

Background: Species-specific differences in WT- and ΔF508-CFTR biology exist.
Results: Ferret WT-CFTR displays enhanced maturation efficiency and post-Golgi stability relative to human. Ferret ΔF508-CFTR maturation was greater than human in certain cell lines; both orthologs lacked function in airway epithelia.
Conclusion: Ferret and human CFTR have unique differences in processing and stability.
Significance: Generation of a ΔF508-CFTR ferret animal model may be useful.
Keywords: Chloride Channels, Cystic Fibrosis, Post-translational Modification, Protein Processing, Protein Stability
Abstract
The most common cystic fibrosis transmembrane conductance regulator (CFTR) gene mutation is ΔF508, and this causes cystic fibrosis (CF). New CF models in the pig and ferret have been generated that develop lung, pancreatic, liver, and intestinal pathologies that reflect disease in CF patients. Species-specific biology in the processing of CFTR has demonstrated that pig and mouse ΔF508-CFTR proteins are more effectively processed to the apical membrane of airway epithelia than human ΔF508-CFTR. The processing behavior of ferret WT- and ΔF508-CFTR proteins remains unknown, and such information is important to predicting the utility of a ΔF508-CFTR ferret. To this end, we sought to compare processing, membrane stability, and function of human and ferret WT- and ΔF508-CFTR proteins in a heterologous expression system using HT1080, HEK293T, BHK21, and Cos7 cells as well as human and ferret CF polarized airway epithelia. Analysis of the protein processing and stability by metabolic pulse-chase and surface On-Cell Western blots revealed that WT-fCFTR half-life and membrane stability were increased relative to WT-hCFTR. Furthermore, in BHK21, Cos7, and CuFi cells, human and ferret ΔF508-CFTR processing was negligible, whereas low levels of processing of ΔF508-fCFTR could be seen in HT1080 and HEK293T cells. Only the WT-fCFTR, but not ΔF508-fCFTR, produced functional cAMP-inducible chloride currents in both CF human and ferret airway epithelia. Further elucidation of the mechanism responsible for elevated fCFTR protein stability may lead to new therapeutic approaches to augment CFTR function. These findings also suggest that generation of a ferret CFTRΔF508/ΔF508 animal model may be useful.
Introduction
Cystic fibrosis (CF)2 is a common life-shortening autosomal recessive multiorgan genetic disorder caused by mutations to the cystic fibrosis transmembrane conductance regulator (CFTR) gene and its protein product. The CF phenotype includes abnormalities in the gastrointestinal tract, pancreas, liver, reproductive tract, sweat ducts, and the lung, with the development of chronic lung disease being the primary cause of morbidity and mortality in these patients (1, 2).
Essential to development of effective therapies for CF are animal models that recapitulate the human disease phenotype. Although mouse models of CF have been very useful, they lack development of spontaneous lung disease with the phenotypic characteristics of humans with CF (3–5). To this end, ferret and pig CF models have been generated, and both develop CF-like lung disease (6, 7). It is clear that species-specific differences in the severity of CF-associated disease in various organs exists in these two species (4, 7–9). Such differences could be the result of species-specific organ physiology and/or CFTR biology. Understanding these differences is imperative to the evaluation of CF animal models, developing and testing therapeutic agents in these models, and elucidating potentially new mechanisms of CFTR processing, stability, regulation, and function.
The CFTR protein contains five structural domains including two membrane-spanning domains, two nucleotide binding domains, and one regulatory domain. These domains act in concert to coordinate cAMP-inducible chloride movement through CFTR at the apical membrane of many types of epithelia (10–13). Like other integral membrane proteins, CFTR is co-translationally inserted into the endoplasmic reticulum (ER) where it either achieves a properly folded conformation or is degraded by ER associated degradation (ERAD). After proper folding of CFTR in the ER, it is transported to the Golgi for glycosylation modifications and finally to the apical membrane. Once at the apical membrane, CFTR primarily acts to regulate cAMP-inducible chloride and bicarbonate movement across the membrane. The level of surface CFTR is regulated by endocytosis, where dependent upon the levels of ubiquitination it is either recycled back to the surface or subjected to endolysosomal degradation (ELAD) (14–16).
As CFTR is being inserted into the ER membrane, it is glycosylated at two highly conserved N-linked glycosylation sites present on the fourth extracellular loop. This glycosylation aids in the proper folding, enhances stability of the protein, and serves as a convenient method for studying protein processing (17–19). On an electrophoretic gel there are three potential CFTR bands (band A, B, and C) that can be seen (18, 20). Band A (∼140 kDa) is present only experimentally after deglycosylation and represents the nascent polypeptide. Band B (∼150 kDa) is found in the ER and represents immature core-glycosylated CFTR. Band C (∼160–170 kDa) represents fully processed, complex-glycosylated CFTR and is found in the Golgi and post-Golgi compartments where the N-linked glycans have been extensively modified by Golgi enzymes (21).
Mutations that disrupt the processing of CFTR are common. The most common disease-causing mutation, present in at least one allele of ∼90% of CF patients, is caused by a deletion of three nucleotides and results in the deletion of the amino acid phenylalanine at position 508 (ΔF508) (22, 23). This mutation is located within nucleotide binding domain 1 and is known to have at least three consequences for human CFTR. 1) Due to protein misfolding, the vast majority of ΔF508-CFTR is degraded by ERAD (18, 24–27). This can be seen by Western blot analysis on an SDS-PAGE gel by the absence of significant processing from the ER core glycosylated band B to the fully mature band C. 2) The stability of the small portion of ΔF508-CFTR protein processed to the cell membrane is greatly decreased relative to wild-type (WT) protein (28). 3) The function of the ΔF508-CFTR protein is greatly reduced relative to wild-type CFTR (29, 30).
Relative to the 1480 amino acids human CFTR protein, the amino acid sequence length, percent amino acid conservation, and percent amino acid identity of CFTR in current CF animal model species is: ferret (1484, 97.0%, 91.9%), pig (1482, 96.8%, 92.3%), and mouse (1476, 92.7%, 79.1%), respectively (4, 31) (supplemental Fig. S1). The processing efficiency of ΔF508-CFTR to band C in polarized and non-polarized cells has been recently demonstrated to be much more efficient for pig and mouse ΔF508-CFTR than the human counterpart (30, 32). It is presently unclear if ferret ΔF508-CFTR also demonstrates differences in processing as seen with pig and mouse orthologs. Such information would help to determine whether generation of a ΔF508-CFTR ferret would be useful to the field and also shed light on species-specific differences in the structure/function of CFTR. Therefore, in this report we sought to comparatively evaluate the processing efficiencies and function of both wild-type and ΔF508-CFTR from human (hCFTR) and ferret (fCFTR) origins.
Results from these studies demonstrated that overexpression of WT-hCFTR and WT-fCFTR gave rise to significantly higher levels fCFTR protein expression relative to hCFTR regardless of cell type. Much of this enhanced expression of fCFTR protein appears to be due to increased steady state levels of fCFTR mRNA caused by cis-acting sequences in the fCFTR cDNA that augment transcription and/or mRNA stability. Interestingly, other post-translational mechanisms also appear to contribute to increased fCFTR protein levels including elevated maturation efficiency and enhanced post-Golgi half-life. By contrast, no differences in ERAD clearance of the two wild-type orthologs were observed. Enhanced half-life of the fCFTR protein was at least in part due to increased surface stability, as determined by surface pulse-chase On-Cell Western blots that detected an extracellular HA-tag in CFTR. Metabolic pulse-chase experiments comparing the human and ferret ΔF508-CFTR orthologs revealed no significant difference in the relative rates of disappearance of band B. However, it was clear that a small portion of ΔF508-fCFTR was processed to band C in two of four cell types tested, unlike ΔF508-hCFTR, which displayed undetectable processing in all cell types but HEK293T. Importantly, despite this observed processing to band C, we found that only the wild-type, but not the ΔF508 mutant CFTR, protein was able to correct the cAMP-inducible chloride transport defect in human and ferret CF airway epithelia. This suggests that generation of a ferret CFTRΔF508/ΔF508 animal model may be useful in modeling the human ΔF508-CFTR mutation.
EXPERIMENTAL PROCEDURES
Generation of Recombinant DNA Constructs and Adenoviral Vectors
Total RNA from ferret airway epithelial cells was isolated and used for generation of the ferret CFTR cDNA using an OneStep RT-PCR kit from Qiagen (Qiagen, Valencia, CA). To eliminate any potential bacterial promoter activity caused by the cryptic bacterial promoter sequence in exon 6b of the CFTR cDNA of both species, silent mutants were introduced by site-specific mutagenesis in the following nucleotide sequence: 794TGATTGAAAATATCC808 with the underlined nucleotides changed from T→C and A→G, which did not alter the coding sequence (base number is identical for human and ferret cDNAs and in reference to the ATG at +1). Additionally, an identical Kozak sequence (GCCGCCACC) was also placed next to the initial codon (ATG) for both human and ferret CFTR cDNAs followed by subcloning to the pacAd5CMV proviral vector backbone for protein expression and generation of adenoviral vectors. A cytomegalovirus promoter and SV40 poly(A) was used to control CFTR expression in these vectors. The ΔF508 mutation was subsequently generated by site-specific mutagenesis of the wild-type CFTR cDNAs in the proviral vectors using the QuikChange II XL site-directed mutagenesis kit from Stratagene (Santa Clara, CA). To assess the surface properties of the CFTR proteins, a 3×HA in tandem tag was inserted in extracellular loop 4 of the CFTR cDNAs by PCR-mediated cloning for all proviral vectors as previously described (7, 16). All plasmids were verified by sequencing. We used these constructs to generate recombinant adenovirus for human and ferret WT- and ΔF508-CFTR with and without the extracellular 3×HA tag. These viruses were clonally purified and amplified to achieve maximum expression of the transgene after infection of polarize airway epithelial cultures and xenografts. In cell line studies, the proviral plasmids were used for transfection.
Cell Culture
HT1080, Cos7, BHK21, and HEK293T cell lines (ATCC: CCL-121, CRL-1651, CCL-10, and CRL-11268, respectively) were cultured using the ATCC recommended culture conditions. CuFi cells were used to generate polarized human CF airway epithelia as previously described (33). In brief, CuFi cells were cultured on collagen-coated plastic cell culture plates using bronchial epithelial growth medium (BEGM) consisting of a 1:1 mixture of Ham's F-12 and DMEM media supplemented with antibiotics and BEGM SingleQuots (Lonza, Basel Switzerland). The cells were then seeded onto collagen-coated Millicell inserts (Millipore, Billerica, MA) with 2 × 105 cells per well using a 1:1 ratio of DMEM and Ham's F-12 media supplemented with 5% FBS. This medium was replaced the next day apically and basolaterally with Ultroser G medium that consists of a 1:1 mixture of Ham's F-12 and DMEM medium supplemented with antibiotics and 2% Ultroser G (Pall Corp., Port Washington, NY). The following day the apical media was removed, and the cells were fed basolaterally every 2–3 days using the Ultroser G media while maintaining an air liquid interface.
Heterologous Transgene Expression
Expression of the CFTR proteins in HT1080, HEK293T, and Cos7 cells was achieved by electroporation of the CFTR expression plasmids as previously described (4). BHK21 cells were transfected with Lipofectamine LTX according to the manufacturer's recommendations. For more details on electroporation and lipid transfection procedures, see the supplemental Methods. Polarized CuFi cells, grown on Millicell plastic inserts, were pretreated apically with 5 mm EGTA for 5 min to break down tight junctions and enhance apical infection with recombinant adenovirus. After washing, the cells were infected apically with adenovirus containing the different transgenes for 2–4 h at 37 °C with 20,000 particles/cell. The cells were washed and incubated at 37 °C until used for immunofluorescence or Ussing chamber analysis. Wild-type and ΔF508 HA-tagged fCFTR adenovirus was used to infect ferret CFTR knock-out tracheal xenografts for transepithelial potential difference measurements as described below.
Immunoprecipitation, in Vitro Phosphorylation, and Electrophoretic Analysis of CFTR
CFTR was immunoprecipitated from 1 mg of protein from whole cell lysates derived from cells expressing CFTR using a mixture of CFTR (M3A7 and MM13-4, Millipore) and HA (HA-High Affinity, Roche Applied Science) antibodies bound to Protein G Dynabeads (Invitrogen) according to the manufacturer's directions. The method of in vitro phosphorylation of immunoprecipitated CFTR has been described elsewhere in detail (4). For more details see the supplemental Methods. After in vitro phosphorylation, proteins were resolved by SDS-PAGE and imaged using a Molecular Dynamics Typhoon 8600 Variable Mode Imager (GE Healthcare). The resulting images were then quantified using Image J software.
Biochemical Glycosidase Studies
Immunoprecipitated and in vitro phosphorylated CFTR was digested with either 1000 units of endoglycosidase H or 5750 units of glycerol-free peptide N-glycosidase F (New England Biolabs, Ipswich, MA) for 2 h at 37 °C. The proteins were then resolved by electrophoresis as described above.
Northern Blot Analysis
HT1080 cells transiently expressing human or ferret WT-CFTR were harvested from tissue culture plates using RNAprotect (Qiagen) at 48 h post-transfection. Total RNA was extracted using the RNesay Plus Mini prep kit (Qiagen). Total RNA (25 μg) samples were resolved on a 1% agarose gel containing 2.2 m formaldehyde in MOPS electrophoresis buffer and transferred to a Nytran membrane using the Whatman Turboblotter system (GE Healthcare). The membranes were hybridized using Rapid-hyb buffer (GE Healthcare) at 65 °C for 2.5 h with 1.65 kb of 32P-labeled DNA probes against the 5′ end of human or ferret CFTR mRNA. The membranes were then exposed to x-ray film to detect the CFTR mRNA. The CFTR probes were then stripped from the membrane by washing with 0.1% SDS twice at 95 °C for 1 h. The membranes were then probed with a human GAPDH probe to serve as an internal RNA loading control and exposed to x-ray film. The resulting Northern blots were quantitated using ImageJ software.
Metabolic [35S]Methionine and [35S]Cysteine Pulse-Chase Experiments
A detailed protocol for metabolic labeling of CFTR has been reported elsewhere (4). Briefly, HT1080 cells expressing the different CFTR proteins were depleted of intracellular pools of methionine and cysteine by gently washing (2×) and incubating the cells with warm starvation medium (DMEM depleted of methionine and cysteine) for 30 min at 37 °C. Newly synthesized CFTR was then labeled by replacing the starvation media with 1 ml of the same media supplemented with 200 μCi of [35S]methionine and [35S]cysteine (PerkinElmer Life Sciences) and then incubated at 37 °C for 15–20 min. The excess label was removed by gently washing the cells three times on ice with cold PBS. This was followed by chasing the label-incorporated CFTR for various amounts of time using full DMEM supplemented with 10% FBS at 37 °C. The cells were harvested on ice by washing 2 times with cold PBS followed by adding 1 ml of radioimmune precipitation assay buffer supplemented with freshly added protease inhibitors (Roche Applied Science) for 30 min. The lysate was then centrifuged at 4 °C for 10 min at 13,000 × g. CFTR was then immunoprecipitated from the supernatant and resolved on an electrophoretic gel as described above.
Ussing Chamber Analysis
Polarized CuFi cells expressing recombinant adenoviral encoding HA-tagged human and ferret WT- and ΔF508-CFTR were prestimulated overnight with 100 μm 3-isobutyl-2-methylxanthine and 10 μm forskolin at 48 h post-infection with adenovirus. The following day short circuit current (ISC) measurements were obtained in the presence of 100 μm amiloride and 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS) as previously described (31) with the following two modifications; 1) the P2300/P2302M Ussing chamber system with Acquire and Analysis rev and 2) acquisition software (Physiologic Instruments, San Diego CA) (2). CFTRINHGlyH-101 (50 μm, Thermo Fisher Scientific, Pittsburgh, PA) was added apically after basolateral bumetanide (100 μm) treatment. The combined bumetanide and CFTRINHGlyH-101 were used to estimate the CFTR-dependent chloride current.
Transepithelial Potential Difference Measurements
All animal protocols were approved by the Institutional Animal Care and Use Committees at the University of Iowa. Ferret CFTR knock-out newborn tracheal xenografts were prepared as previously described (4, 7). To express ferret HA-tagged WT- and ΔF508-CFTR, the xenografts were pretreated with 5 mm EGTA for 10 min to breakdown tight junctions. This was followed by washing of the xenograft with Ham's F-12 media to remove the EGTA. The xenografts were then infected with 1 × 1011 adenoviral particles for 16–20 h. This was followed by flushing the xenografts with Ham's F-12 and then air to return to air liquid interface conditions. Transepithelial potential difference (TEPD) measurements were then made 4–8 days post-infection to measure CFTR activity. For details on these procedures, see the supplemental Methods.
Immunofluorescence
BHK21 and HT1080 cells expressing the different CFTR constructs were split onto glass coverslips 24 h post-transfection. The following day the cells were washed at 4 °C with cold HBSS media supplemented with 10 mm HEPES and 1× essential amino acids. The surface CFTR was stained with a mouse anti-HA antibody (Covance, Princeton, NJ) diluted 1:1000 in the same HBSS solution for 1 h at 4 °C. The cells were then washed with cold HBSS and fixed at room temperature with 4% paraformaldehyde (PFA) for 15 min followed by washing 3 times for 5 min each with PBS. The cells were then blocked and permeabilized in PBS containing 5% donkey serum and 0.1% Triton X-100 for 1 h. A rat anti-CFTR (1:200, 3G11) antibody obtained from CFTR Folding Consortium was used to stain total CFTR. The cells were then washed and stained using donkey anti-mouse AlexaFluor 594 and donkey anti-rat AlexaFluor 488 antibodies (Invitrogen). The cells were then washed, mounted in VectaShield mounting solution containing DAPI (Vector Laboratories, Burlingame, CA), and imaged using a Lieca spinning disk confocal microscope with Metamorph acquisition software.
Recombinant surface CFTR staining was achieved in polarized CuFi cells grown on Millicells (Millipore) expressing adenoviral-delivered HA-tagged human and ferret WT- and F508-CFTR. The surface HA-tagged CFTR was captured by cold binding a mouse anti-HA antibody as described for BHK21 and HT1080 cells. The cells were then fixed in 4% PFA followed by blocking and permeabilization using SuperBlock in PBS (Thermo Fisher Scientific) supplemented with 5% donkey serum and 0.1% Triton X-100 for 1 h. Total CFTR staining was achieved using a rat anti-HA antibody (1:200, HA-High Affinity, Roche Applied Science) in addition to the rat anti-CFTR (3G11). ZO-1 staining was used to mark the apical boundary of the polarized cells with a rabbit anti-ZO-1 primary (1:200, Invitrogen). We then incubated cultures with donkey anti-rat AlexaFluor 488, donkey anti-mouse AlexaFluor 568 (1:200, Invitrogen), and donkey anti-rabbit DyLight 649 (1:200, Jackson ImmunoResearch, West Grove, PA) secondary antibodies. The Millicell membranes were then excised, mounted on glass slides using VectaShield mounting solution containing DAPI (Vector Laboratories), and imaged using a Zeiss laser scanning microscope. Images were processed using Zeiss acquisition and ImageJ software.
Surface CFTR On Cell Western Blots
HT1080 and BHK21 cells expressing the different CFTR proteins were split to 24- or 48-well plates at 24 h post-transfection. The next day the live cells were quickly washed once with cold HBSS containing 10 mm HEPES and 1× essential amino acids. The cells, including the negative control wells (cells expressing a non-HA-tagged WT- or ΔF508-CFTR), were incubated with a mouse anti-HA antibody (Covance) diluted 1:1000 in the same HBSS media for 1 h at 4 °C. The excess antibody was washed away by washing the cells 4× in cold HBSS media at 4 °C, and the cells were fixed for 15 min at room temperature in 4% PFA. The cells were then permeabilized and blocked in PBS containing 0.1% Triton X-100 and 5% goat serum for 1 h at room temperature. Finally, the cells were stained with a goat anti-mouse AlexaFluor 680 IR-conjugated antibody (1:200, Invitrogen) for 1 h, washed, and imaged using a Li-Cor Infrared Odyssey imager.
Surface CFTR Pulse-Chase
Surface CFTR pulse-chase experiments were carried out to determine the total amount of CFTR that made it to the plasma membrane during a 1-h experiment independent of whether it was endocytosed during that period. In these experiments HA-CFTR presented at the cell surface of transfected HT1080 cells was captured by labeling viable cells for 1 h at 37 °C with a mouse anti-HA antibody (Covance) diluted 1:1000 in warm DMEM containing 10% FBS. The unbound anti-HA antibody was removed by washing the cells 3 times with DMEM containing 10% FBS. The cells were then replenished with DMEM (10% FBS) and returned to the incubator for 0, 4, 24, or 48 h. To determine the stability of the surface and intracellular labeled CFTR, the cells were washed using PBS and fixed with 4% PFA for 30 min, blocked, permeabilized in PBS containing 5% goat serum and 0.1% Triton X-100 for 1 h, and stained with a secondary goat anti-mouse AlexaFluor 680 IR-conjugated antibody (1:200 for 1 h, Invitrogen) as described in the On Cell Western blotting protocol. The stability of only the cell surface CFTR-labeled pool (i.e. excluding the intracellular pools) was achieved under non-permeabilization conditions (i.e. no Triton X-100).
Statistical Methods
Statistical comparisons for quantified data were made using the two-tailed Student's t test for the given N reported in each figure legend. Non-quantified autoradiographic and immunolocalization studies are representative of at least three independent experiments.
RESULTS
Steady State Heterologous Expression of fCFTR Is Significantly Greater Than hCFTR
Presentation of a HA tag into the fourth extracellular loop of WT- and ΔF508-hCFTR has been extremely useful in characterizing CFTR processing defects. Importantly, this insertion has been shown to preserve the native-like processing of the human CFTR protein (16, 19, 34). Because fCFTR processing has yet to be evaluated, it was important to first demonstrate that insertion of a HA tag into the fourth extracellular loop of WT- and ΔF508-fCFTR did not alter expression patterns of band B and C. To this end, HT1080 cells (derived from a human fibrosarcoma) were transfected with human and ferret CFTR expression constructs with or without the HA tag, and CFTR protein levels were evaluated using an IP kinase assay (Fig. 1, A and B). As expected, introduction of the 3×HA tag caused a slight shift in migration of both bands B and C for each of the constructs. Nevertheless, the processing of fCFTR remained unchanged between the untagged and tagged proteins, consistent with other reports on hCFTR (16, 19). Several interesting findings emerged from these studies. First, it was clearly evident that ΔF508-fCFTR expressed a small amount of band C that was not observed with ΔF508-hCFTR constructs. Second, the fCFTR steady state levels of band C and, to a lesser extent band B, were significantly greater than that observed in cells transfected with the hCFTR counterparts. This enhanced expression of fCFTR was consistently observed using multiple DNA preparations and identical transfection and autoradiographic exposure conditions. It is also important to point out that with the exception of the coding cDNA sequence, expression vectors and junctional sequences were identical between all constructs.
FIGURE 1.
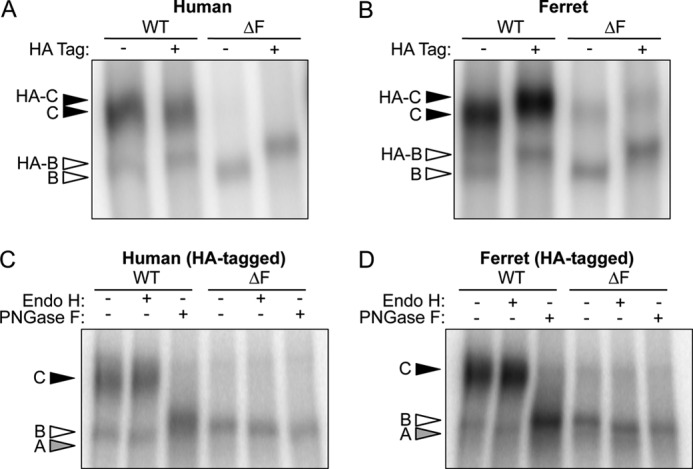
Expression of HA-tagged and non-tagged ferret and human CFTR. A and B, human fibrosarcoma cells (HT1080) were transiently transfected by electroporation with expression plasmids containing either the HA- and non-tagged (A) human and (B) ferret WT- and ΔF508-CFTR cDNAs. The cells were harvested 48 h post-transfection, and the recombinant proteins were immunoprecipitated with a combination of both anti-CFTR and anti-HA antibodies. The protein was then in vitro phosphorylated by PKA in the presence of [γ-32P]ATP and resolved by SDS-PAGE. The gels were dried and exposed to a phosphoscreen, and the autoradiographs were developed using phosphorimaging. HA-tagged and untagged bands B and C are represented by empty and solid arrows, respectively. C and D, cell lysate from the HA-tagged samples (A and B) was subjected to the IP kinase assay. However, before SDS-PAGE analysis, the samples were treated with endoglycosidase H (Endo H) or peptide N-glycosidase (PNGase F) for 2 h at 37 °C. Bands A, B, and C are shown in gray, empty, and black, respectively. Note that the peptide N-glycosidase F-sensitive band (Band A) runs slightly slower than band B in the other lanes as seen in other reports (19, 25).
Biochemical deglycosylation assays were performed to confirm normal processing of both human and ferret CFTR (Fig. 1, C and D). All band B forms of both human and ferret WT- and ΔF508-CFTR were susceptible to endoglycosidase H treatment, which cleaves high mannose sugars as seen in the ER. Furthermore, both ferret and human WT-CFTR band C was resistant to endoglycosidase H, demonstrating that the channel underwent complex glycosylation in the Golgi compartment. The processing of band C was further verified by peptide N-glycosidase F cleavage that removes all N-linked glycans including those resistant to endoglycosidase H. These findings confirmed that glycosylation patterns of ferret and human CFTR are extremely similar. It should be noted that wild-type CFTR proteins treated with peptide N-glycosidase F migrated slower than the expected apparent molecular weight for band A, as also observed by others to varying extents (19, 25). One potential explanation for this could be that the change in net charge after removal of sugars or change in protein conformation influences migration of band A. Using a glycerol-free peptide N-glycosidase F enzyme improved, but did not restore, migration of the WT-CFTR band A to that seen with endoglycosidase H treatment.
The finding that fCFTR protein was expressed at higher levels than hCFTR in HT1080 cells was intriguing (Fig. 1, A and B). To determine whether this difference would be consistently observed in other cell lines, we compared human to ferret CFTR expression in three additional cell lines including BHK21 (hamster kidney fibroblasts), Cos7 (monkey kidney fibroblasts), and HEK293T (human embryonic kidney fibroblasts). Results from these experiments demonstrated that wild-type fCFTR expression was enhanced relative to wild-type hCFTR in each of these cell lines (Fig. 2, A–C). These differences in human and ferret CFTR expression did not appear to be due to differences in antibody affinities used for the immunoprecipitation (4), as multiple individual antibodies gave similar results (data not shown) as the combination of three antibodies presented here (Fig. 2, A–C). Importantly, this increase was also seen using Western blot analysis with 12 commonly used hCFTR antibodies (supplemental Fig. S2). Furthermore, transfection efficiencies for the various CFTR constructs were similar, as determined using co-transfection with an internal control luciferase expression construct (data not shown). There was evidence in HEK293T cells that some of ferret and human ΔF508-CFTR was processed to band C (Fig. 2A). However, in contrast to HT1080 and HEK293T cells, both ferret and human ΔF508-CFTR produced negligible band C in BHK21 and Cos7 cells (Fig. 2, B and C). These results suggest that under overexpression conditions, a small fraction of ΔF508-fCFTR is processed to band C in a cell type-specific fashion.
FIGURE 2.

Multiple cell lines have increased steady state expression of ferret WT-CFTR band C, whereas only certain cell lines process ferret ΔF508-CFTR to band C. A–C, shown are representative autoradiographs from immunoprecipitated and in vitro phosphorylated CFTR protein derived from human embryonic kidney (HEK293T) (A), baby hamster kidney (BHK21) (B), and monkey kidney fibroblast (Cos7) cells (C) transiently overexpressing HA-tagged human and ferret WT- and ΔF508-CFTR proteins. D, Northern blot analysis was performed on HT1080 cells transiently expressing HA-tagged ferret and human WT-CFTR to evaluate the relative abundance of the CFTR mRNAs relative to GAPDH. The mRNA was harvested at 48 h after transfection, and a Northern blot was performed using 25 μg of total RNA. The human and ferret blots were probed with homologous regions to their respective CFTR ortholog cDNA, and the specific activities of the probes varied less than 5%. After CFTR probing, the blots were stripped and reprobed for GAPDH. E, quantification of the CFTR mRNA levels relative to the GAPDH is shown. Data represent the mean ± S.E. (n = 3). Marked comparison (*) demonstrates a significant difference relative to fCFTR as determined by Student's t test (p < 0.0001).
Increased abundance of wild-type fCFTR band C, relative to hCFTR, could be a consequence of increased transcription, enhanced mRNA stability, enhanced translation, increased efficiency of maturation of band B to C, or increased protein stability (i.e. reduced ERAD and ELAD). Although vector construction attempted to control for differences in transcription, we first evaluated whether mRNA expression levels between the two orthologs differed, thus accounting for differences in both transcription and mRNA stability. Northern blot analysis revealed a 3.5-fold increase in fCFTR mRNA levels relative to hCFTR (Fig. 2, D and E). This data suggests that the fCFTR cDNA contains a sequence(s) that acts to either enhance transcription of a heterologous promoter and/or mRNA stability. Thus, it is possible that the increased steady state expression of fCFTR band C (Figs. 1 and 2, A–C) relative to the hCFTR may be due to enhanced steady state levels of fCFTR mRNA.
Ferret Wild-type CFTR Protein Synthesis, Processing Efficiency, and Half-life Are Increased Relative to Human CFTR
Before comparing the biology of ferret and human ΔF508-CFTR, we first sought to determine if the wild-type CFTR orthologs maintained similar protein processing biology. To this end, metabolic [35S]methionine pulse-chase experiments were performed in transiently transfected HT1080 cells (Fig. 3A). Initial labeling efficiency of band B was 35.2 ± 10.4% higher (p < 0.006) for fCFTR relative to hCFTR (Fig. 3B), and this was likely due to enhanced mRNA production from the fCFTR cDNA (Fig. 2, D and E). It is unclear why this difference did not reflect the magnitude of increase in mRNA (3.5-fold) expressed from the fCFTR cDNA, but this suggests that the fCFTR mRNA may have decreased translational efficiency relative to hCFTR mRNA. Furthermore, the observed similar rates of band B disappearance between human and ferret CFTR (Fig. 3C) suggest that species-specific difference in ERAD does not contribute to enhanced expression of fCFTR protein (Fig. 3C). Interestingly, there was a 45.3% increase (p < 0.03) in the maturation efficiency of fCFTR (62.1 ± 6.7%) relative to hCFTR (42.7 ± 4.4%) (Fig. 3C). Similar results for initial band B labeling and maturation efficiency were seen in BHK21 cells transiently expressing human and ferret WT-CFTR (supplemental Fig. S3, A and B).
FIGURE 3.

The half-life of complex-glycosylated ferret WT-CFTR is elevated relative to its human ortholog in HT1080 cells. A, metabolic [35S]methionine pulse-chase experiments were performed on HT1080 cells transiently expressing HA-tagged ferret and human WT-CFTR to evaluate the rates of protein synthesis, processing, maturation, and stability. Top and bottom panels are representative autoradiographs for ferret and human WT-CFTR, respectively. B, shown is quantification of the average intensity of band B at 0 h (Band B0) for ferret and human CFTR normalized to total protein. C, shown is quantification of the disappearance of band B (lower graph) over the course of the experiment relative to B and B0 (Band BT and B0). The upper graph represents quantification of the maturation efficiency as calculated by the percentage of band C4 h to band B0. D, shown is quantification of the stability of band C over the course of the experiment relative to B and C4 h (band CT/band C4 h). Data for initial protein labeling and maturation efficiency (B and C, inset, respectively) represent the mean ± S.E. (n = 9 from 3 independent experiments labeled in triplicate). Time course graphs in C and D represent the mean ± S.E. with n = 6. Marked comparisons demonstrate significant differences as determined by Student's t test (*, p < 0.03; †, p < 0.007).
The kinetics of band C stability were also quantified for ferret and human CFTR. In these studies, maximal band C accumulation peaked at 4 h post-chase for both fCFTR and hCFTR. Band C stability kinetics was calculated by measuring the intensity of the remaining band C over time relative to the band C intensity at t = 4 h post-chase (Fig. 3D). By 24–48 h post-maturation, species-specific differences in the stability of CFTR band C began to emerge. The calculated half-life of fCFTR band C (57.9 h) was ∼2.2 fold greater than that for hCFTR (28.2 h) (Fig. 3D). This was further evident by the % band C remaining at 48 h for fCFTR (58.4 ± 13.0%) and hCFTR (22.1 ± 3.9%). Several biologic mechanisms for the observed increased half-life of fCFTR band C could include 1) mislocalization of fully processed fCFTR (i.e. absence at the cell surface by retention in the Golgi or other post Golgi compartments) allowing it to avoid cell surface protein quality control that turns over hCFTR, 2) reduced rates of endocytosis and/or enhanced recycling of fCFTR at the cell surface, 3) reduced efficiency of fCFTR processing through ELAD, and/or 4) enhanced conformational stability of fCFTR that would influence turnover by a number of these previous processes.
Ferret ΔF508-CFTR Processes More Efficiently to Band C Than Human ΔF508-CFTR in HT1080 Cells
Among the cell lines tested, HT1080 cells demonstrated the most consistent processing of a small fraction of ΔF508-fCFTR to band C. To evaluate the kinetics of ΔF508-fCFTR processing in this cell line relative to ΔF508-hCFTR, we performed metabolic pulse-chase experiments (Fig. 4A). Similar to the WT proteins, an increase (61.2 ± 6.5%, p < 0.0001) in the initial amount of ΔF508-fCFTR band B was synthesized during the labeling period compared with ΔF508-hCFTR (Fig. 4B). The half-life of band B for ΔF508-fCFTR (1.7 h) was not significantly different from ΔF-hCFTR (1.4 h), although there was a trend toward increased stability of ΔF508-fCFTR band B (Fig. 4C). Furthermore, the maturation efficiency of band B to band C was more efficient for ΔF508-fCFTR (10.4 ± 1.3%) than it was for ΔF508-hCFTR (2.3 ± 0.6%) at 4 h post-chase in HT1080 cells (Fig. 4D). These findings confirm that in HT1080 cells, ΔF508-fCFTR is more efficiently processed to band C than ΔF508-hCFTR.
FIGURE 4.
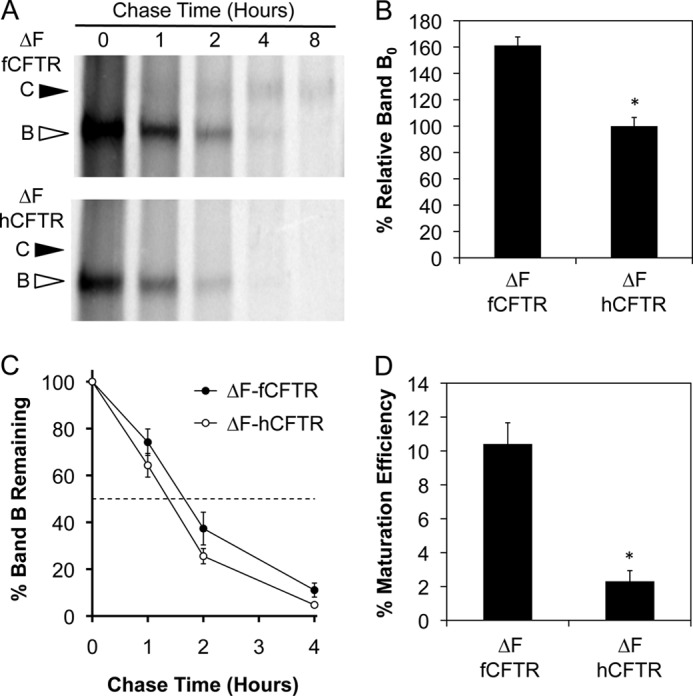
Maturation of ferret ΔF508-CFTR band B to band C is more efficient than the human ortholog in HT1080 cells. A, metabolic [35S]methionine pulse-chase experiments were performed on HT1080 cells transiently expressing HA-tagged ferret and human ΔF508-CFTR to evaluate the rate of band B disappearance and maturation efficiency of the proteins as done in Fig. 3. Top and bottom panels are representative autoradiographs for ferret and human ΔF508-CFTR, respectively. B, shown is quantification of the average intensity of band B at 0 h (Band B0) for ferret and human ΔF508-CFTR normalized to total protein. C, shown is quantification of the disappearance of band B over the course of the experiment relative to Band B0 (Band BT/Band B0). D, shown is quantification of the maturation efficiency as calculated by the percentage of band C4 h relative to band B0. Data in all graphs represent the mean ± S.E. with n = 4 for panel C and n = 9 for panels B and D derived from three independent experiments done in triplicate. Marked comparisons (*) demonstrate significant differences relative to ferret ΔF508-CFTR as determined by Student's t test (p < 0.0001).
Analysis of Ferret and Human WT- and ΔF508-CFTR Abundance at Cell Surface
The expression of CFTR band C is suggestive of its processing to the plasma membrane. However, other factors including the rates of endosomal recycling and ELAD can also control the steady state level of fully processed band C at the plasma membrane. To directly evaluate the extent to which ΔF508-fCFTR moves to the plasma membrane, we used the alternative approach of detecting an extracellular HA epitope present in the fourth extracellular loop of CFTR. To this end, BHK21 and HT1080 cells were transfected with the various CFTR constructs and anti-HA staining to detect surface CFTR performed under non-permeabilized conditions when endocytosis was blocked by reduced temperature. Anti-HA staining was then followed by permeabilization and staining for total CFTR using an anti-CFTR antibody. Using this approach, we observed significant surface membrane localization for both human and ferret WT-CFTR proteins in BHK21 cells (Fig. 5A). As anticipated from CFTR IP kinase assays, there was significantly more membrane staining for WT-fCFTR as compared with WT-hCFTR. By contrast, no surface staining for CFTR was noted in the ΔF508-CFTR expressing BHK21 for either species of CFTR. Of note, the total CFTR staining was significantly lower for both human and ferret ΔF508-CFTR expressing cells. This is believed to be due to the folding defect of the ΔF508-CFTR that prevents the channel conformational maturation at the ER, leading to ERAD. The lack of surface staining for both ΔF508-CFTR orthologs correlated with the lack of significant band C seen in the in vitro phosphorylation data for BHK21 (Fig. 2B). Surface HA immunofluorescent studies in HT1080 cells yielded similar findings for surface WT-CFTR staining as seen in BHK21 cells (supplemental Fig. S4). However, unlike studies in the BHK21 cells, there was a small amount of detectable surface ΔF508-fCFTR staining in HT1080 cells that was not observed in ΔF508-hCFTR samples (supplemental Fig. S4). These findings correlate with the abundance of ΔF508-fCFTR band C detected using the in vitro phosphorylation assay on HT1080 cells expressing ΔF508-fCFTR (Fig. 1, B and D) and suggest that indeed a small fraction of ΔF508-fCFTR band C is at the membrane in HT1080 cells.
FIGURE 5.

Ferret and human ΔF508-CFTR lack significant cell surface expression relative to the wild-type counterparts. A, BHK21 cells grown on coverslips were transfected with the indicated plasmid expression constructs. At 48 h post-transfection, the cells were brought to 4 °C, and a mouse anti-HA antibody was applied for 1 h to stain surface HA-tagged CFTR (red). The unbound antibody was washed away, and the cells were then fixed and permeabilized. The cells were then stained for total CFTR using a rat anti-CFTR antibody (green) and DAPI to mark nuclei (blue). Immunofluorescent images of ferret and human WT-CFTR (left panels) and ΔF508-CFTR (right panels) are shown with the single anti-HA channel shown to the right of the merged color images. Boxed insets show a second example to the larger image. Scale bars represent 10 μm. B–E, surface On Cell Western blots were performed by binding an anti-HA antibody at 4 °C to cell surface CFTR on live BHK21 (B and C) and HT1080 (D and E) cells that were transfected with the indicated constructs as in A and a non-HA tagged CFTR control construct. Excess anti-HA antibody was washed away at 4 °C followed by fixation and staining with an IR secondary antibody. Wells were then imaged using an Odyssey IR scanner. The resulting example images (B and D) were quantified using ImageJ software to give the data shown in (C and E). Only regions containing cells, as indicated by the hashed circles on the WT-fCFTR samples (B and D) were evaluated for the average intensity of staining (which is independent of the area of the field quantified). This was necessary because variable sloughing of cells occurred during the assay washings. Data in all graphs represent the control background subtracted mean ± S.E. with n = 16 from 4 independent experiments. All comparisons with the exception of human ΔF508-CFTR versus ferret ΔF508-CFTR were statistically significant as determined by the Student's t test (p < 0.001). Human ΔF508-CFTR versus ferret ΔF508-CFTR was not significant for BHK21 cells but was significant for HT1080 (p < 0.008). With the exception of ferret ΔF508-CFTR in HT1080 cells, all other ΔF508-CFTR analyses were not significantly above control background levels of cells transfected with a non-HA-tagged version of CFTR.
To better quantify differences in surface CFTR expression using HA-epitope detection, we adapted our methods to evaluate large populations of cells using a modified infrared detection On Cell Western blot assay in non-permeabilized cell monolayers. To this end, BHK21 and HT1080 cells expressing the HA-tagged WT- and ΔF508-CFTR orthologs were cooled to 4 °C to prevent endocytosis of surface CFTR and then labeled with an anti-HA antibody. The cells were then fixed and stained with infrared-labeled secondary antibodies before scanning and quantification. Results from these experiments in BHK21 cells demonstrate that surface WT-fCFTR was ∼3-fold higher than WT-hCFTR (Fig. 5, B and C). Minimal surface ΔF508-CFTR staining was observed for both the human (5.4% that seen for WT-hCFTR) and ferret (7.2% that seen for WT-fCFTR) mutant proteins in BHK21 cells, with no significant difference between the orthologs. However, neither ferret nor human ΔF508-CFTR surface staining was significantly above background levels of control cells transfected with a non-HA-tagged version of CFTR. These findings are consistent with other studies performed in this cell line suggesting that BHK21 cells inefficiently process ΔF508-fCFTR to band C and the plasma membrane. HT1080 cells demonstrate a similar trend in relative levels of surface ferret and human WT-CFTR as seen in BHK21 cells (Fig. 5, D and E). Furthermore, although the total amount of surface ΔF508-fCFTR was significantly greater (∼5-fold) than that for ΔF508-hCFTR in HT1080 cells relative to the total amount of WT-CFTR, the fraction of surface expression of ΔF508-fCFTR (12.8% that seen for WT-fCFTR) and ΔF508-hCFTR (8.1% that seen for WT-hCFTR) was remarkably similar (Fig. 5, D and E).
WT-fCFTR Has Greater Stability at Plasma Membrane Than WT-hCFTR
Metabolic pulse-chase experiments in HT1080 cells demonstrate that WT-fCFTR band C maturation efficiency and half-life is greater than WT-hCFTR by 45 and 220%, respectively (Fig. 3, C and D). When combined with the 35% enhanced level of band B synthesis for WT-fCFTR (Fig. 3B), these three factors together likely explain most of the 360% enhancement in membrane surface staining for WT-fCFTR as compared with WT-hCFTR (Fig. 5E). The major factor contributing to the enhanced surface staining of WT-fCFTR, however, appears to be it enhanced half-life. Several mechanisms could exist for the observed increased half-life of WT-fCFTR including reduced plasma membrane recycling and/or ELAD. To this end, we sought to test the hypothesis that fCFTR may be more stable at the plasma membrane relative to hCFTR. To address this hypothesis, we performed On Cell Western blots following a pulse-chase with anti-HA antibody. We labeled surface HA-tagged human and ferret WT-CFTR with HA antibody for 1 h at 37 °C. The excess antibody was then removed, and cells were washed followed by a chase at 37 °C for various lengths of time. Cells were then fixed at time points ranging from 0 to 48 h before staining with an IR secondary antibody (Fig. 6A). The cells were stained either under permeabilization (Fig. 6, A and B) or non-permeabilization (Fig. 6, C and D) conditions. Results from these experiments demonstrated no significant differences between orthologs in % of HA immunoreactivity remaining on the cell at early stages of the pulse-chase experiment (<8 h). However, by 24 and 48 h after cell surface labeling, fCFTR-expressing cells demonstrated a 26.7% increased abundance in total fCFTR label (Fig. 6B) and a 25.9% increase in labeled fCFTR at the surface (Fig. 6, C and D) as compared with hCFTR. The t½ for cell surface fCFTR and hCFTR was 15.0 and 11.9 h, respectively, whereas the t½ for total fCFTR and hCFTR in post-Golgi compartments was 22.6 and 17.9 h, respectively. These observations suggest that the mature, complex-glycosylated fCFTR has an attenuated metabolic turnover and degradation relative to its human CFTR counterpart. Surface CFTR half-life studies performed in BHK21 cells also showed a significantly elevated t½ for fCFTR relative to hCFTR (supplemental Fig. S3C). This data, in concert with the metabolic pulse-chase data (Fig. 3D), support a model where the relative increase in ferret wild-type protein compared with human is at least in part due to differences in the metabolic stability in post-Golgi compartments.
FIGURE 6.

Cell surface ferret WT-CFTR is more stable than human WT-CFTR in HT1080 cells. A, HT1080 cells were transfected with human or ferret WT-CFTR HA-tagged expression constructs. After 48 h the medium was replaced with warm medium containing an anti-HA antibody and incubated for 1 h at 37 °C, thus labeling any CFTR that made it to the cell surface during the labeling time. The unbound antibody was removed by washing, and the cells were returned to 37 °C for 0, 4, 24, and 48 h. The cells were then quickly washed at room temperature, fixed in 4% PFA, permeabilized with 0.1% Triton X-100, stained with an infrared secondary antibody, and imaged using an Odyssey IR imager. Permeabilization allowed access of the secondary antibody to all CFTR that was labeled during the 1-h labeling period including the endocytosed intracellular pools. B, shown is quantification of data from multiple experiments as shown in A using ImageJ software. Data represent the mean ± S.E. with n = 12 individual wells from three independent experiments; the marked comparison (*) demonstrates a significant difference as determined by Student's t test of the comparison (p < 0.012). C and D, surface ferret or human WT-CFTR protein on HT1080 cells was labeled and stained as in A but under non-permeabilization conditions (i.e. no Triton X-100). The quantified surface CFTR data contained within these two graphs are identical but are depicted on a linear (C) or logarithmic (D) scale with ferret and human WT-CFTR represented by closed and open circles, respectively. These data represent the mean ± S.E. with each time point represented by 6–8 individual wells; marked comparisons demonstrate significant differences as determined by the Student's t test (**, p < 0.022; † p < 0.001).
ΔF508-fCFTR Does Not Efficiently Traffic to Apical Membrane or Produce cAMP-inducible Chloride Currents in Human and Ferret CF Airway Epithelia
Given the differences in ΔF508-fCFTR processing between cell lines, it was important to evaluate both its localization and function in polarized airway epithelial cells. To this end, we first evaluated surface HA immunofluorescent staining of CFTR in polarized human CF airway epithelia infected with recombinant adenovirus expressing the HA-tagged WT- or ΔF508-CFTR orthologs. Results from these experiments demonstrated significant plasma membrane localization for both human and ferret WT-CFTR proteins but undetectable plasma membrane staining in the vast majority of cells expressing the ΔF508-CFTR mutants (Fig. 7). Although 100% of WT-fCFTR-expressing cells demonstrated robust apical membrane staining, less than 6% of ΔF508-fCFTR-expressing cells demonstrated detectable apical membrane staining (>50 cells imaged for each group).
FIGURE 7.

Ferret and human ΔF508-CFTR lack significant apical surface CFTR in human CF polarized airway cells. Polarized human CF airway epithelial cells (CuFi) grown on Millicell membrane inserts were infected with human and ferret HA-tagged WT- and ΔF508-CFTR adenoviral vectors. At 48 h post-infection, the cells were brought to 4 °C, and a mouse anti-HA antibody was applied for 1 h to stain the surface HA-tagged CFTR (red). The unbound antibody was removed, and the cells were then fixed and permeabilized. Total CFTR (green) was stained using rat anti-CFTR (3G11) and rat anti-HA antibodies. The tight junctions (white) were stained using a rabbit anti-ZO-1 antibody. The membranes were excised and mounted with VectaShield containing DAPI (blue). Confocal immunofluorescent images of ferret and human WT-CFTR and ΔF508-CFTR are shown in the XY, XZ, and YZ planes with the single anti-HA channel shown to the right of the merged color images. The dotted lines represent the location of the Millicell membrane. Scale bars represent 10 μm.
We next compared the ability of human and ferret WT- and ΔF508-CFTR to produce functional cAMP-inducible chloride current by measuring short circuit currents (ISC) in polarized CF epithelia. To this end, polarized human CF epithelia grown at the air-liquid interface were infected with recombinant adenovirus expressing each of the CFTR proteins, and ISC measurements were made. Expression of human and ferret WT-CFTR produced significant cAMP-inducible chloride current relative to mock-infected controls (Fig. 8, A and B). Interestingly, fCFTR produced a significantly (p < 0.001) greater peak and plateau cAMP-inducible chloride currents than hCFTR (peak, 5.2 ± 0.5 μA/cm2 versus 2.6 ± 0.5 μA/cm2; plateau, 4.0 ± 0.4 μA/cm2 and 2.5 ± 0.4 μA/cm2, respectively) as might be expected from the increased stability of the fCFTR protein in cell lines and greater protein staining in CF airway epithelia. However, the extent of inhibition by bumetanide/GlyH101 was not significantly different for hCFTR (3.5 ± 0.8 μA/cm2) and fCFTR (4.6 ± 0.4 μA/cm2), suggesting hCFTR may have slightly higher constitutive activity than fCFTR. Importantly, under these conditions, cAMP-mediated changes in Isc were lacking for both of the human and ferret ΔF508-CFTR proteins (Fig. 8, A and B). These data are consistent with significantly reduced human and ferret ΔF508-CFTR localization at the apical surface of airway epithelia (Fig. 7).
FIGURE 8.

Ferret and human ΔF508-CFTR lacks the ability to correct chloride transport and permeability in CF human and ferret polarized airway epithelia. A, polarized human CF airway epithelial cells (CuFi) grown under air liquid interface (ALI) on Millicell inserts were infected with the indicated adenoviral vectors. At 48 h post infection, the inserts were mounted in Ussing chambers, and short circuit current (ISC) measurements were made. Representative tracings are shown for the indicated samples in response to serial addition of 3-isobutyl-2-methylxanthine (IBMX, 100 μm)/forskolin (Forsk, 10 μm), Bumet (100 μm), and GlyH101 (50 μm). B, shown is quantification of the ΔISC (A). Data in this graph represent plateau value (not peak) mean ± S.E. with n = 12 Millicells for WT-CFTR and n = 10 for ΔF508-CFTR. Mock refers to uninfected controls. No significant differences between human and ferret ΔF508-CFTR values were observed by Student's t test. However, human and ferret WT-CFTR values in response to 3-isobutyl-2-methylxanthine/forskolin were significantly greater for ferret (p < 0.017); this difference and its significance was greater when peak currents were evaluated (mean data not shown). C, ferret CFTR-knock-out tracheal xenografts were infected with either ferret WT- or ΔF508-CFTR adenoviruses overnight. Transepithelial potential difference measurements were made as described under “Experimental Procedures.” Representative TEPD tracings before and after infection are shown. D, quantification of the average cAMP/forskolin and GlyH101 responses is shown. Data in this graph represent the mean ± S.E. (n = 7 measurements from 5 WT-fCFTR infected xenografts and n = 8 from 6 ΔF508-fCFTR infected xenografts). Significant differences between WT- and ΔF508-CFTR responses to both cAMP/forskolin and GlyH101 were observed by Student's t test (p < 0.0055).
Although the above studies demonstrate that ΔF508-fCFTR lacks function in human CF airway epithelia, it remained unclear if ferret ΔF508-CFTR might have altered processing in human cells. To formally address this possibility, we generated newborn ferret CFTR−/− tracheal xenografts in nu/nu mice and evaluated the ability of both WT-fCFTR and ΔF508-fCFTR adenoviral vectors to complement cAMP-inducible defects in chloride permeability as assessed by TEPD. Base-line TEPD recordings were first measured in each xenograft and then after infection with recombinant CFTR adenovirus. Results from these studies (Fig. 8, C and D) demonstrated significant cAMP/forskolin-induced changes in TEPD under chloride secretory conditions in xenografts after infection with WT-fCFTR-expressing recombinant adenovirus. These WT-fCFTR-dependent changes in TEPD were also inhibited by the CFTR channel inhibitor GlyH101. Such changes were not observed in xenografts infected with ΔF508-fCFTR adenoviral vectors. These findings conclusively demonstrate that ΔF508-fCFTR lacks function in differentiated ferret airway epithelium.
DISCUSSION
Cystic fibrosis results from the lack of functional CFTR protein at the apical membrane of epithelial cells. The demonstration that CFTR knock-out ferrets develop the multiorgan phenotypes seen in human CF patients (4), including lung disease, suggests that this species may also have utility for modeling various CFTR mutations such as ΔF508. However, because pig and mouse ΔF508-CFTR have been shown to retain some level of processing in airway epithelia (30, 32), it was important to similarly evaluate ΔF508-CFTR biogenesis before generating the ferret animal model. Through a comparative study of human and ferret WT- and ΔF508-CFTR protein processing and function, this study has discovered several biologic differences between human and ferret CFTR. 1) The ferret cDNA sequence influences transcription or mRNA stability, leading to an increased steady state level of fCFTR mRNA relative to hCFTR, 2) Despite the enhanced levels of fCFTR mRNA, fCFTR protein synthesis rates were only marginally increased over hCFTR, suggesting that the translational efficiency of the fCFTR transcript is significantly lower than that for hCFTR. 3) Fully processed fCFTR protein accumulates in cells to a much greater extent than hCFTR, and this is predominantly due to enhanced stability of the fully processed fCFTR protein. 4) The fCFTR protein has enhanced maturation efficiency and increased stability at the plasma membrane and in post-Golgi compartments. 5) Cell line-specific differences in the processing of band C and movement of ΔF508-fCFTR to the plasma membrane are evident. 6) Similar to ΔF508-hCFTR, ΔF508-fCFTR is inefficiently processed to the plasma membrane and does not generate functional chloride channels in human- or ferret-polarized airway epithelia.
Differences in the level of CFTR mRNA produced by ferret and human expression constructs are intriguing given the fact that all expression elements other than the cDNA were identical (promoter, Kozak sequences, and polyA). The 3.5-fold elevation in fCFTR mRNA relative to hCFTR could be due to enhanced transcription or mRNA stability. The finding that fCFTR and hCFTR band B protein synthesis rates did not mirror differences in mRNA abundance suggests that the hCFTR protein is translated more efficiently than fCFTR by a factor of approximately three. These differences in the behavior of ortholog mRNAs could reflect species-specific factors that control mRNA stability and/or translation such as microRNAs. Although the biologic significance of these findings is unclear, because expression constructs did not include the endogenous 5′- and 3′-untranslated regions, the findings warrant further investigation.
Heterologous overexpression of human and ferret WT-CFTR demonstrated consistently elevated levels of fCFTR band C and surface membrane staining. Based on metabolic [35S]methionine pulse-chase experiments, the difference in WT-fCFTR accumulations reflected enhanced maturation efficiency and increased stability of the fully processed band C form. Surface On Cell Western blot pulse-chase experiments confirmed that the enhanced stability of WT-fCFTR was due to reduced turnover in post-Golgi compartments, including at the plasma membrane. Mechanistically, this could be due to reduced rate of endocytosis of WT-fCFTR, enhanced recycling of internalized WT-fCFTR, and/or impeded lysosomal delivery from the endosomal compartment. It is plausible that the primary sequence difference is translated into an increased conformational stability of the WT-fCFTR that confers higher resistance to ubiquitination by the cellular protein quality control machinery at the ER and in post-Golgi compartments.
The enhanced cell surface density of the fully processed WT-fCFTR protein was also reflected in greater cAMP-mediated chloride currents in CF airway epithelia. Such novel characteristics of the WT-fCFTR protein suggest new strategies for potentially enhancing functions of mutant CFTR proteins at the plasma membrane. Currently, CFTR potentiators (which restore gating defects) and correctors (which restore protein folding and plasma membrane trafficking) have great therapeutic potential as CF therapies. Current CFTR potentiators do not enhance the function of WT-CFTR. Our finding that WT-fCFTR has enhanced plasma membrane retention over WT-hCFTR suggest the potential for new drug targets that either reduce hCFTR membrane turnover or targeting to ELAD. Such drugs that essentially target mutant- or WT-hCFTR to adopt a conformation similar to WT-fCFTR could conceivably be used to enhance the functional potential of potentiators/correctors on CFTR mutants or also increase membrane retention of WT-CFTR in conjunction with gene therapy approaches. Although the specific amino acids that control differences in membrane stability of fCFTR and hCFTR remain to be determined, one can assume that these changes alter the confirmation of CFTR and/or association of effectors that control membrane recycling and/or degradation through ELAD.
Our findings using IP in vitro phosphorylation, immunolocalization, and surface On Cell Western blotting demonstrate that in two of the four cell lines tested the majority of ΔF508-fCFTR protein, like ΔF508-hCFTR, fails to undergo maturation to the fully processed band C form and fails to accumulate at the plasma membrane in detectable levels. This was true for BHK21 and COS7 cells. However, using these same methods, HT1080 cells demonstrated a small amount of ΔF508-fCFTR protein that was processed to band C and to the plasma membrane. In the context of HEK293T cells, band C was also observed for both ferret and human ΔF508-CFTR. The enhanced levels of ΔF508-fCFTR to band C accumulation relative to ΔF508-hCFTR may in part be due to enhanced fCFTR stability at the plasma membrane for both WT- and ΔF508-fCFTR. In support of this hypothesis, the fraction of surface expression of ΔF508-CFTR relative to WT-CFTR was quite similar for ferret (12.8%) and human (8.1%) in HT1080 cells (Fig. 5, D and E). Importantly, ΔF508-fCFTR did not produce functional chloride channels in either human or ferret CF-polarized airway epithelia, and this was consistent with significantly reduced trafficking of ΔF508-fCFTR to the plasma membrane. Cumulatively, results of this study and those of others support the notion that the inherent ER-to-Golgi processing defect of ΔF508 mutant CFTR exists on a continuum with human > ferret > pig and mouse (8, 30, 32).
In conclusion, comparisons of ferret and human WT-CFTR processing and function have uncovered a unique biologic difference in fCFTR that enhances stability in post-Golgi compartments. Determination of the structural characteristics that control these differences may aid in the development of new drug targets to enhance CFTR function by retaining a greater fraction at the plasma membrane. Additionally, our studies demonstrating that ΔF508-fCFTR is not functional in polarized airway epithelia suggest that generation of a ferret CFTRΔF508/ΔF508 animal model could be of particular value for testing new drugs that target this mutant.
This work was supported, in whole or in part, by National Institutes of Health Grants DK047967, HL108902, HL091842, DK075302, and DK54759. This work was also supported by the Cystic Fibrosis Foundation and the Canadian Institute of Health Research.

This article contains supplemental Methods and Figs. S1–S4.
- CF
- cystic fibrosis
- CFTR
- cystic fibrosis transmembrane conductance regulator
- hCFTR
- human CFTR
- fCFTR
- ferret CFTR
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation
- ELAD
- endolysosomal associated degradation
- ΔF508
- deletion of phenylalanine 508
- ISC
- short circuit current
- TEPD
- transepithelial potential difference
- PFA
- paraformaldehyde
- HBSS
- Hanks' balanced salt solution.
REFERENCES
- 1. Rosenstein B. J., Zeitlin P. L. (1998) Cystic fibrosis. Lancet 351, 277–282 [DOI] [PubMed] [Google Scholar]
- 2. Rowe S. M., Miller S., Sorscher E. J. (2005) Cystic fibrosis. N. Engl. J. Med. 352, 1992–2001 [DOI] [PubMed] [Google Scholar]
- 3. Egan M. E. (2009) How useful are cystic fibrosis mouse models? Drug Discov. Today 6, 35–41 [Google Scholar]
- 4. Fisher J. T., Zhang Y., Engelhardt J. F. (2011) Comparative biology of cystic fibrosis animal models. Methods Mol. Biol. 742, 311–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grubb B. R., Boucher R. C. (1999) Pathophysiology of gene-targeted mouse models for cystic fibrosis. Physiol. Rev. 79, S193–S214 [DOI] [PubMed] [Google Scholar]
- 6. Stoltz D. A., Meyerholz D. K., Pezzulo A. A., Ramachandran S., Rogan M. P., Davis G. J., Hanfland R. A., Wohlford-Lenane C., Dohrn C. L., Bartlett J. A., Nelson G. A., 4th, Chang E. H., Taft P. J., Ludwig P. S., Estin M., Hornick E. E., Launspach J. L., Samuel M., Rokhlina T., Karp P. H., Ostedgaard L. S., Uc A., Starner T. D., Horswill A. R., Brogden K. A., Prather R. S., Richter S. S., Shilyansky J., McCray P. B., Jr., Zabner J., Welsh M. J. (2010) Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci. Transl. Med. 2, 29ra31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sun X., Sui H., Fisher J. T., Yan Z., Liu X., Cho H. J., Joo N. S., Zhang Y., Zhou W., Yi Y., Kinyon J. M., Lei-Butters D. C., Griffin M. A., Naumann P., Luo M., Ascher J., Wang K., Frana T., Wine J. J., Meyerholz D. K., Engelhardt J. F. (2010) Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J. Clin. Invest. 120, 3149–3160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ostedgaard L. S., Meyerholz D. K., Chen J. H., Pezzulo A. A., Karp P. H., Rokhlina T., Ernst S. E., Hanfland R. A., Reznikov L. R., Ludwig P. S., Rogan M. P., Davis G. J., Dohrn C. L., Wohlford-Lenane C., Taft P. J., Rector M. V., Hornick E., Nassar B. S., Samuel M., Zhang Y., Richter S. S., Uc A., Shilyansky J., Prather R. S., McCray P. B., Jr., Zabner J., Welsh M. J., Stoltz D. A. (2011) The ΔF508 mutation causes CFTR misprocessing and cystic fibrosis-like disease in pigs. Sci. Transl. Med. 3, 74ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rogers C. S., Stoltz D. A., Meyerholz D. K., Ostedgaard L. S., Rokhlina T., Taft P. J., Rogan M. P., Pezzulo A. A., Karp P. H., Itani O. A., Kabel A. C., Wohlford-Lenane C. L., Davis G. J., Hanfland R. A., Smith T. L., Samuel M., Wax D., Murphy C. N., Rieke A., Whitworth K., Uc A., Starner T. D., Brogden K. A., Shilyansky J., McCray P. B., Jr., Zabner J., Prather R. S., Welsh M. J. (2008) Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 321, 1837–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Drumm M. L., Pope H. A., Cliff W. H., Rommens J. M., Marvin S. A., Tsui L. C., Collins F. S., Frizzell R. A., Wilson J. M. (1990) Correction of the cystic fibrosis defect in vitro by retrovirus-mediated gene transfer. Cell 62, 1227–1233 [DOI] [PubMed] [Google Scholar]
- 11. Gregory R. J., Cheng S. H., Rich D. P., Marshall J., Paul S., Hehir K., Ostedgaard L., Klinger K. W., Welsh M. J., Smith A. E. (1990) Expression and characterization of the cystic fibrosis transmembrane conductance regulator. Nature 347, 382–386 [DOI] [PubMed] [Google Scholar]
- 12. Riordan J. R. (1993) The cystic fibrosis transmembrane conductance regulator. Annu. Rev. Physiol. 55, 609–630 [DOI] [PubMed] [Google Scholar]
- 13. Riordan J. R., Rommens J. M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J. L. (1989) Identification of the cystic fibrosis gene. Cloning and characterization of complementary DNA. Science 245, 1066–1073 [DOI] [PubMed] [Google Scholar]
- 14. Bradbury N. A., Cohn J. A., Venglarik C. J., Bridges R. J. (1994) Biochemical and biophysical identification of cystic fibrosis transmembrane conductance regulator chloride channels as components of endocytic clathrin-coated vesicles. J. Biol. Chem. 269, 8296–8302 [PubMed] [Google Scholar]
- 15. Prince L. S., Workman R. B., Jr., Marchase R. B. (1994) Rapid endocytosis of the cystic fibrosis transmembrane conductance regulator chloride channel. Proc. Natl. Acad. Sci. U.S.A. 91, 5192–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sharma M., Pampinella F., Nemes C., Benharouga M., So J., Du K., Bache K. G., Papsin B., Zerangue N., Stenmark H., Lukacs G. L. (2004) Misfolding diverts CFTR from recycling to degradation. Quality control at early endosomes. J. Cell Biol. 164, 923–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chang X. B., Mengos A., Hou Y. X., Cui L., Jensen T. J., Aleksandrov A., Riordan J. R., Gentzsch M. (2008) Role of N-linked oligosaccharides in the biosynthetic processing of the cystic fibrosis membrane conductance regulator. J. Cell Sci. 121, 2814–2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cheng S. H., Gregory R. J., Marshall J., Paul S., Souza D. W., White G. A., O'Riordan C. R., Smith A. E. (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63, 827–834 [DOI] [PubMed] [Google Scholar]
- 19. Glozman R., Okiyoneda T., Mulvihill C. M., Rini J. M., Barriere H., Lukacs G. L. (2009) N-Glycans are direct determinants of CFTR folding and stability in secretory and endocytic membrane traffic. J. Cell Biol. 184, 847–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Farinha C. M., Mendes F., Roxo-Rosa M., Penque D., Amaral M. D. (2004) A comparison of 14 antibodies for the biochemical detection of the cystic fibrosis transmembrane conductance regulator protein. Mol. Cell Probes 18, 235–242 [DOI] [PubMed] [Google Scholar]
- 21. Amaral M. D. (2004) CFTR and chaperones. Processing and degradation. J. Mol. Neurosci. 23, 41–48 [DOI] [PubMed] [Google Scholar]
- 22. Kerem B., Rommens J. M., Buchanan J. A., Markiewicz D., Cox T. K., Chakravarti A., Buchwald M., Tsui L. C. (1989) Identification of the cystic fibrosis gene. Genetic analysis. Science 245, 1073–1080 [DOI] [PubMed] [Google Scholar]
- 23. Zielenski J., Tsui L. C. (1995) Cystic fibrosis. Genotypic and phenotypic variations. Annu. Rev. Genet. 29, 777–807 [DOI] [PubMed] [Google Scholar]
- 24. Lukacs G. L., Mohamed A., Kartner N., Chang X. B., Riordan J. R., Grinstein S. (1994) Conformational maturation of CFTR but not its mutant counterpart (ΔF508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 13, 6076–6086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sharma M., Benharouga M., Hu W., Lukacs G. L. (2001) Conformational and temperature-sensitive stability defects of the ΔF508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J. Biol. Chem. 276, 8942–8950 [DOI] [PubMed] [Google Scholar]
- 26. Skach W. R. (2000) Defects in processing and trafficking of the cystic fibrosis transmembrane conductance regulator. Kidney Int. 57, 825–831 [DOI] [PubMed] [Google Scholar]
- 27. Ward C. L., Kopito R. R. (1994) Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J. Biol. Chem. 269, 25710–25718 [PubMed] [Google Scholar]
- 28. Lukacs G. L., Chang X. B., Bear C., Kartner N., Mohamed A., Riordan J. R., Grinstein S. (1993) The ΔF508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J. Biol. Chem. 268, 21592–21598 [PubMed] [Google Scholar]
- 29. Dalemans W., Barbry P., Champigny G., Jallat S., Dott K., Dreyer D., Crystal R. G., Pavirani A., Lecocq J. P., Lazdunski M. (1991) Altered chloride ion channel kinetics associated with the ΔF508 cystic fibrosis mutation. Nature 354, 526–528 [DOI] [PubMed] [Google Scholar]
- 30. Ostedgaard L. S., Rogers C. S., Dong Q., Randak C. O., Vermeer D. W., Rokhlina T., Karp P. H., Welsh M. J. (2007) Processing and function of CFTR-ΔF508 are species-dependent. Proc. Natl. Acad. Sci. U.S.A. 104, 15370–15375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu X., Luo M., Zhang L., Ding W., Yan Z., Engelhardt J. F. (2007) Bioelectric properties of chloride channels in human, pig, ferret, and mouse airway epithelia. Am. J. Respir. Cell Mol. Biol. 36, 313–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu Y., Wang Y., Jiang Y., Zhu N., Liang H., Xu L., Feng X., Yang H., Ma T. (2008) Mild processing defect of porcine ΔF508-CFTR suggests that ΔF508 pigs may not develop cystic fibrosis disease. Biochem. Biophys. Res. Commun. 373, 113–118 [DOI] [PubMed] [Google Scholar]
- 33. Zabner J., Karp P., Seiler M., Phillips S. L., Mitchell C. J., Saavedra M., Welsh M., Klingelhutz A. J. (2003) Development of cystic fibrosis and noncystic fibrosis airway cell lines. Am. J. Physiol. Lung Cell Mol. Physiol. 284, L844–L854 [DOI] [PubMed] [Google Scholar]
- 34. Pedemonte N., Lukacs G. L., Du K., Caci E., Zegarra-Moran O., Galietta L. J., Verkman A. S. (2005) Small-molecule correctors of defective ΔF508-CFTR cellular processing identified by high-throughput screening. J. Clin. Investig. 115, 2564–2571 [DOI] [PMC free article] [PubMed] [Google Scholar]