

Background: The structure-specific endonuclease ERCC1-XPF has multiple DNA binding domains.
Results: Mutations in two individual domains abolish activity on model substrates, but mutations in multiple domains are needed to affect NER activity.
Conclusion: Multiple weak DNA binding interactions mediate the role of ERCC1-XPF in NER.
Significance: DNA binding domains regulate the activity of ERCC1-XPF in NER and ICL repair.
Keywords: DNA-binding Protein, DNA Damage, DNA Enzymes, DNA Nucleotide Excision Repair, DNA Repair, ERCC1-XPF
Abstract
ERCC1-XPF is a heterodimeric, structure-specific endonuclease that cleaves single-stranded/double-stranded DNA junctions and has roles in nucleotide excision repair (NER), interstrand crosslink (ICL) repair, homologous recombination, and possibly other pathways. In NER, ERCC1-XPF is recruited to DNA lesions by interaction with XPA and incises the DNA 5′ to the lesion. We studied the role of the four C-terminal DNA binding domains in mediating NER activity and cleavage of model substrates. We found that mutations in the helix-hairpin-helix domain of ERCC1 and the nuclease domain of XPF abolished cleavage activity on model substrates. Interestingly, mutations in multiple DNA binding domains were needed to significantly diminish NER activity in vitro and in vivo, suggesting that interactions with proteins in the NER incision complex can compensate for some defects in DNA binding. Mutations in DNA binding domains of ERCC1-XPF render cells more sensitive to the crosslinking agent mitomycin C than to ultraviolet radiation, suggesting that the ICL repair function of ERCC1-XPF requires tighter substrate binding than NER. Our studies show that multiple domains of ERCC1-XPF contribute to substrate binding, and are consistent with models of NER suggesting that multiple weak protein-DNA and protein-protein interactions drive progression through the pathway. Our findings are discussed in the context of structural studies of individual domains of ERCC1-XPF and of its role in multiple DNA repair pathways.
Introduction
Structure-specific endonucleases are widespread enzymes that incise DNA as components of most DNA repair and recombination pathways. The activity of these enzymes needs to be tightly regulated since they might otherwise inadvertently fragment DNA (1). One of the most important pathways depending on the action of endonucleases is nucleotide excision repair (NER),3 which addresses lesions induced by UV light, environmental mutagens and certain cancer chemotherapeutic agents. In NER, an oligonucleotide of 24–32 nucleotides in length containing the damage is removed and the original DNA sequence restored using the non-damaged strand as a template (2). NER can be initiated in two ways: Transcription coupled NER (TC-NER) is triggered when RNA polymerase II is stalled by a bulky DNA lesion during transcription (3); Global genome NER (GG-NER) occurs anywhere in the genome and is initiated by the damage sensor XPC-RAD23B, in some cases with the help of the UV-DDB-ubiquitin ligase complex (4–6). Subsequently, TFIIH verifies the presence of the lesion and opens up the DNA helix, allowing the formation of a pre-incision complex containing the endonuclease XPG, the single-stranded DNA binding protein RPA and the architectural protein XPA (7–9). Finally, the second endonuclease, ERCC1-XPF is recruited to the pre-incision complex (10, 11) and incises the DNA 5′ to the lesion, triggering initiation of repair synthesis, incision 3′ to the lesion by XPG, completion of repair synthesis and ligation (12–14).
The two structure-specific endonucleases involved in NER, XPG and ERCC1-XPF, are multifunctional proteins, with diverse roles in NER and other pathways. XPG is a latent endonuclease, fulfilling first a structural and subsequently a catalytic role in NER. It has additional roles in transcription in conjunction with TFIIH (1). The roles of XPG outside of NER are manifest in the severe phenotypes of many XP-G patients (15, 16). By contrast, most of the known XP-F patients present with a mild XP phenotype and have significant residual NER activity due to the presence of low levels of active XPF protein (17). However, a subset of patients and mice with deficiencies in XPF or ERCC1 are much more severely affected and suffer symptoms not caused by NER deficiency alone including developmental abnormalities, premature aging and early death, (18–22). It is believed that these additional symptoms are due to the roles of ERCC1-XPF in interstrand crosslink (ICL) repair, homologous recombination, and possibly telomere maintenance (23–27).
ERCC1-XPF forms an obligate heterodimer and contains a number of distinct domains that contribute to various aspects of its function (Fig. 1). Both proteins contain helix-hairpin-helix (HhH) domains at their C termini that are required for heterodimer formation (28, 29). The active site with the conserved nuclease motif is located adjacent to the HhH domain in XPF (30). The central domain of ERCC1 is structurally homologous to the XPF nuclease domain (29, 31), however, instead of the active site rich in acidic residues, it contains a groove lined with basic and aromatic residues that interact with the XPA protein, connecting ERCC1-XPF to the NER machinery (10, 32–34). XPF, the larger of the two proteins, contains an N-terminal SF2-helicase like domain (HLD), which has lost the ability to bind ATP and to unwind duplex DNA (35). This domain has been implicated in DNA binding and protein-protein interactions, possibly mediating an interaction with SLX4 in ICL repair and other pathways (36–39).
FIGURE 1.

Structure of the ERCC1-XPF HhH domains and scheme of the DNA binding domains in ERCC1-XPF. A, HhH domains of ERCC1 (gray) and XPF (yellow) contribute to dimerization. The residues shown in atom color are believed to contribute to DNA binding and were mutated in our study. The figure is adapted from Ref. 29. B, five domains of ERCC1-XPF that may contribute to DNA binding are shown. Residues in the central and HhH domains of ERCC1 and in the nuclease and HhH domains of XPF mutated to alanine in our study are highlighted in red.
Interestingly, these five domains have been implicated in DNA binding, but evidence to date has been based on analysis of individual domains or on studies of archaeal XPF proteins (10, 29, 34, 40–43). We investigated the role of four C-terminal DNA binding domains by mutational analysis in the context of full-length ERCC1-XPF. Our studies show that DNA binding mutations in any single domain are insufficient to abolish NER in vitro and in vivo. Instead, we report that mutations in multiple domains are necessary to disrupt NER and that there is a hierarchy of importance of the individual domains. Our studies are consistent with the notion that multiple weak interactions among proteins and DNA substrates drive progress through the NER reaction (2, 44).
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Site-directed mutagenesis of pFastBac-XPF and pFastBac-ERCC1-His (30) was performed using the QuikChange site-directed mutagenesis kit (Stratagene) to generate the following mutations: XPFR678A, XPFK850A/R853A, XPFR678A/K850A/K853A, ERCC1K247A/K281A, ERCC1N110A, and ERCC1N110A/K247A/K281A. Bacmid DNA was generated in DH10Bac cells and transfected into the Sf9 insect cells to obtain baculoviruses according to a standard protocol (Bac-to-Bac, Invitrogen). The following combination of ERCC1 and XPF proteins were co-expressed in Sf9 cells for 60 to 65 h with an MOI of 5: ERCC1-XPF, ERCC1-XPFR678A, ERCC1-XPFK850A/R853A, ERCC1-XPFR678A/K850A/R853A, ERCC1K247A/K281A-XPF, ERCC1N110A-XPF, ERCC1N110A/K247A/K281A-XPF, ERCC1K247A/K281A-XPFR678A, ERCC1N110A-XPFR678A, and ERCC1K247A/K281A-XPFK850A/K853A. Cells were lysed and proteins purified over a 1 ml Nickel (His Trap) column (Amersham Biosciences), a HiLoad 16/60 Superdex 200 column (Amersham Biosciences) and a 1 ml Hitrap Heparin column (Amersham Biosciences) as described in (30). The proteins eluted at 650 mm NaCl from the Heparin column and were in some cases concentrated on an Amicon Ultra-4 Centrifugal Filter (Millipore). Proteins were frozen in aliquots in liquid N2 and stored at −80 °C.
Endonuclease Assays
10 pmol of a stem-loop oligonucleotide (GCCAGCGCTCGG(T)22CCGAGCGCTGGC) labeled with fluorescent dye Cy5 at the 3′-end (IDT) were annealed in 200 μl solution (10 mm Tris, pH 8.0, 50 mm NaCl, 1 mm MgCl2) by heating at 90 °C for 10 min and allowing to cool at room temperature for 2 h. 100 fmol of the substrate were incubated in 25 mm Tris, pH 8.0, 2 mm MgCl2, 10% glycerol, 0.5 mm β-mercaptoethanol, 0.1 mg/ml BSA, 40 mm NaCl with various amounts of protein in a volume of 15 μl. The reactions were incubated at 30 °C for 30 min and quenched by adding 10 μl of 80% formamide/10 mm EDTA. After heating at 95 °C for 5 min and cooling on ice, 3 μl of each sample were analyzed on a 12% denaturing polyacrylamide gel. Bands were visualized by fluorescence imaging on a Typhoon 9400 imaging system (Amersham Biosciences).
In Vitro NER Assay
Extracts derived from XPF-deficient XP2YO cells and plasmids containing dG-acetylaminofluorene (dG-AAF) or 1,3-intrastrand cisplatin (cis-Pt) lesions were prepared as described previously (45–47). For each reaction, 2 μl of repair buffer (200 mm Hepes-KOH (pH 7.8), 25 mm MgCl2, 2.5 mm DTT, 10 mm ATP, 110 mm phosphocreatine (di-Tris salt, Sigma), and 1.8 mg/ml BSA), 0.2 μl of creatine phosphokinase (2.5 mg/ml, sigma), 3 μl of XPF-deficient cell extract (about 10 mg/ml), NaCl (to a final concentration of 70 mm), and different amounts of purified ERCC1-XPF in a total volume of 9 μl were pre-warmed at 30 °C for 10 min. 1 μl plasmid containing dG-AAF or cis-Pt (50 ng/μl) was added to each reaction, and the samples were incubated at 30 °C for 45 min. After adding 0.5 μl of 1 μm of an oligonucleotide complementary to the excision product with a 4G overhang for either dG-AAF (5′ GGGGCATGTGGCGCCGGTAATAGCTACGTAGCTCp-3′) or cis-Pt (5′-GGGGGAAGAGTGCACAGAAGAAGACCTGGTCGACCp-3′), the mixtures were denatured by heating at 95 °C for 5 min. After cooling at room temperature for 15 min, 1 μl of sequenase mixture (0.13 units of sequenase (USB), and 2.0 μCi [α-32P]dCTP for each reaction) was added and incubated at 37 °C for 3 min, followed by addition of 1.2 μl dNTP mixture (50 μm dCTP, 100 μm dTTP, 100 μm dATP, and 100 μm dGTP). The reactions were incubated at 37 °C for 12 min and stopped by addition of 8 μl of 80% formamide/10 mm EDTA. After heating to 95 °C for 5 min, samples were placed on ice and analyzed on a 14% denaturing polyacrylamide gel. Gels were exposed to a phosphor screen and visualized by Phosphorimager (Typhoon 9400, Amersham Biosciences Biosciences).
DNA Binding Measurements by Fluorescent Anisotropy
For the anisotropy experiments, the protein storage buffer was exchanged to 25 mm KH2PO4 pH 7.6, 5 mm β-mercaptoethanol, 10% glycerol, 150 mm NaCl. Increasing concentrations of protein were incubated with the splayed arm substrate (10 nm) which was made by annealing the following oligonucleotides: 5′-CTTTCGAACATCCAGGAGAGCACGGCCTTTTTTTTTTTTTTTTTTTT with an FAM label at the 3′-end and 5′-TTTTTTTTTTTTTTTTTTTTGGCCGTGCTCTCCTGGATGTTCGAAAG. 100 nm of competitor double-stranded DNA (5′-TCAAAGTCACGACCTAGACACTGCGAGCTCGAATTCACTGGAGTGACCTC and 5′-GAGGTCACTCCAGTGAATTCGAGCTCGCAGTGTCTAGGTCGTGACTTTGA) were used in each reaction. Each reaction was incubated in 20 μl of buffer (25 mm Hepes-KOH (pH 8.0), 15% glycerol, 0.1 mg/ml BSA, 2 mm CaCl2, 0.5 mm β -mercaptoethanol, and 40 mm NaCl) at 30 °C for 5 min. The experiments were conducted at least four times and measured on Infinite M1000 plate reader (Tecan). The data were fitted using the grafit4 program to the equation f = y + a × x^b/(c^b + x^b), where x is the protein concentration, f is the fluorescent anisotropy and c is the Kd value.
Generation of Mutant Cell Lines using Lentiviral Transduction
Human XP-F mutant fibroblasts XP2YO, Chinese hamster ovary cells UV20 (with a mutation in ERCC1), and 293T cells were cultured in Dulbecco's modified Eagle's medium high glucose 1× (GIBCO), 10% fetal bovine serum, 100 units/ml penicillin, and 0.1 mg/ml streptomycin at 37 °C in a 5% CO2 humid incubator. Wild-type or mutant XPF cDNA with an hemaglutinin (HA) tag at C-terminal was inserted into the pWPXL vector, which was co-transfected with the packaging plasmid psPAX2 and the envelop plasmid pMD2G into 293T cells to generate lentiviral particles. The particles were transduced into XP2YO cells to produce cell lines stably expressing wild-type or mutant XPF proteins according to established procedures (48, 49). The same procedure was applied to generate cell lines expressing wild-type or mutant ERCC1 in UV20 cells.
Local UV Irradiation and Immunofluorescence
About 50,000 cells were plated on a coverslip in 6-well plates, grown for 2–3 days and irradiated through a polycarbonate membrane with 5 μm pores (Millipore) with UV light (254 nm) with a dose of 150 J/m2 (XP2YO cells) or 120 J/m2 (UV20 cells). Cells were incubated at 37 °C, 5% CO2 for 30 min to 24 h. They were washed first with PBS and then with PBS plus 0.05% Triton X-100 and fixed with 3% paraformaldehyde plus 0.1% Triton X-100 (XP2YO cells) or washed first with PBS and then with PBS plus 0.1% triton X-100, and fixed by 3% paraformaldehyde plus 0.2% triton X-100 (UV20 cells). After fixation, cells were washed with PBS containing 0.2% Triton X-100. To stain for (6–4)PPs, cells were treated with 0.07 m NaOH in PBS for 5 min, followed by washing with PBS plus 0.2% Triton X-100. After blocking with PBS plus 5 mg/ml BSA and 1.5 mg/ml glycine, cells were stained with mouse monoclonal anti-(6–4)PP antibody (Cosmo Bio) 1:400, rabbit polyclonal anti-ERCC1 antibody (FL-297, Santa Cruz Biotechnology) 1:100, or rabbit polyclonal anti-HA antibody (ChIP grade, Abcam) 1:2000 for 1.5 h, and washed with PBS containing 0.2% Triton X-100. Cells were then incubated with secondary antibodies: Cy3-conjugated affinipure goat anti-mouse IgG(H+L) (Jackson ImmunoResearch) 1:1000 and Alexa Fluor 488-labeled F(ab′)2 fragment of goat anti-rabbit IgG (H+L) (Invitrogen) 1:800 for 1 h, followed by washing with PBS with 0.2% Triton X-100. Samples were washed with PBS, embedded in Vectashield Mounting Medium with 1.5 μg/ml of DAPI (Vector Laboratories) and analyzed using a confocal microscope (Zeiss LSM 510). About 100 cells were counted in at least three independent experiments for quantification.
Clonogenic Survival Assay
Exponentially growing cells were plated in 6 cm dishes in triplicate at a density of 1–20 × 103-cells/plate for human cells or 250–5,000 cells/plate for Chinese hamster ovary cells, depending on the dose of genotoxin used and the predicted phenotype of the cells. For the hamster cells, wild-type cells were plated as follows: 3 plates at 250 cells/plate (untreated) 3 plates at 250 cells/plate (dose 1), 3 plates at 250 cells/plate (dose 2), 3 plates at 500 cells/plate (dose 3), and 3 plates at 500 cells/plate (dose 4). For hamster cells, knock-out cells were plated as follows: 3 plates at 250 cells/plate (untreated), 3 plates at 250 cells/plate (dose 1), 3 plates at 500 cells/plate (dose 2), 3 plates at 1000 cells/plate (dose 3), and 3 plates at 5,000 cells/plate (dose 4). The next day, the cells were transiently exposed to genotoxins. For UV treatment, the media was aspirated from the plates; the cells were washed with PBS; irradiated with UV (254 nm, Spectroline X-15) and the cells were replenished with fresh medium. For mitomycin C (MMC) treatment, the cells were treated with medium containing MMC (Fisher) at 37°C for 1 h. The plates were then washed twice with PBS and replenished with fresh medium. 7–10 days after exposure to genotoxins, when colonies are visible to the naked eye, the cells were fixed and stained with 50% methanol, 7% acetic acid and 0.1% Coomassie Brilliant Blue. The colonies (defined as containing >10 cells) were counted using an Olympus SZ61 stereomicroscope with a 10× eyepiece. The data were plotted as the number of colonies on treated plates divided by the number of colonies on untreated plates ± S.E. for a minimum of two independent experiments, both done in triplicate.
RESULTS
Generation of ERCC1-XPF with Mutations in Four DNA Binding Domains
We used information from structural and functional studies to design ERCC1-XPF proteins with mutations in four DNA binding domains in the C-terminal halves of the two proteins. Previous studies have shown that a protein containing only the two HhH, XPF nuclease, and ERCC1 central domains can still cleave ss/dsDNA junctions, albeit with much reduced efficiency (29). HhH domains have been shown to contribute to DNA binding in ERCC1-XPF (29, 43) and other proteins (50). Based on structural studies, we chose two basic residues in each HhH domain, K247 and K281 in ERCC1, and K850 and R853 in XPF, which have been suggested to play a role in DNA binding (Fig. 1) (29, 43). NMR studies revealed that the HhH domain in ERCC1 appears to be more important for DNA binding (43). We expected the nuclease active site in XPF to interact with DNA directly and chose to include R678 (Fig. 1B), as we have previously shown that mutating this residue to alanine resulted in a protein with only marginal activity on model substrate, while being active in NER in the presence of the other NER proteins. We suggest that this residue contributes to positioning of the active site on the substrate rather than the catalytic mechanism (14, 30). Although the central domain of ERCC1 can bind DNA (29, 34), its main function is likely to interact with XPA. We included the N110A mutant, which has a weakened interaction with XPA (Fig. 1B) (10, 34), to determine to what extent defects in the interaction with XPA synergize with defects in DNA binding. We mutated all of these residues to alanine and combined the different mutations to study how these four domains contribute to the DNA binding and NER activity of ERCC1-XPF.
Mutations in the HhH Domain of ERCC1 and the Nuclease Domain of XPF Lead to the Loss of Incision Activity on Stem Loop Substrates
Heterodimeric ERCC1-XPF containing mutations in the HhH, nuclease and central domains were expressed in insect cells and purified using our established protocol (30). All the proteins, including those containing mutations in two domains formed heterodimers similar to the wild-type protein. Only fractions eluting at 60–70 ml, representing heterodimeric ERCC1-XPF were collected during the gel filtration chromatography step (supplemental Fig. S1A). We have previously shown that these fractions contain the active protein (30). Furthermore, the CD spectrum of selected mutants were identical to that of wild-type ERCC1-XPF suggesting that they were properly folded (supplemental Fig. S1B). The endonuclease activity of ERCC1-XPF in wild-type and mutant form was tested on a 3′ fluorescently labeled stem-loop model substrate, in which the protein cut at the ss and dsDNA junction in the presence of Mg2+ (51). ERCC1-XPF in wild-type form and with mutations in the HhH domain of XPF or the central domain of ERCC1, cleaved the stem-loop substrate with similar efficiencies as the wild-type protein in a concentration-dependent manner (Fig. 2). By contrast, no such incision products were observed for proteins containing mutations in the HhH domain of ERCC1 (ERCC1K247A/K281A, Fig. 2A, lanes 4–5) or in the nuclease domain of XPF (XPFR678A, Fig. 2B, lanes 4–5). From these results we conclude that the HhH domain of ERCC1 and the nuclease domain of XPF are more important than the ERCC1 central and XPF HhH domains in mediating nuclease activity.
FIGURE 2.
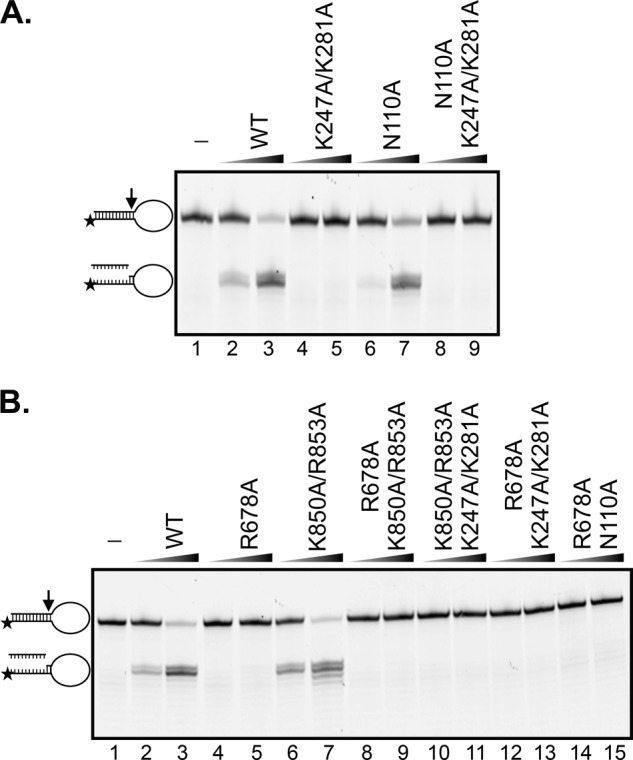
Mutations in the HhH domain of ERCC1 and the nuclease domain of XPF impact nuclease activity on a stem-loop substrate. ERCC1-XPF in wild-type form or with mutations in ERCC1 (A), XPF or both (B) were incubated with 3′-Cy5-labeled stem-loop DNA substrate (6.7 nm) at 37 °C for 30 min, and products were visualized on 12% denaturing polyacrylamide gels. Concentrations of ERCC1-XPF: lanes 1: no protein; lanes 2, 4, 6, 8, and 10, 12 and 14: 3.3 nm; lanes 3, 5, 7, 9, and 11: 26.7 nm. The position of the substrate and product are indicated on the left side of the gel.
The HhH Domain of ERCC1 Is a Key Contributor for DNA Binding Affinity
We wanted to gain insight into whether the reduced nuclease activity caused by mutations in the ERCC1 HhH and XPF nuclease domains correlated with reduced DNA binding activity. We used fluorescence anisotropy to measure the binding affinity of ERCC1-XPF to model DNA substrates. We titrated 3′-labeled splayed arm, stem loop or duplex DNA substrates with wild-type ERCC1-XPF and measured the increase in anisotropy as a function of the protein concentration. Surprisingly, ERCC1-XPF-bound dsDNA with similar affinity as the splayed arm or stem-loop substrates (data not shown), although ERCC1-XPF did not incise dsDNA (51). However, addition of a 10-fold excess of non-labeled dsDNA abolished the binding of ERCC1-XPF to dsDNA, but not to the splayed arm and stem loop substrates, confirming that ERCC1-XPF specifically bound ss/dsDNA junctions.
To determine the effect of mutations in the four domains on DNA binding quantitatively, 3′-FAM-labeled spayed arm substrates were incubated with increasing amounts of protein in the presence of a 10-fold excess of non-labeled dsDNA competitor. The anisotropy binding curves for the WT, XPFR678A, ERCC1N110A, and XPFK850A/K853A proteins displayed a similar shape, while the anisotropy signal of ERCC1K247A/K281A was about 40% lower, suggesting that the architecture of this protein-DNA complex was different from that of the wild-type protein (Fig. 3). Quantification of the data revealed that the ERCC1K247A/K281A also displayed the lowest binding affinity for the splayed arm substrate (Kd = 219 ± 9 nm), about 2-fold lower than the WT, XPFR678A and ERCC1N110A proteins. The Kd of XPFK850A/K853A was about 1.6-fold lower than that of the wild-type protein. The results suggest that the HhH domains of both ERCC1 and XPF contribute to substrate binding by ERCC1-XPF, with a more prominent contribution from the ERCC1 HhH domain. Our data also show that mutations in a single domain alone are not sufficient to completely abolish DNA binding by ERCC1-XPF.
FIGURE 3.
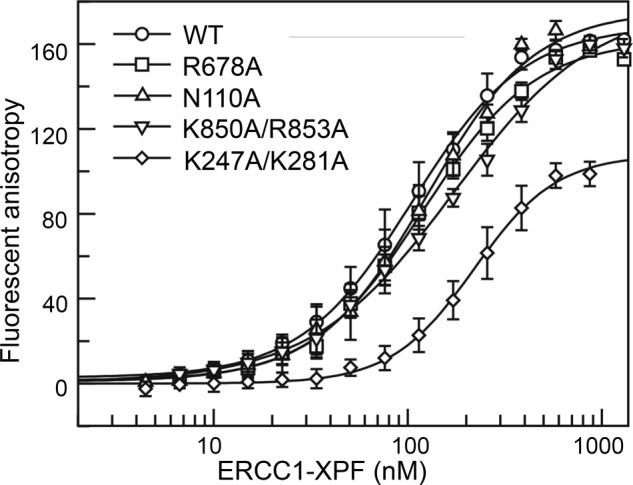
Mutations in the HhH domain of ERCC1 lead to reduced DNA binding affinity. Fluorescein-labeled splayed arm DNA substrate (10 nm) in the presence of 100 nm non-labeled competitor double-stranded DNA was titrated with wild-type and mutant ERCC1-XPF. DNA affinities were measured by fluorescence anisotropy. Error bars represent the standard deviation from a minimum of four experiments.
Mutations in Multiple DNA Binding Domains Synergistically Affect NER Activity in Vitro
We then investigated the effect of these mutations on the overall NER reaction in the context of cell-free extracts. Plasmids containing site-specific bulky DNA substrates, either 1,3-intrastrand cisplatin crosslinks (cis-Pt) or acetylaminofluorene adducts of dG (dG-AAF), were incubated with XPF-deficient cell-free extract and wild-type or mutant ERCC1-XPF to observe the ability of the protein to excise DNA in the context of other NER proteins (45, 46, 52). XP-F extracts are devoid of ERCC1 and XPF and did not display any NER activity, but addition of wild-type ERCC1-XPF restored NER activity as shown previously (Fig. 4, A and B, lanes 1–3) (14, 33). Mutations in the HhH domain of ERCC1 (ERCC1K247A/K281A) led to a mild reduction in NER activity, at least on the cisplatin substrates, while the analogous mutation in XPF (XPFR850A/R853A) had no effect (Fig. 4, A and B, lanes 4–5, and 12–13). Therefore, a defect in the intrinsic DNA binding activity can be overcome at least partially in the context of the full NER complex. XPFR678A was similarly active in NER, although with a slightly shifted incision pattern (Fig. 4A, compare lanes 10–11 to lanes 2–3). This suggests that R678 contributes to the proper positioning of active site residues to carry out the incision. As observed previously, ERCC1N110A displayed reduced NER activity, due to a weakened interaction with XPA (Fig. 4, A and B, lanes 6–7 (33).
FIGURE 4.

Mutations in multiple domains are needed to affect NER in vitro. Plasmids containing site-specific cis-Pt (A) or dG-AAF (B) lesions were incubated with XPF-deficient cell extracts and recombinant purified ERCC1-XPF in wt or mutant form. The excised 25–32mer oligonucleotide was annealed to a complementary strand with a 4G overhang followed by a fill-in reaction with [α-32P]dCTP. Products were analyzed on 14% denaturing polyacrylamide gels and visulized by phosphorimaging. ERCC1-XPF concentrations: lane 1: no protein; even lanes: 5 nm; odd lanes except lane 1: 40 nm. An asterisk denotes the unique band observed with XPFR678A.
Because mutations in the individual domains led to only partial inhibition of NER activity, we tested if a combination of multiple mutations would lead to a more severe defect. We found that, in all cases, a combination of mutations in multiple domains led to reduced NER activity compared with the respective mutations in any single domain. Combining the ERCC1K247A/K281A mutation with ERCC1N110A (Fig. 4A, lanes 8–9), XPFK850A/R853A (Fig. 4A, lanes 16–17), or XPFR678A (Fig. 4A, lanes 18–19) led to a loss of detectable NER activity on the cis-Pt substrate, while some activity was preserved for the dG-AAF substrate for the combination of ERCC1K247A/K281A with ERCC1N110A (Fig. 4B, lanes 8–9) and XPFK850A/R853A (Fig. 4B, lanes 16–17). Similarly, a combination of XPFR678A with XPFK850A/R853A (Fig. 4, A and B, lanes 14–15) or ERCC1N110A (Fig. 4, A and B, lanes 20–21) led to decreased NER activity with both the cis-Pt and dG-AAF substrates. These studies show that combined mutations in multiple DNA binding domains of ERCC1-XPF synergistically affected NER activity and that many domains simultaneously contributed to the DNA binding and cleavage activity of ERCC1-XPF. Interestingly, the reactions of cis-Pt substrates were generally more severely affected than those of the dG-AAF substrates. This could be due to differences in the structure of the substrates in the NER complex and/or because dG-AAF were more efficiently processed by NER under our reaction conditions.
ERCC1-XPF with Impaired DNA Binding Ability Associates with NER Complexes in Cells
We wished to define how the DNA binding mutations of ERCC1-XPF affected NER activity in vivo, by studying the recruitment of the protein to sites of UV damage and the repair kinetics of 6–4 photoproducts ((6–4)PPs). ERCC1-deficient UV20 and XPF-deficient XP2YO cells were transduced with lentiviral vectors expressing wild-type or mutant ERCC1 and XPF proteins, respectively (14, 33). Western blot analysis of the transduced cells revealed that wild-type (ERCC1WT, XPFWT) and mutant proteins (ERCC1K247A/R281A, ERCC1N110A/K247A/K281A, XPFR678A, XPFK850A/R853A, and XPFR678A/K850A/R853A) were expressed at or above endogenous level (supplemental Fig. S2). As it has been shown that only about 35% of the cellular ERCC1-XPF is engaged in NER following UV irradiation (53), we believe that slight differences in expression levels do not influence the outcome of the experiments described here. Cells were irradiated with UV light (254 nm) through a filter with 5 μm pores to generate sites of local UV damage in the cell nuclei. After incubation for 30 min following UV irradiation, cells were fixed and stained with antibodies to detect (6–4)PPs and ERCC1-XPF. As previously shown (33), around 90% of wild-type protein co-localized with (6–4)PPs (Fig. 5). Interestingly, similar amounts of co-localization were observed for the proteins with mutations affecting DNA binding (ERCC1K247A/R281A, XPFR678A, XPFK850A/R853A, and XPFR678A/K850A/R853A), indicating that the DNA binding activity of ERCC1-XPF was not required for the stable association of ERCC1-XPF with NER complexes. The only variant that showed decreased co-localization with (6–4)PPs was ERCC1N110A/K247A/K281A (Fig. 5), but this was expected, as we have shown previously that the N110A mutation affected binding to XPA (33).
FIGURE 5.

Mutations in DNA binding domains of ERCC1-XPF do not affect the recruitment to sites of UV damage. ERCC1-deficient UV20 cells were transduced with wild-type or mutant ERCC1, and XPF-deficient XP2YO with wild-type or mutant XPF. Cells were irradiated with UV light (254 nm, 120 J/m2 for UV20 cells; 150 J/m2 for XP2YO cells) through a polycarbonate filter with 5 μm pores and incubated at 37 °C for 30 min. Cells were fixed and stained with antibodies against (6–4)PPs (red), ERCC1-XPF (green, using an anti-HA antibody in UV20 cells (A), and an anti-ERCC1 antibody in XP2YO cells (B)). Nuclear DNA was visualized by DAPI staining. C, graphical representation of the percentage of co-localization of ERCC1-XPF with (6–4)PPs in UV20 cells. D, graphical representation of the percentage of co-localization of ERCC1-XPF with (6–4)PPs in XP2YO cells. The quantification is based on at least three independent experiments, and error bars represent the standard deviation.
The Repair of (6–4)PPs Is Delayed by DNA Binding Mutations
We next measured how the mutations in ERCC1 and XPF affected the rate of repair of UV lesions. Cells were incubated for various times after local UV irradiation and fixed. The percentage of cells containing (6–4)PPs was determined. 30 min after UV irradiation, about 40% of all UV 20 cells contained sites of (6–4)PPs. At 24 h 17% of the non-transduced UV20 cells contained 6–4PPs, establishing the rate of removal rate in the absence of NER. (Fig. 6, A and C). In ERCC1WT-expressing cells, the damage was almost all gone at 2 h and detectable at only marginal levels at later time points. The rate of repair was significantly reduced in ERCC1K247A/R281A-expressing cells with 24 and 18% of the cells containing (6–4)PPs at 2 h and 4 h, respectively. Repair appeared to be mostly completed however at later time points (8 h, 24 h), indicating that this mutation in the HhH domain of ERCC1 slowed down, but did not abrogate NER. Adding the XPA-interacting mutation N110A to K247A/K281A led to further delay in repair, although repair was still completed at the 24 h time point (Fig. 6, A and C).
FIGURE 6.

Mutations in DNA binding domains of ERCC1-XPF affect repair kinetics of (6–4)PPs in vivo. ERCC1-deficient UV20 cells were transduced with wild-type or mutant ERCC1 (A), XPF-deficient XP2YO cells with wild-type or mutant XPF (B). Cells are irradiated with UV light (254 nm, 120 J/m2 for UV20; 150 J/m2 for XP2YO) through a polycarbonate filter with 5 μm pores, incubated at 37 °C for 30 min, 2 h, 4 h, 8 h, 24 h, fixed and stained with antibody against (6–4)PPs (red) and with DAPI (blue). C, graphical representation of the percentage of cells containing (6–4)PPs in (transduced) UV20 cells. D, graphical representation of the percentage of cells containing (6–4)PPs in (transduced) XP2YO. Quantification is based on at least three independent experiments, and the standard deviation is indicated by error bars.
In the XP-F deficient XP2YO cells, significant levels (∼16%) of (6–4)PPs also persisted at 24 h, and the observed damage levels at 30 min was slightly higher than in the UV20 cells. Transduction of XPFWT restored the complete removal of (6–4)PPs and the repair rate in XPFK850A/R853A-expressing cells was only marginally slower than in wild-type cells. However, the damage removal was impaired in XPFR678A-expressing cells, and a combination of the R678A and K850A/R853A mutations led to a further reduction in the repair rate (Fig. 6, B and D). The cellular studies are thus consistent with the in vitro results showing that DNA binding mutations in the nuclease domain of XPF and the HhH domain of ERCC1 synergistically affected NER.
Mutations in Multiple DNA Binding Domains Render Cells Hypersensitive to UV and Mitomycin C
Having shown that mutations in DNA binding domains in ERCC1-XPF synergistically affected NER in vitro and in vivo, we wanted to test whether these mutations also render cells hypersensitive to UV irradiation as well as mitomycin C (MMC), an agent that causes DNA interstrand crosslinks (ICLs). Cells were first treated with different doses of UV light (254 nm) and the sensitivity was assessed using clonogenical survival assays. Consistent with the results from the NER studies above, mutations in single domains (XPFR678A, XPFK850A/R853A, and ERCC1K247A/K281A) led to mild UV sensitivity, while mutations in two domains (XPFR678A/K850A/R853A or ERCC1N110A/K247A/K281A) led to a more pronounced effect. The hypersensitivity to MMC was even more prominent: cells expressing ERCC1-XPF with mutations in single domains (XPFR678A, XPFK850A/R853A, and ERCC1K247A/K281A) were already associated with dramatic sensitivity, especially XPFR678A. Cells expressing XPFR678A/K850A/R853A or ERCC1N110A/K247A/K281A were as sensitive as non-transfected cells to MMC. These results demostrate that proper DNA binding is even more important in the context of ICL repair.
DISCUSSION
Multiple DNA- and Protein-binding Domains of ERCC1-XPF Control Its Function in NER
In our study, we investigated the role of four C-termial DNA binding domains in ERCC1-XPF (Fig. 1) and show that there is a hierarchy of importance of these domains in mediating the interaction with its DNA substrates. Mutations of DNA binding residues located in the HhH domain of ERCC1 and the nuclease domain of XPF have a more deleterious effect on nuclease and NER activity than mutations in the HhH domain of XPF or the central domain of ERCC1. Importantly, mutations in multiple DNA binding domains are required to significantly suppress NER activity in cultured cells and cell-free extracts, whereas mutations in the ERCC1 HhH or XPF nuclease domain alone are enough to deplete nuclease activity of the purified heterodimer on model substrates. These findings are consistent with current models of NER positing that this process is driven by the dynamic assembly and disassembly of the factors involved (7, 53). Accordingly, multiple protein-protein and protein-DNA interactions drive NER and enable the ordered hand-off of the substrates in damage recognition, damage verification, pre-incision complex assembly, dual incision, repair synthesis, and ligation (2, 44). Our studies allow us to propose how multiple interactions between ERCC1-XPF, XPA, and DNA control incision 5′ to the damage during NER. ERCC1-XPF is recruited to NER complexes by interaction of the central domain of ERCC1 with XPA, in the context of a pre-incision complex consisting of XPA, RPA, TFIIH, and XPG (10, 33). Our cellular studies suggest that DNA binding of ERCC1-XPF is not required to localize the endonuclease to NER complexes (Fig. 5). DNA binding domains therefore anchor the protein at the junction following recruitment, allowing the nuclease domain of XPF to be positioned at the incision site and to cleave the DNA, triggering dual incision and repair synthesis (14). It is likely that interactions with other NER proteins, for example RPA and TFIIH (54, 55), contribute to the fine-tuning of the incision and coordination with other steps in the NER pathway.
The HhH Domains of ERCC1 and XPF Have Distinct Roles in NER
In accordance with NMR studies of individual domains of ERCC1-XPF (43), our results show that mutations in the HhH domain of ERCC1 affect DNA binding, nuclease and NER activity more severely than analogous mutations in the HhH domain of XPF. XPF and ERCC1 are believed to have evolved from a common ancestor (56), which has been studied in the form of archaeal XPF homologs (57). During divergent evolution, XPF retained the nuclease active site and ERCC1 conserved a typical double HhH motif. ERCC1 now harbors an XPA-interacting domain instead of a nuclease domain and the HhH in XPF has lost some typical features. In accordance with this observation and with structural studies, our work demonstrates that the HhH domain of ERCC1 is more important for substrate binding and NER activity than the analogous domain in XPF (43). We know less about how the HLD of XPF contributes to the interaction with DNA. Based on studies of the homologous domain in the Hef helicase, the HLD is expected to contribute to the interaction with ss/dsDNA junctions. Consistent with this observation, human ERCC1-XPF lacking the HLD has been shown to cut model substrates only with marginal efficiency (29).
How Does ERCC1-XPF Bind Substrates in Different Pathways?
ERCC1-XPF has been known for a long time to have roles outside of NER in the repair of interstrand crosslinks and in homologous recombination (23, 24, 58, 59). Deficiencies in these processes have been implicated to cause the severe phenotypes of select ERCC1- or XPF-mutant patients and mice that cannot be explained by NER deficiency (18–22). Recent evidence suggests that an interaction between XPF and the Fanconi anemia protein SLX4/FANCP, rather than with XPA, is crucial for ICL repair (36–39, 60–62).
With respect to our present study, it will be of great interest to determine whether DNA binding by ERCC1-XPF in ICL repair is similar as in NER or whether it follow a distinct pattern. We have shown that even within the NER pathway the repair efficiency of cis-Pt and dG-AAF adducts is affected to different extents by mutations in DNA binding domains of ERCC1-XPF (compare Fig. 4, A and B, lanes 8–9, 16–17), perhaps reflecting a different conformation of the substrates in the NER complex. These differences may be even bigger between substrates for NER and ICL repair, as reflected by the sensitivities to UV light and MMC: cells expressing XPFR678A or ERCC1K247A/K281A are more sensitive to MMC than to UV (Fig. 7).
FIGURE 7.

Mutations in DNA binding residues render cells more sensitive to MMC than to UV. Cells were challenged with UV (254 nm) to determine the impact of the mutations on NER or with MMC to determine the impact on ICL repair. Top row: WT (AA8), ERCC1-deficient (UV20) and complement Chinese hamster ovary cell lines. Bottom row: WT (SV-40, SV40-transformed human fibroblasts), XPF-deficient (XP2YO) and complemented cell lines. The data were plotted as the percentage of colonies that were formed on the treated plates relative to the untreated plates ± S.E. (error bars) for a minimum of two independent experiments each done in triplicate.
High resolution structures of other structure-specific endonucleases such as FEN1 or EXO1 in complex with DNA substrates have shown that these proteins firmly bind the double-stranded portion of the DNA and induce a kink of about 90° in the non-cleaved DNA strand at the junction (63, 64). The cleaved strand undergoes local unpairing of the dsDNA at the incision site, and the ssDNA overhang of the cleaved strand may be bound at different sites of the protein. Based on the structures of archaeal homologs of XPF (40, 41) a similar arrangement can be envisioned for ERCC1-XPF. Interestingly, the residue equivalent to R678 in XPF in the Aeropyrum pernix XPF homolog (R26) is located at a site that possibly binds the ssDNA overhang. Mutation of R678 of XPF affects ICL repair more severely than NER, showing that this residue is more crucial for the positioning of the endonuclease in the context of ICL repair.
We therefore believe that further studies of the five DNA binding domains in ERCC1-XPF will provide valuable insight into how this protein operates in multiple DNA repair pathways. Furthermore, introduction of specific mutations in those domains may lead to pathway-specific defects, enabling new approaches to study the importance of ERCC1-XPF in maintaining genome stability.
Acknowledgments
We thank Drs. Jill Fuss and John Tainer for help with the fluorescence anisotropy experiments, Dr. Jung-Eun Yeo for the gift of dG-AAF-containing plasmid, Dr. Arthur J. Campbell for help with Fig. 1 and Drs. Tom Ellenberger and Oleg Tsodikov for a critical reading of the manuscript and helpful discussions.
This work was supported, in whole or in part, by National Institute of Health Grants GM080454 and CA092584.

This article contains supplemental Figs. S1 and S2.
- NER
- nucleotide excision repair
- ICL
- interstrand crosslink
- HhH
- helix-hairpin-helix
- HLD
- helicase like domain.
REFERENCES
- 1. Fagbemi A. F., Orelli B., Schärer O. D. (2011) Regulation of endonuclease activity in human nucleotide excision repair. DNA Repair 10, 722–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gillet L. C., Schärer O. D. (2006) Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev. 106, 253–276 [DOI] [PubMed] [Google Scholar]
- 3. Hanawalt P. C., Spivak G. (2008) Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 9, 958–970 [DOI] [PubMed] [Google Scholar]
- 4. Min J. H., Pavletich N. P. (2007) Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature 449, 570–575 [DOI] [PubMed] [Google Scholar]
- 5. Sugasawa K., Ng J. M., Masutani C., Iwai S., van der Spek P. J., Eker A. P., Hanaoka F., Bootsma D., Hoeijmakers J. H. (1998) Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol. Cell 2, 223–232 [DOI] [PubMed] [Google Scholar]
- 6. Sugasawa K., Okuda Y., Saijo M., Nishi R., Matsuda N., Chu G., Mori T., Iwai S., Tanaka K., Tanaka K., Hanaoka F. (2005) UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 121, 387–400 [DOI] [PubMed] [Google Scholar]
- 7. Riedl T., Hanaoka F., Egly J. M. (2003) The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J. 22, 5293–5303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tapias A., Auriol J., Forget D., Enzlin J. H., Schärer O. D., Coin F., Coulombe B., Egly J. M. (2004) Ordered conformational changes in damaged DNA induced by nucleotide excision repair factors. J. Biol. Chem. 279, 19074–19083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wakasugi M., Sancar A. (1998) Assembly, subunit composition, and footprint of human DNA repair excision nuclease. Proc. Natl. Acad. Sci. U.S.A. 95, 6669–6674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tsodikov O. V., Ivanov D., Orelli B., Staresincic L., Shoshani I., Oberman R., Schärer O. D., Wagner G., Ellenberger T. (2007) Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA. EMBO J. 26, 4768–4776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Volker M., Moné M. J., Karmakar P., van Hoffen A., Schul W., Vermeulen W., Hoeijmakers J. H., van Driel R., van Zeeland A. A., Mullenders L. H. (2001) Sequential assembly of the nucleotide excision repair factors in vivo. Mol. Cell 8, 213–224 [DOI] [PubMed] [Google Scholar]
- 12. Moser J., Kool H., Giakzidis I., Caldecott K., Mullenders L. H., Fousteri M. I. (2007) Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III α in a cell-cycle-specific manner. Mol. Cell 27, 311–323 [DOI] [PubMed] [Google Scholar]
- 13. Ogi T., Limsirichaikul S., Overmeer R. M., Volker M., Takenaka K., Cloney R., Nakazawa Y., Niimi A., Miki Y., Jaspers N. G., Mullenders L. H., Yamashita S., Fousteri M. I., Lehmann A. R. (2010) Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol. Cell 37, 714–727 [DOI] [PubMed] [Google Scholar]
- 14. Staresincic L., Fagbemi A. F., Enzlin J. H., Gourdin A. M., Wijgers N., Dunand-Sauthier I., Giglia-Mari G., Clarkson S. G., Vermeulen W., Schärer O. D. (2009) Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 28, 1111–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lehmann A. R. (2003) DNA repair-deficient diseases, Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 85, 1101–1111 [DOI] [PubMed] [Google Scholar]
- 16. Schärer O. D. (2008) XPG: its products and biological roles. Adv. Exp. Med. Biol. 637, 83–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ahmad A., Enzlin J. H., Bhagwat N. R., Wijgers N., Raams A., Appledoorn E., Theil A. F., JH J. H., Vermeulen W., NG J. J., Schärer O. D., Niedernhofer L. J. (2010) Mislocalization of XPF-ERCC1 nuclease contributes to reduced DNA repair in XP-F patients. PLoS Genet. 6, e1000871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Niedernhofer L. J., Garinis G. A., Raams A., Lalai A. S., Robinson A. R., Appeldoorn E., Odijk H., Oostendorp R., Ahmad A., van Leeuwen W., Theil A. F., Vermeulen W., van der Horst G. T., Meinecke P., Kleijer W. J., Vijg J., Jaspers N. G., Hoeijmakers J. H. (2006) A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 444, 1038–1043 [DOI] [PubMed] [Google Scholar]
- 19. Jaspers N. G., Raams A., Silengo M. C., Wijgers N., Niedernhofer L. J., Robinson A. R., Giglia-Mari G., Hoogstraten D., Kleijer W. J., Hoeijmakers J. H., Vermeulen W. (2007) First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Am. J. Hum. Genet. 80, 457–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weeda G., Donker I., de Wit J., Morreau H., Janssens R., Vissers C. J., Nigg A., van Steeg H., Bootsma D., Hoeijmakers J. H. (1997) Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr. Biol. 7, 427–439 [DOI] [PubMed] [Google Scholar]
- 21. McWhir J., Selfridge J., Harrison D. J., Squires S., Melton D. W. (1993) Mice with DNA repair gene (ERCC-1) deficiency have elevated levels of p53, liver nuclear abnormalities and die before weaning. Nat. Genet. 5, 217–224 [DOI] [PubMed] [Google Scholar]
- 22. Tian M., Shinkura R., Shinkura N., Alt F. W. (2004) Growth retardation, early death, and DNA repair defects in mice deficient for the nucleotide excision repair enzyme XPF. Mol. Cell. Biol. 24, 1200–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Niedernhofer L. J., Odijk H., Budzowska M., van Drunen E., Maas A., Theil A. F., de Wit J., Jaspers N. G., Beverloo H. B., Hoeijmakers J. H., Kanaar R. (2004) The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol. Cell. Biol. 24, 5776–5787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ahmad A., Robinson A. R., Duensing A., van Drunen E., Beverloo H. B., Weisberg D. B., Hasty P., Hoeijmakers J. H., Niedernhofer L. J. (2008) ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol. Cell. Biol. 28, 5082–5092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bhagwat N., Olsen A. L., Wang A. T., Hanada K., Stuckert P., Kanaar R., D'Andrea A., Niedernhofer L. J., McHugh P. J. (2009) XPF-ERCC1 participates in the Fanconi anemia pathway of cross-link repair. Mol. Cell. Biol. 29, 6427–6437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhu X. D., Niedernhofer L., Kuster B., Mann M., Hoeijmakers J. H., de Lange T. (2003) ERCC1/XPF removes the 3′ overhang from uncapped telomeres and represses formation of telomeric DNA-containing double minute chromosomes. Mol. Cell 12, 1489–1498 [DOI] [PubMed] [Google Scholar]
- 27. Gregg S. Q., Robinson A. R., Niedernhofer L. J. (2011) Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease. DNA Repair 10, 781–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. de Laat W. L., Sijbers A. M., Odijk H., Jaspers N. G., Hoeijmakers J. H. (1998) Mapping of interaction domains between human repair proteins ERCC1 and XPF. Nucleic Acids Res. 26, 4146–4152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tsodikov O. V., Enzlin J. H., Schärer O. D., Ellenberger T. (2005) Crystal structure and DNA binding functions of ERCC1, a subunit of the DNA structure-specific endonuclease XPF-ERCC1. Proc. Natl. Acad. Sci. U.S.A. 102, 11236–11241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Enzlin J. H., Schärer O. D. (2002) The active site of the DNA repair endonuclease XPF-ERCC1 forms a highly conserved nuclease motif. EMBO J. 21, 2045–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nishino T., Komori K., Ishino Y., Morikawa K. (2003) X-ray and biochemical anatomy of an archaeal XPF/Rad1/Mus81 family nuclease: similarity between its endonuclease domain and restriction enzymes. Structure 11, 445–457 [DOI] [PubMed] [Google Scholar]
- 32. Li L., Peterson C. A., Lu X., Legerski R. J. (1995) Mutations in XPA that prevent association with ERCC1 are defective in nucleotide excision repair. Mol. Cell Biol. 15, 1993–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Orelli B., McClendon T. B., Tsodikov O. V., Ellenberger T., Niedernhofer L. J., Schärer O. D. (2010) The XPA-binding domain of ERCC1 is required for nucleotide excision repair but not other DNA repair pathways. J. Biol. Chem. 285, 3705–3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tripsianes K., Folkers G. E., Zheng C., Das D., Grinstead J. S., Kaptein R., Boelens R. (2007) Analysis of the XPA and ssDNA-binding surfaces on the central domain of human ERCC1 reveals evidence for subfunctionalization. Nucleic Acids Res. 35, 5789–5798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sgouros J., Gaillard P. H., Wood R. D. (1999) A relationship betweena DNA-repair/recombination nuclease family and archaeal helicases. Trends Biochem. Sci. 24, 95–97 [DOI] [PubMed] [Google Scholar]
- 36. Fekairi S., Scaglione S., Chahwan C., Taylor E. R., Tissier A., Coulon S., Dong M. Q., Ruse C., Yates J. R., 3rd, Russell P., Fuchs R. P., McGowan C. H., Gaillard P. H. (2009) Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 138, 78–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Muñoz I. M., Hain K., Déclais A. C., Gardiner M., Toh G. W., Sanchez-Pulido L., Heuckmann J. M., Toth R., Macartney T., Eppink B., Kanaar R., Ponting C. P., Lilley D. M., Rouse J. (2009) Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol. Cell 35, 116–127 [DOI] [PubMed] [Google Scholar]
- 38. Svendsen J. M., Smogorzewska A., Sowa M. E., O'Connell B. C., Gygi S. P., Elledge S. J., Harper J. W. (2009) Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 138, 63–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Andersen S. L., Bergstralh D. T., Kohl K. P., LaRocque J. R., Moore C. B., Sekelsky J. (2009) Drosophila MUS312 and the vertebrate ortholog BTBD12 interact with DNA structure-specific endonucleases in DNA repair and recombination. Mol. Cell 35, 128–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Newman M., Murray-Rust J., Lally J., Rudolf J., Fadden A., Knowles P. P., White M. F., McDonald N. Q. (2005) Structure of an XPF endonuclease with and without DNA suggests a model for substrate recognition. EMBO J. 24, 895–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nishino T., Komori K., Ishino Y., Morikawa K. (2005) Structural and functional analyses of an archaeal XPF/Rad1/Mus81 nuclease: asymmetric DNA binding and cleavage mechanisms. Structure 13, 1183–1192 [DOI] [PubMed] [Google Scholar]
- 42. Nishino T., Komori K., Tsuchiya D., Ishino Y., Morikawa K. (2005) Crystal structure and functional implications of Pyrococcus furiosus hef helicase domain involved in branched DNA processing. Structure 13, 143–153 [DOI] [PubMed] [Google Scholar]
- 43. Tripsianes K., Folkers G., Ab E., Das D., Odijk H., Jaspers N. G., Hoeijmakers J. H., Kaptein R., Boelens R. (2005) The structure of the human ERCC1/XPF interaction domains reveals a complementary role for the two proteins in nucleotide excision repair. Structure 13, 1849–1858 [DOI] [PubMed] [Google Scholar]
- 44. Stauffer M. E., Chazin W. J. (2004) Structural mechanisms of DNA replication, repair, and recombination. J. Biol. Chem. 279, 30915–30918 [DOI] [PubMed] [Google Scholar]
- 45. Shivji M. K., Moggs J. G., Kuraoka I., Wood R. D. (1999) Dual-incision assays for nucleotide excision repair using DNA with a lesion at a specific site. Methods Mol. Biol. 113, 373–392 [DOI] [PubMed] [Google Scholar]
- 46. Gillet L. C., Alzeer J., Schärer O. D. (2005) Site-specific incorporation of N-(deoxyguanosin-8-yl)-2-acetylaminofluorene (dG-AAF) into oligonucleotides using modified ultra-mild DNA synthesis. Nucleic Acids Res. 33, 1961–1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Biggerstaff M., Wood R. D. (1999) Assay for nucleotide excision repair protein activity using fractionated cell extracts and UV-damaged plasmid DNA. Methods Mol. Biol. 113, 357–372 [DOI] [PubMed] [Google Scholar]
- 48. Salmon P., Kindler V., Ducrey O., Chapuis B., Zubler R. H., Trono D. (2000) High-level transgene expression in human hematopoietic progenitors and differentiated blood lineages after transduction with improved lentiviral vectors. Blood 96, 3392–3398 [PubMed] [Google Scholar]
- 49. Salmon P., Trono D. (2006) Current Protocols in Neuroscience, Chapter 4, Unit 4, page 21 [DOI] [PubMed] [Google Scholar]
- 50. Doherty A. J., Serpell L. C., Ponting C. P. (1996) The helix-hairpin-helix DNA-binding motif: a structural basis for non-sequence-specific recognition of DNA. Nucleic Acids Res. 24, 2488–2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. de Laat W. L., Appeldoorn E., Jaspers N. G., Hoeijmakers J. H. (1998) DNA structural elements required for ERCC1-XPF endonuclease activity. J. Biol. Chem. 273, 7835–7842 [DOI] [PubMed] [Google Scholar]
- 52. Dunand-Sauthier I., Hohl M., Thorel F., Jaquier-Gubler P., Clarkson S. G., Schärer O. D. (2005) The spacer region of XPG mediates recruitment to nucleotide excision repair complexes and determines substrate specificity. J. Biol. Chem. 280, 7030–7037 [DOI] [PubMed] [Google Scholar]
- 53. Houtsmuller A. B., Rademakers S., Nigg A. L., Hoogstraten D., Hoeijmakers J. H., Vermeulen W. (1999) Action of DNA repair endonuclease ERCC1/XPF in living cells. Science 284, 958–961 [DOI] [PubMed] [Google Scholar]
- 54. Coin F., Auriol J., Tapias A., Clivio P., Vermeulen W., Egly J. M. (2004) Phosphorylation of XPB helicase regulates TFIIH nucleotide excision repair activity. EMBO J. 23, 4835–4846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. de Laat W. L., Appeldoorn E., Sugasawa K., Weterings E., Jaspers N. G., Hoeijmakers J. H. (1998) DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes Dev. 12, 2598–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gaillard P. H., Wood R. D. (2001) Activity of individual ERCC1 and XPF subunits in DNA nucleotide excision repair. Nucleic Acids Res. 29, 872–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rouillon C., White M. F. (2011) The evolution and mechanisms of nucleotide excision repair proteins. Res. Microbiol. 162, 19–26 [DOI] [PubMed] [Google Scholar]
- 58. Bardwell A. J., Bardwell L., Tomkinson A. E., Friedberg E. C. (1994) Specific cleavage of model recombination and repair intermediates by the yeast Rad1-Rad10 DNA endonuclease. Science 265, 2082–2085 [DOI] [PubMed] [Google Scholar]
- 59. Hoy C. A., Thompson L. H., Mooney C. L., Salazar E. P. (1985) Defective DNA cross-link removal in Chinese hamster cell mutants hypersensitive to bifunctional alkylating agents. Cancer Res. 45, 1737–1743 [PubMed] [Google Scholar]
- 60. Crossan G. P., van der Weyden L., Rosado I. V., Langevin F., Gaillard P. H., McIntyre R. E., Sanger Mouse Genetics Project, Gallagher F., Kettunen M. I., Lewis D. Y., Brindle K., Arends M. J., Adams D. J., Patel K. J. (2011) Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat. Genet. 43, 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kim Y., Lach F. P., Desetty R., Hanenberg H., Auerbach A. D., Smogorzewska A. (2011) Mutations of the SLX4 gene in Fanconi anemia. Nat. Genet. 43, 142–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stoepker C., Hain K., Schuster B., Hilhorst-Hofstee Y., Rooimans M. A., Steltenpool J., Oostra A. B., Eirich K., Korthof E. T., Nieuwint A. W., Jaspers N. G., Bettecken T., Joenje H., Schindler D., Rouse J., de Winter J. P. (2011) SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat. Genet. 43, 138–141 [DOI] [PubMed] [Google Scholar]
- 63. Tsutakawa S. E., Classen S., Chapados B. R., Arvai A. S., Finger L. D., Guenther G., Tomlinson C. G., Thompson P., Sarker A. H., Shen B., Cooper P. K., Grasby J. A., Tainer J. A. (2011) Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell 145, 198–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Orans J., McSweeney E. A., Iyer R. R., Hast M. A., Hellinga H. W., Modrich P., Beese L. S. (2011) Structures of human exonuclease 1 DNA complexes suggest a unified mechanism for nuclease family. Cell 145, 212–223 [DOI] [PMC free article] [PubMed] [Google Scholar]