triethylamine (1:0.05:0.01). Yield: 28%, 14 mg as a pale yellow solid. 1H NMR (CDCl3) δ 7.81 (d, J = 1.9 Hz, 1H), 7.72 (d, J = 8.6 Hz, 1H), 7.69 (m, 1H), 7.51 (dm, J = 6.1 Hz, 1H), 7.40 (dd, J = 8.6, 1.9 Hz, 1H), 7.16 (m, 1H), 7.14 (m, 1H), 6.91 (br s, 1H), 6.65 (dm, J = 6.1 Hz, 1H), 3.91 (s, 3H), 1.43, (s, 9H); 13C NMR (CDCl3/CD3OD, 5:1) δ 170.3, 158.6, 157.4, 156.9, 146.6, 140.5, 134.5, 129.7, 128.9, 127.8, 127.6, 127.2, 126.4, 119.8, 111.7, 107.5, 106.0, 55.6, 33.2, 29.6 (one carbon signal was not detected).21 ESI-MS (m/z) 373 (M + H+). HRMS (M + H+): 373.2037, C23H24N4O requires 373.2028.
Acknowledgments
We thank Johan Gustavsson for preparative work and Dr. Aleh Yahorau, Department of Pharmaceutical Biosciences, Uppsala University, Sweden, for conducting HRMS analyses. The Swedish Foundation for Strategic Research (SSF), the Swedish Research Council (VR), and the EU Sixth Framework Program NM4TB (Grant CT:018 923) provided financial support.
Glossary
Abbreviations Used
- MtGS
glutamine synthetase from Mycobacterium tuberculosis
- MIC
minimum inhibitory concentration
- PDB
Protein Data Bank
- WHO
World Health Organization
- HTS
high throughput screening
- MSO
l-methionine-S-sulfoximine
- MSO-P
l-methionine-S-sulfoximine phosphate
- MW
microwave
Supporting Information Available
Additional experimental details concerning synthesis of all compounds, protein expression, purification, activity/inhibition studies, spectroscopic data, and structural studies. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Accession Codes
† PDB codes for the MtGS-3 and MtGS-11a structures are 3ZXR and 3ZXV, respectively.
Author Contributions
⊥ J.G. and M.T.N. contributed equally to this work.
Supplementary Material
References
- www.who.org.
- Raviglione M. C.; Smith I. M. XDR tuberculosis: implications for global public health. N. Engl. J. Med. 2007, 356, 656–659. [DOI] [PubMed] [Google Scholar]
- Migliori G. B.; Lange C.; Centis R.; Sotgiu G.; Muetterlein R.; Hoffmann H.; Kliiman K.; De laco G.; Lauria F. N.; Richardson M. D.; Spanevello A.; Cirillo D. M.; Grp T. S. Resistance to second-line injectables and treatment outcomes in multidrug-resistant and extensively drug-resistant tuberculosis cases. Eur. Respir. J. 2008, 31, 1155–1159. [DOI] [PubMed] [Google Scholar]
- Liaw S. H.; Eisenberg D. Structural model for the reaction mechanism of glutamine synthetase, based on 5 crystal structures of enzyme substrate complexes. Biochemistry 1994, 33, 675–681. [DOI] [PubMed] [Google Scholar]
- Gordon A. H.; Darcyhart P.; Young M. R. Ammonia inhibits phagosome lysosome fusion in macrophages. Nature 1980, 286, 79–80. [DOI] [PubMed] [Google Scholar]
- Clemens D. L.; Horwitz M. A. Characterization of the Mycobacterium tuberculosis phagosome and evidence that phagosomal maturation is inhibited. J. Exp. Med. 1995, 181, 257–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harth G.; Clemens D. L.; Horwitz M. A. Glutamine-synthetase of Mycobacterium tuberculosis: extracellular release and characterization of its enzymatic-activity. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 9342–9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harth G.; Zamecnik P. C.; Tang J. Y.; Tabatadze D.; Horwitz M. A. Treatment of Mycobacterium tuberculosis with antisense oligonucleotides to glutamine synthetase mRNA inhibits glutamine synthetase activity, formation of the poly-l-glutamate/glutamine cell wall structure, and bacterial replication. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harth G.; Horwitz M. A. An inhibitor of exported Mycobacterium tuberculosis glutamine synthetase selectively blocks the growth of pathogenic mycobacteria in axenic culture and in human monocytes: extracellular proteins as potential novel drug targets. J. Exp. Med. 1999, 189, 1425–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harth G.; Horwitz M. A. Inhibition of Mycobacterium tuberculosis glutamine synthetase as a novel antibiotic strategy against tuberculosis: demonstration of efficacy in vivo. Infect. Immun. 2003, 71, 456–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski W. W.; Jones T. A.; Mowbray S. L. Structure of Mycobacterium tuberculosis glutamine synthetase in complex with a transition-state mimic provides functional insights. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 10499–10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson M. T.; Krajewski W. W.; Yellagunda S.; Prabhumurthy S.; Chamarahally G. N.; Siddamadappa C.; Srinivasa B. R.; Yahiaoui S.; Larhed M.; Karlén A.; Jones T. A.; Mowbray S. L. Structural basis for the inhibition of Mycobacterium tuberculosis glutamine synthetase by novel ATP-competitive inhibitors. J. Mol. Biol. 2009, 393, 504–513. [DOI] [PubMed] [Google Scholar]
- Odell L. R.; Nilsson M. T.; Gising J.; Lagerlund O.; Muthas D.; Nordqvist A.; Karlén A.; Larhed M. Functionalized 3-amino-imidazo[1,2-a]pyridines: a novel class of drug-like Mycobacterium tuberculosis glutamine synthetase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 4790–4793. [DOI] [PubMed] [Google Scholar]
- Nordqvist A.; Nilsson M. T.; Rottger S.; Odell L. R.; Krajewski W. W.; Andersson C. E.; Larhed M.; Mowbray S. L.; Karlén A. Evaluation of the amino acid binding site of Mycobacterium tuberculosis glutamine synthetase for drug discovery. Biorg. Med. Chem. 2008, 16, 5501–5513. [DOI] [PubMed] [Google Scholar]
- Larhed M.; Hallberg A. Microwave-assisted high-speed chemistry: a new technique in drug discovery. Drug. Discovery Today. 2001, 6, 406–416. [DOI] [PubMed] [Google Scholar]
- Erdélyi M.; Gogoll A. Rapid homogeneous-phase Sonogashira coupling reactions using controlled microwave heating. J. Org. Chem. 2001, 66, 4165–4169. [DOI] [PubMed] [Google Scholar]
- Wannberg J.; Sabnis Y. A.; Vrang L.; Samuelsson B.; Karlén A.; Hallberg A.; Larhed M. A new structural theme in C-2-symmetric HIV-1 protease inhibitors: ortho-substituted P1/P1′ side chains. Bioorg. Med. Chem. 2006, 14, 5303–5315. [DOI] [PubMed] [Google Scholar]
- Srinivasan N. S.; Lee D. G. Preparation of 1,2-diketones: oxidation of alkynes by potassium-permenganate in aqueous acetone. J. Org. Chem. 1979, 44, 1574–1574. [Google Scholar]
- Legion; Tripos: St. Louis, MO, 1998. [Google Scholar]
- Georgsson J.; Hallberg A.; Larhed M. Rapid palladium-catalyzed synthesis of esters from aryl halides utilizing Mo(CO)6 as a solid carbon monoxide source. J. Comb. Chem. 2003, 5, 350–352. [DOI] [PubMed] [Google Scholar]
- Fedorov L. A.; Savarino P.; Viskardi G.; Rebrov A. I.; Barni E. 13C NMR spectroscopy of tautomeric conversions in imidazole compounds. Russ. Chem. Bull. 1992, 41, 223–230. [Google Scholar]
- O’Brien J.; Wilson I.; Orton T.; Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.