

Background: The cystic fibrosis transmembrane conductance regulator (CFTR), a chloride channel critical for ionic homeostasis in epithelial cells, is mutated in cystic fibrosis.
Results: FKBP8 stabilizes WT and ΔF508 CFTR in the ER and appears to act downstream of Hsp90.
Conclusion: FKBP8 is critical for the biogenesis of WT and ΔF508 CFTR.
Significance: Our findings suggest that FKBP8 is a late acting chaperone for WT and ΔF508 CFTR.
Keywords: CFTR, Cystic Fibrosis, Heat Shock Protein, Hsp90, Immunophilin, Protein Misfolding
Abstract
Cystic fibrosis (CF) is caused by mutations in the apical chloride channel cystic fibrosis transmembrane conductance regulator (CFTR) with 90% of patients carrying at least one deletion of the F508 (ΔF508) allele. This mutant form of CFTR is characterized by a folding and trafficking defect that prevents exit from the endoplasmic reticulum. We previously reported that ΔF508 CFTR can be recovered in a complex with Hsp90 and its co-chaperones as an on-pathway folding intermediate, suggesting that Δ508 CF disease arises due to a failure of the proteostasis network (PN), which manages protein folding and degradation in the cell. We have now examined the role of FK506-binding protein 8 (FKBP8), a component of the CFTR interactome, during the biogenesis of wild-type and ΔF508 CFTR. FKBP8 is a member of the peptidylprolyl isomerase family that mediates the cis/trans interconversion of peptidyl prolyl bonds. Our results suggest that FKBP8 is a key PN factor required at a post-Hsp90 step in CFTR biogenesis. In addition, changes in its expression level or alteration of its activity by a peptidylprolyl isomerase inhibitor alter CFTR stability and transport. We propose that CF is caused by the sequential failure of the prevailing PN pathway to stabilize ΔF508-CFTR for endoplasmic reticulum export, a pathway that can be therapeutically managed.
Introduction
The ability of a protein to fold is dictated both by the energetic minimization of amino acids in the peptide sequence (1) and by the chaperoning properties of the protein homeostasis, or proteostasis, network (PN)5 (2). The PN includes the stress-inducible heat shock proteins (Hsp) and constitutively expressed homologs, as well as chaperonins and protein refolding complexes. Other components of the PN direct misfolded or slowly folding proteins to the ubiquitin/proteasome and autophagy/lysosomal degradation systems (2–7). Misfolding diseases can occur as a result of alterations of the protein fold in response to inherited and sporadic causes leading to either loss-of-function or gain-of-toxic function conformers that trigger human pathophysiology (7).
Cystic fibrosis (CF), the most common lethal genetic disease in the Caucasian population, is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) protein. CFTR is a cAMP-regulated chloride channel expressed at the apical surface of epithelial cells. Over 90% of patients carry at least one Phe-508 deletion allele (ΔF508-CFTR), which leads to impaired folding and rapid degradation of the mutant protein by endoplasmic reticulum-associated degradation (ERAD) (3, 9–11). The absence of CFTR at the cell surface contributes to loss of hydration of the epithelial lining of the lung and other tissues, triggering the progressive pathology characteristic of the disease (12).
CFTR folding is critically dependent on the activity of multiple chaperone proteins, including the cytosolic heat shock protein (Hsp) 70 and 90 systems (13–19). The folding defect associated with ΔF508-CFTR results in a prolonged association with the Hsp40-Hsp70 chaperone system, resulting in its degradation via the ubiquitin proteasome pathway (9, 17, 19–24). Hsp90 is a key member of the PN (2) and also plays a critical role in the folding of multiple client proteins, including CFTR (18, 25–29). Hsp90 is not generally considered to function during co-translational folding of de novo synthesized proteins (28). Rather, the chaperone activity of Hsp90 and its associated co-chaperones are thought to regulate the structure of more mature clients, which occupy multiple folded states, to mediate function (27). The ATPase activity of Hsp90 can be slowed by silencing the expression of the accelerator of Hsp90 ATPase, Aha1 (18, 30, 31). We previously showed that Aha1 silencing promotes the maturation and trafficking of ΔF508-CFTR to the cell surface and re-establishes channel activity (18, 30). This indicates that the misfolded ΔF508 channel is recognized by components of the PN (18, 32–36).
To begin to understand, mechanistically, the operation of the Hsp70/90 system in the folding of WT- and ΔF508-CFTR, we have now investigated the role of the FK506-binding protein (FKBP) isoform 8 (FKBP8). FKBP8 is the only FKBP family member recovered in the CFTR interactome that preferentially associated with ΔF508-CFTR (18), suggesting that it functions at a critical step in the folding of CFTR. FKBPs define a family of enzymes that mediate the cis/trans conversion of peptidyl-prolyl bonds through their peptidylprolyl isomerase activity (PPIase), a critical step in folding of both de novo synthesized (37, 38) and mature proteins (39–41). The integrating feature of this family is the presence of a PPIase domain. This subfamily of PPIases is further characterized by their ability to bind to the immunosuppressive drugs, FK506 and rapamycin, that act as inhibitors of isomerase activity. FKBP12 represents the prototypical member of this enzyme family. FKBP12 contains a single FK506-binding domain (FBD) (Fig. 1), and its binding to immunosuppressive drugs results in the inhibition of calcineurin phosphatase activity and subsequent inhibition of the immune cascade (42–44). Higher molecular weight members of this family, such as FKBP51, -52, and -8, contain additional domains, such as tetratricopeptide (TPR) and calmodulin binding domains (Fig. 1) (45). These TPR domain-containing family members also harbor a leucine zipper motif (LZ) spanning residues 278–306 of human FKBP8, which overlaps with its TPR domain and is involved in mediating protein-protein interactions (45).
FIGURE 1.
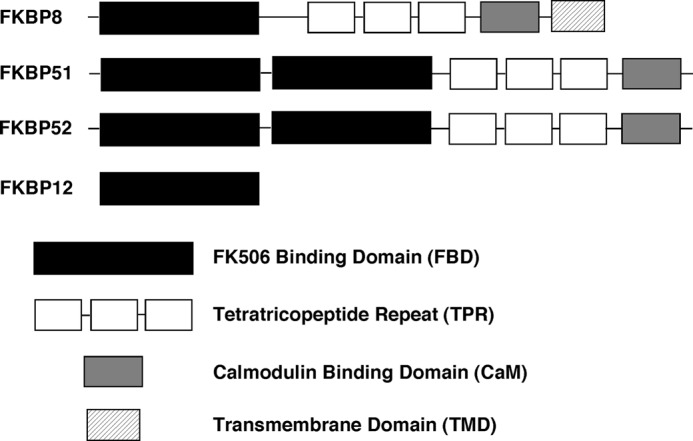
Schematic diagram of the domain arrangements of FKBP8, -12, -51, and -52.
FKBP8 represents a unique member of the FKBP family in that it is localized to both ER and mitochondrial membranes through its C-terminal transmembrane domain, and its N-terminal functional regions reside in the cytosol (46). FKBP8 is able to bind to Hsp90 through its tripartite TPR motif (47), consistent with what has been shown for related family members, such as FKBP51 and -52 (48–51). However, unlike what is seen with FKBP51 and -52, which facilitate delivery of client proteins through their ability to bind Hsp90 and client simultaneously, Hsp90 binding prevents the ability of FKBP8 to interact with client proteins (47). This raises the possibility that FKBP8 has an additional independent role in the PN. In fact, this hypothesis is supported by data showing that FKBP8 exhibits Hsp90-independent chaperoning activity that determines the stability and anti-apoptotic activity of Bcl-2 (52) and that FKBP8 is required for the Hsp90-independent stability and function of the voltage-dependent potassium channel, HERG (53). In the case of CFTR, one possibility is that FKBP8 exhibits an independent function that mediates the ER retention of the ΔF508 mutant. Alternatively, FKBP8 could be a component of an “on-pathway” folding intermediate that the ΔF508 mutant cannot resolve. The latter possibility is in agreement with recent data showing that FKBP8 is required for the trafficking of WT-CFTR (54).
Herein, we demonstrate the mechanism of FKBP8 activity in CFTR biogenesis. We find that silencing of FKBP8 results in the accumulation of ΔF508- and WT-CFTR in a trapped folding intermediate. This results in destabilization of the protein and a concomitant loss of channel activity. Thus, in the absence of FKBP8 even WT-CFTR becomes prone to degradation, highlighting the need for a critical PPIase-dependent step in CFTR maturation. Moreover, we provide evidence that FKBP8 is able to bind to CFTR independently of Hsp90 and exhibits a preference for the export-competent pool of CFTR, suggesting that FKBP8 is a late acting chaperone and functions downstream of the Hsp90-dependent folding step. Consequently, FKBP8 may fine-tune or complete the folding of the CFTR conformation that is subject to ER export. We therefore suggest that FKBP8 is a key component of the on-pathway PN, mediating the proper folding and maturation of both WT- and ΔF508-CFTR. Therefore, FKBP8 is a new and important target to consider in the therapeutic management of cystic fibrosis.
MATERIALS AND METHODS
Reagents and Antibodies
Antibodies for CFTR used in this study are as follows: the M3A7 antibody (NBD2) from Dr. John Riordan (University of North Carolina), and the 3G11 antibody (NBD1) obtained from the CF Folding Consortium. The FKBP8 antibody was a gift from Dr. Michiko Shirane (Kyushu University). The N-(N′,N′-dimethylcarboxamidomethyl)cycloheximide (DM-CHX) was a kind gift from Dr. Gunther Fisher (Max Planck Research Unit for Enzymology of Protein Folding). The FKBP51 and FKBP52 antibodies were a kind gift from Dr. David Smith (Mayo Clinic, Scottsdale, AZ).
DNA Constructs
Full-length FKBP8 was PCR-amplified using the following forward (5′-CCG GTA TCT AGA ATG GCA TCG TGT GCT GAA-3′) and reverse (5′-GCG CAA GGA TCC TCA GTT CCT GGC AGC GAT-3′) primers and cloned into the XbaI/BamHI restriction sites of pcDNA3.1 containing an N-terminal Myc tag.
Immunofluorescence
CFBE41o− cells stably expressing ΔF508-CFTR were grown on glass coverslips and fixed in PBS containing 4% paraformaldehyde at room temperature for 15 min. Cells were then washed with three changes of PBS and incubated with blocking buffer (PBS containing 1% BSA, 2% normal goat serum, and 0.4% saponin) for 20 min. Coverslips were then incubated for 1 h with primary antibodies (M3A7 at 1:100 (mouse) and FKBP8 at 1:300 (rabbit)) diluted in blocking buffer. Coverslips were washed with five changes of PBS + 0.1 m glycine and reblocked as before. Cells were then incubated for 1 h with secondary antibodies (goat anti-mouse Alexa488 and goat anti-rabbit Alexa596) diluted at 1:300 in blocking buffer and subsequently washed with PBS + 0.1 m glycine as before. Coverslips were mounted and imaged.
siRNA Treatment of HBE and CFBE Cells
Cells were seeded in 12-well or 60-mm dishes at a density of 1 or 5 × 105 cells, respectively, 1 day prior to transfection. Transfection complexes were prepared by combining 600 nm siRNA diluted in Opti-MEM and 2 μl (12-well) or 8 μl (60 mm) of RNAiMax transfection reagent (Invitrogen) diluted in Opti-MEM. This mixture was then added to the dish containing Opti-MEM media to a final siRNA concentration of 50 nm and gently mixed to ensure proper distribution of the transfection mixture. Opti-MEM was replaced with growth media (α-minimal essential medium containing 10% FBS, 2 mm l-glutamine, and 2 μg/ml puromycin (CFBE) or 1 μg/ml blasticidin (HBE)) 24 h after transfection. Cells were then transfected as above for a second time and cultured for an additional 48 h. Prior to the completion of the second incubation, temperature-corrected CFBE cells were transferred to a 30 °C/5% CO2 incubator 24 h prior to harvesting cells. Double siRNA transfections were performed as above but using 25 nm final of each pair of siRNAs (siFKBP8 + scramble, siAha1 + scramble, or siFKBP8 + siAha1).
Treatment of HBE and CFBE Cells with DM-CHX
Cells were seeded as for siRNA treatment. Once the cells had reached confluency, they were treated with DM-CHX at the indicated concentration or Hepes, pH 7.8, at a final concentration of 78.9 μm for 24 h at 37 °C/5% CO2 prior to harvesting. Temperature-corrected CFBE cells were cultured for 24 h at 30 °C/5% CO2 prior to harvesting.
Preparation of Cell Lysate and Western Blotting
Cells were washed twice with ice-cold PBS, and 50 μl of lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100, and 2 mg/ml of complete protease inhibitor mixture) was added to each well. Cells were lysed on ice for 30 min with occasional mixing. The lysates were collected and spun at 14,000 × g for 20 min at 4 °C. The protein concentration in the supernatants was assessed by the Bradford assay using the Coomassie Protein Assay Reagent (Pierce catalog no. 1856209). 15 μg of total protein were separated by 8% SDS-PAGE, transferred to nitrocellulose, and incubated with primary antibody at the indicated dilutions overnight at 4 °C in TBS + 0.1% Tween 20 + 5% milk. Nitrocellulose were subsequently incubated with the appropriate HRP-conjugated secondary antibody diluted at 1:10,000 in TBS + 0.1% Tween 20 + 5% milk at room temperature for 1 h. The detection was performed by chemiluminescence using ECL reagent.
Immunoprecipitation
Lysates were prepared as described for Western blotting, and 1.0 mg of total protein was pre-cleared by incubating with γ-bind beads (GE Healthcare) for 1 h at 4 °C and subsequently incubated for 16 h at 4 °C with 20 μl of M3A7 CFTR antibody cross-linked to γ-bind beads (GE Healthcare). The beads were washed with two changes of lysis buffer without protease inhibitor mixture. Beads were pelleted at 2000 × g for 1 min, and bound proteins were eluted by incubation with 50 mm Tris-HCl, pH 6.8, containing 1% SDS at room temperature for 30 min and at 37 °C for 5 min. Western blots of eluted proteins were analyzed for CFTR recovery (M3A7), Hsp90 (Assay Designs (SPA-846)), Hsp70 (Assay Designs (SPA-812)), and FKBP8. The blots were visualized as described above.
Iodide Efflux Assay
CFBE and HBE cells were cultured in 60-mm dishes and treated as described above. Cells were washed five times with loading buffer (136 mm NaI, 3 mm KNO3, 2 mm Ca(NO3)2, 20 mm Hepes, 11 mm glucose, and pH adjusted to 7.4) and subsequently incubated with 2.5 ml of loading buffer for 1 h at room temperature. Cells were washed 15 times with efflux buffer (136 mm NaNO3, 3 mm KNO3, 2 mm Ca(NO3)2, 20 mm Hepes, 11 mm glucose, and pH adjusted to 7.4) followed by two 1-min incubations with 2.5 ml of efflux buffer that was collected for iodide concentration determination. Cells were then incubated with stimulation buffer (efflux buffer containing 10 μm forskolin (Sigma) and 50 μm genistein (Sigma)) four times for 1 min, and the buffer was collected for iodide concentration determination. Stimulation buffer was washed out by incubating cells with efflux buffer six times for 1 min, and the buffer was collected for iodide concentration determination. The concentration of iodide at each time point was determined using an iodide selective electrode (Analytical Sensors and Instruments Ltd.) connected to a Beckman 390 pH/temperature/mV/ISE meter.
Analysis of FKBP8 on CFTR Stability in Yeast
The yeast strain RSY620 (MATa, ade2-1, trp1-1, leu2-3,112, ura3-1, his3-11,15, PEP4::TRP1) was propagated, and yeast growth medium was prepared using established methods (55). To create a copper-inducible vector for the controlled expression of FKBP8, the FKBP8 coding sequence was amplified by PCR using the following forward (5′-CCG GTA GGA TCC ATG GCA TCG TGT GCT GAA CC-3′) and reverse (5′-GGC CAA AAG CTT TCA GTT CCT GGC AGC GAT GAC C-3′) primers, and the resulting isolated fragment was inserted into the BamHI and HindIII restriction sites in plasmid pCu425-CUP1 (56). RSY620 cells expressing HA epitope-tagged CFTR (57) were then transformed with pCu-425-CUP1-FKBP8 or a vector control (pCu425-CUP1). Double transformants were selected and grown to early logarithmic phase (A600 ∼0.20) in synthetic complete medium lacking uracil and leucine but supplemented with glucose to a final concentration of 2%. The cells were incubated with 1 mm copper sulfate for 4 h to induce maximal expression of FKBP8, and cycloheximide was added to a final concentration of 100 μg/ml. A total of 2–2.5 ODs of cells were removed (t = 0); the culture was shifted to 37 °C, and the same number of cells was taken at the indicated times. The yeast were washed, and total protein was precipitated as described (57). Proteins were resolved on 10% SDS-polyacrylamide gels, transferred to nitrocellulose, and probed with mouse monoclonal anti-HA antibody (12CA5, Roche Applied Science). Signals were visualized using horseradish peroxidase-conjugated secondary antiserum, and the results were quantified using the Kodak 440CF Image Station and the associated Kodak 1D (Version 3.6) software (Rochester, NY).
Data Analysis
The values represent densitometric analysis of Western blots using an Alpha Innotech Fluorochem SP. In all cases, the densitometry was performed below the saturation limit of the antibody. For CFTR band C immunoblot analysis, a doublet, representing the intermediate and C forms of CFTR, was used for quantitation and is referred to as band C. For FKBP8 analysis, the slower migrating band seen in overexpression experiments represents the Myc-tagged form. Both bands in the doublet were used for the quantitation of FKBP8 overexpression. In cases where the band C signal was very low, densitometry was performed on longer exposures, and the value normalized to the expected intensity on the band B quantified exposure using a reference band that is below saturation on both exposures. The values are expressed as a percentage of the most intense band (% of maximum) and averaged. The error bars represent the means ± S.E. (n ≥ 3).
RESULTS
Modulating FKBP8 Expression Alters CFTR Folding in the ER
Our previous data revealed that FKBP8 is preferentially recovered in association with ΔF508-CFTR when compared with WT-CFTR (18). Because FKBP8 is the only FKBP family member recovered in CFTR immunoprecipitates, we considered the possibility that it could have a very specialized role in CFTR biogenesis. In addition, FKBP8 has been localized to the ER (58–60). We therefore assessed whether ΔF508 and FKPB8 co-localized in our human bronchial epithelial cell line, CFBE41o−, expressing ΔF508-CFTR (referred to henceforth as CFBE cells). Fig. 2A shows a high degree of co-localization between the stably expressed ΔF508-CFTR and the endogenous FKBP8 in this lung epithelial cell line.
FIGURE 2.

FKBP8 co-localizes with ΔF508-CFTR and its silencing reduces the stability of CFTR. A, CFBE41o− cells stably expressing ΔF508-CFTR were stained with M3A7 anti-CFTR antibody (left) and FKBP8-N1 anti-FKBP8 antibody (middle). The merged image (right) reveals significant co-localization. Immunoblot analysis of CFTR, Hsp90, and Hsp70 following the silencing of FKBP8 (siFK8), FKBP51 (siFK51), or FKBP52 (siFK52) in CFBE41o− cells expressing ΔF508-CFTR at 37 °C (B), or WT-CFTR (D), or ΔF508-CFTR at 30 °C (F). The knockdown was confirmed by immunoblot analysis of the respective FKBPs. Quantitative analysis of ΔF508-CFTR band B (B) and C (C) glycoforms following knockdown of FKBP8, -51, or -52 in CFBE41o− cells expressing ΔF508-CFTR at 37 °C (C), or WT-CFTR (E) or ΔF508-CFTR at 30 °C (G). C, E, and G data were normalized to the percent of the maximal signal and are shown as a mean ± S.E., n ≥ 3. p values were determined by two-tailed t test using the scramble control as the reference point; asterisks indicate p < 0.05.
Given that FKBP8 and ΔF508-CFTR co-localize in the ER and interact (18), we sought to define the potential role of FKBP8 in the folding, stability, and trafficking of CFTR. For this purpose, transport of CFTR from the ER to the cell surface was monitored by a change in migration on SDS-PAGE. ER-acquired N-linked oligosaccharides (band B) (Fig. 2B) are processed during trafficking through the Golgi to generate the slower migrating band C glycoform, as shown for wild type (WT) (Fig. 2D). In contrast, ΔF508 is misfolded at physiological temperature (37 °C) and is transiently retained in the ER in the band B glycoform prior to rapid degradation by the proteasome. However, when ΔF508-expressing CFBE cells are cultured at reduced temperature (30 °C), ΔF508-CFTR folds more efficiently, resulting in the accumulation of band B glycoform and a significant level of export from the ER. Therefore, band C is formed in the Golgi prior to its delivery to cell surface (Fig. 2F).
Because FKBP8 is found preferentially associated with ΔF508-CFTR (18), we examined whether it is part of an on-pathway productive folding complex, which ΔF508 is unable to progress beyond at physiological temperature, or that it represents part of an off-pathway unproductive complex, mediating the specific retention of mutant CFTR in the ER. If the latter is accurate, then siRNA-mediated silencing of FKBP8 might dissolve this complex and alleviate the ER retention of ΔF508. Silencing of FKBP8 in CFBE cells or in CFBE41o− stably expressing WT-CFTR (referred to henceforth as HBE cells) resulted in a decrease in the level of both WT and mutant CFTR (Fig. 2, B–E), consistent with a recent report showing destabilization of WT-CFTR following silencing of FKBP8 (54). We also examined whether the increased stability and trafficking of ΔF508-CFTR seen at the permissive temperature of 30 °C (61) is dependent upon FKBP8. We observed that silencing of FKBP8 had the same effect on ΔF508 at the permissive temperature (30 °C) as it did at physiological temperature (Fig. 2, F and G). These data suggest that FKBP8 is required for the folding, stability, and/or maturation of both WT- and ΔF508-CFTR. Thus, FKBP8 performs an essential role in an on-pathway folding complex, leading to the productive maturation of CFTR.
To address the specificity of FKBP8 function, we took advantage of the observation that FKPB8 is most closely related to FKBP51 and -52, based on its domain arrangements and cytosolic exposure (Fig. 1). Therefore, we monitored the effect of silencing these related FKBPs on the stability and trafficking of both WT- and ΔF508-CFTR. A 96 and 94% reduction in the respective levels of FKBP51 or -52 had no effect on the stability of the band B pool of ΔF508-CFTR at physiological temperature (Fig. 2, B and C). These data suggest that the dependence of CFTR on FKBP8 for folding and stability in the ER is specific, relative to closely related family members. In contrast, we observed that silencing of FKBP8, FKBP51, or FKPB52 did affect the extent of maturation of CFTR to band C of both WT and ΔF508 at the permissive temperature of 30 °C (Fig. 2, D–G). Specifically, the silencing of FKBP8 resulted in a 62 and 83% reduction in band C for WT- and ΔF508-CFTR at 30 °C, respectively. In contrast, silencing of FKBP51 and -52 resulted in a 45 and 40% respective reduction in the processing of WT- and ΔF508-CFTR to band C. These results suggest that FKBP51, -52, and -8 all contribute to the post-ER stability and/or trafficking of both WT and ΔF508-CFTR, with the loss of FKBP8 causing the strongest effect on post-ER stability. These data again support a critical role for FKBP8 in the management of CFTR folding. Finally, it is important to note that none of the FKBP siRNAs altered the levels of the core Hsp70 and Hsp90 chaperones (Fig. 2, B, D, and F). Therefore, the observed effects by siRNA silencing of FKBP8, -51, and -52 do not reflect a major change in the general PN environment of the cell.
Overexpression of FKBP8 Restores ΔF508-CFTR Folding and Export
Given that the critical event in correcting CF disease is prevention/correction of ΔF508-CFTR misfolding in the ER, we initially focused our attention on the role of FKBP8 at this step in the exocytic pathway. Because we observed a destabilizing effect when FKBP8 was depleted, we monitored the effect of FKBP8 overexpression on the stability and trafficking of ΔF508- or WT-CFTR. A 3-fold increase in the expression of Myc-tagged FKBP8 resulted in an ∼2-fold correction of the trafficking defect of ΔF508-CFTR at 37 °C, consistent with the previously observed effects of FKBP8 on a mutant form of the HERG potassium channel (53). Although we observed an increase in both band B and C for ΔF508-CFTR at 37 °C (Fig. 3, A and B), we did not observe an effect on the stability or trafficking of WT (Fig. 3, C and D) or ΔF508-CFTR at 30 °C (Fig. 3, E and F). These data emphasize that the steady-state pool of FKBP8 supports the biogenesis of the more stably folded forms of CFTR, whereas increased FKBP8 benefits the maturation of the otherwise destabilized ΔF508-CFTR protein at physiological temperature. Similar to siRNA-mediated silencing of FKBP8, overexpression of FKBP8 had no major effect on the levels of Hsp70 and -90, indicating a direct correlation between the level of FKBP8 expression and the stability of ΔF508-CFTR that most likely reflects an on-pathway effect.
FIGURE 3.
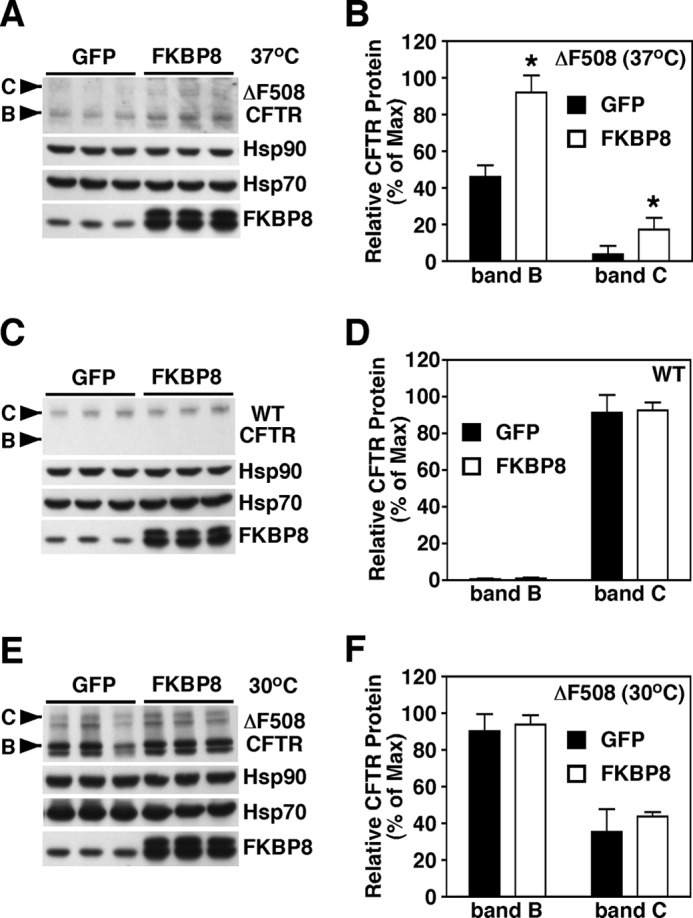
Overexpression of FKBP8 increases the stability of ΔF508-CFTR at physiological temperature. Immunoblot analysis of CFTR, Hsp90, Hsp70, and FKBP8 following lentiviral expression of FKBP8 in CFBE41o− cells expressing ΔF508-CFTR cultured at 37 °C (A) or WT-CFTR (C) or ΔF508-CFTR cultured at 30 °C (E). Quantitative analysis of ΔF508-CFTR band B (B) and C (C) glycoforms following overexpression of FKBP8 in CFBE41o− cells expressing ΔF508-CFTR cultured at 37 °C (B), or WT-CFTR (D) or ΔF508-CFTR cultured at 30 °C (F). B, D, and F data were normalized to the percent of the maximal signal. Data are shown as a mean ± S.E., n ≥ 3. p values were determined by two-tailed t test using the control as the reference point; asterisks indicate p < 0.05.
FKPB8 Stabilizes WT-CFTR in Yeast
In yeast, WT-CFTR is unable to exit the ER and traffic to the cell surface (57, 62); thus, it behaves like ΔF508-CFTR in mammalian cells. In fact, factors isolated from yeast screens that impact CFTR only affect ΔF508-CFTR biogenesis (20). Given that FKBP8 overexpression selectively stabilized ΔF508-CFTR in CFBE cells (Fig. 3, A and B), we wanted to address whether the expression of human FKBP8 (hFKBP8) would stabilize human WT-CFTR in yeast. Indeed, the expression of hFKBP8 resulted in a dramatic increase in the stability of WT-CFTR (Fig. 4). In yeast lacking hFKBP8, a t½ of 69 min was observed for CFTR, whereas only 10% of the CFTR was degraded at this same time point when hFKBP8 was expressed. In fact, the degradation rate was nearly five times faster in the absence of hFKBP8 (compare −0.0073 min−1 for mock and −0.0015 min−1 for FKBP8). Of note, FKBP8 overexpression had no effect on the growth of yeast or on the levels of Hsp70 or Hsp90 (data not shown). These data further support a key role for FKBP8 in the folding of CFTR prior to export from the ER.
FIGURE 4.
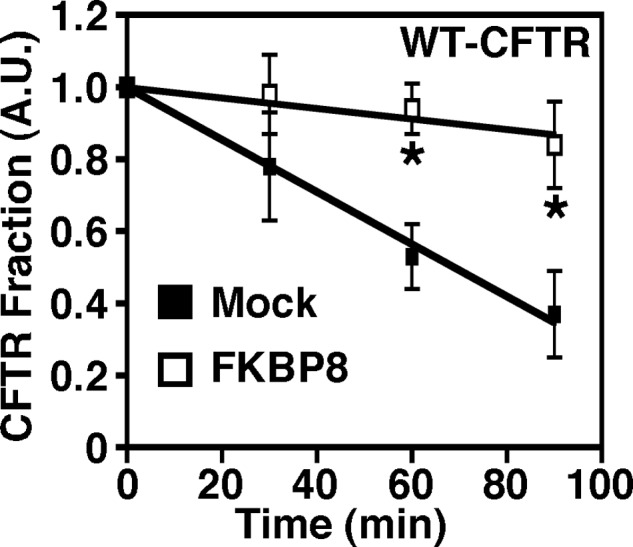
Overexpression of hFKBP8 in yeast increases the stability of CFTR. Quantitative analysis of WT-CFTR remaining over time under mock transformed conditions (closed squares) or following expression of hFKBP8 (open squares) is shown. FKBP8 expression was induced with copper sulfate for 4 h and subsequently stopped with cycloheximide prior to analysis of CFTR stability. The data were expressed as the fraction of CFTR remaining at t = 0. Data are shown as a mean ± S.E., n ≥ 3. p values were determined by two-tailed t test using the mock transformed yeast as the reference point; asterisks indicate p < 0.05. A.U., arbitrary units.
Modulating the Expression of FKBP8 Alters the Channel Activity of CFTR
Given the effect of modulating the expression levels of FKBP8 on the stability and trafficking of CFTR, we wanted to see if these effects translated into a change in the channel activity of CFTR at the cell surface as measured by iodide efflux assay. As expected, silencing of FKBP8 did not result in recovery of ΔF508-CFTR channel activity at 37 °C (Fig. 5, A and B). Moreover, we observed a decrease in the level of activity of both ΔF508-CFTR at 30 °C and WT-CFTR following FKBP8 silencing (Fig. 5, A–D), a result consistent with the observed decrease in CFTR protein expression seen following the knockdown of FKBP8 (Fig. 2) and in agreement with recent results on the effect of FKBP8 silencing on WT-CFTR activity in Calu3 cells (54). Surprisingly, the overexpression of FKBP8, which yielded a 2-fold increase in ΔF508-CFTR band B as well as a weak but significant appearance of band C (Fig. 3, A and B), did not result in the appearance of CFTR channel activity at the cell surface (data not shown). Interestingly, FKBP8 expression also failed to promote CFTR trafficking to the plasma membrane in yeast (data not shown). Thus, although a modest elevation of FKBP8 expression accelerates a rate-limiting ER folding event, it is apparently insufficient to generate a fully functional channel that resides at the cell surface.
FIGURE 5.
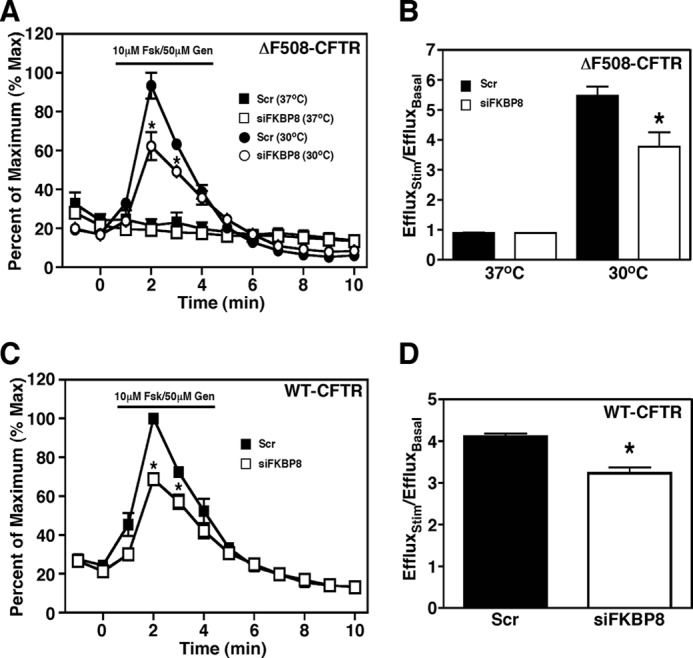
FKBP8 knockdown decreases CFTR channel activity. Iodide efflux analysis in CFBE41o− cells expressing ΔF508-CFTR (A) or WT-CFTR (C) following treatment with siFKBP8 (open symbols) or scramble (Scr) control (closed symbols). Fold increase in iodide efflux at maximal stimulation for CFBE41o− cells expressing ΔF508-CFTR (B) or WT-CFTR (D) following treatment with siFKBP8 (white bars) or scramble control (black bars), The data are expressed as a ratio of the efflux upon stimulation divided by efflux under pre-stimulation (basal) conditions. A and C, data were normalized to the percent of the maximal signal. Data are shown as a mean ± S.E., n ≥ 3. p values were determined by two-tailed t test using the scramble control at the same time as the reference point; asterisks indicate p < 0.05.
FKBP8 Inhibitor DM-CHX Stabilizes ΔF508-CFTR
Given our observations that siRNA-mediated knockdown of FKBP8 decreases CFTR stability in the ER, we investigated whether FKPB8 plays an important role in the folding and/or maturation of the protein through either its PPIase activity, associated with the FBD (Fig. 1), or its chaperoning activity, associated with its TPR/LZ domain, or both. To distinguish between these possibilities, we treated CFBE and HBE cells with known inhibitors of FKBP PPIase activity, namely FK506 and rapamycin, as well as cyclosporin A, which targets the related cyclophilin family of PPIases (63). These compounds had no effect on the stability or trafficking of either WT- or ΔF508-CFTR (Fig. 6A). Because FK506, rapamycin and cyclosporin A will inhibit the PPIase activity of all FKBP or cyclophilin family members, respectively, the results represent the sum total effect of inhibiting all PPIase family members. Therefore, to selectively target FKBP8, we examined the effect of a previously characterized FKBP8-specific PPIase inhibitor DM-CHX (64) on CFTR stability and trafficking in CFBE and HBE cells. Surprisingly, and in contrast to the effect of FKBP8 silencing, DM-CHX treatment resulted in a dose-dependent increase in the steady-state stability of both ΔF508- and WT-CFTR (Fig. 6, B and C). Thus, the phenotype of FKPB8 silencing is distinct from inhibition of its enzymatic activity. One possibility is that the slowing of the FKBP8 prolyl isomerase activity by DM-CHX could provide an additional degree of stability of transient, dynamic folding intermediates. This is similar to what we previously showed for the coupling of the co-chaperone Aha1 to Hsp90; here, reducing Hsp90 ATP turnover resulted in more efficient trafficking of ΔF508 (18). Alternatively, inhibition of the enzymatic activity of FKBP8 might trap the WT- and ΔF508-CFTR clients in a stable intermediate, isolating CFTR in a step insensitive to the degradation machinery and thereby generating a stable pool of CFTR available to engage the ER export machinery.
FIGURE 6.
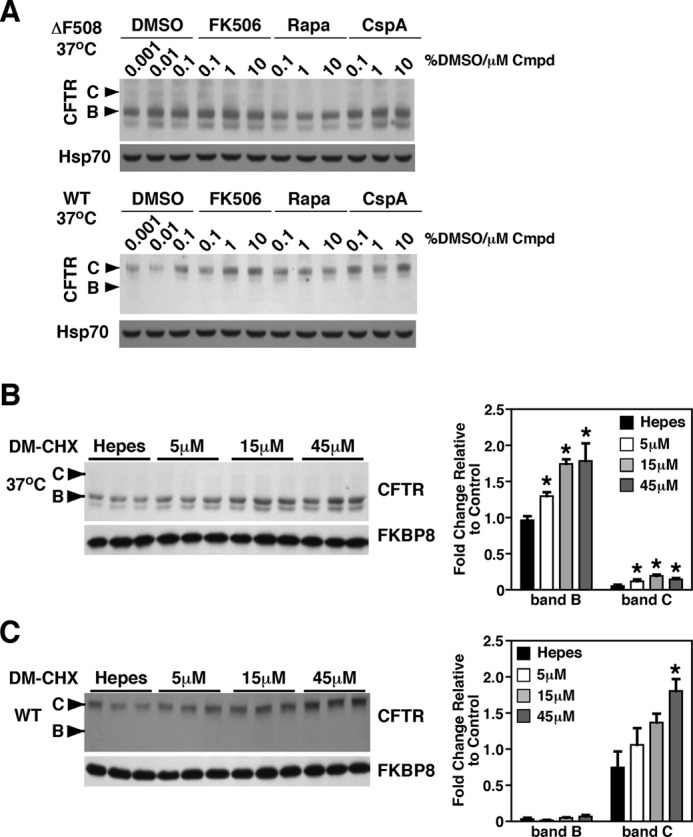
FKBP8 inhibitor increased the stability of CFTR. A, immunoblot analysis of ΔF508 (top) and WT-CFTR (bottom), following treatment of CFBE41o− cells with the immunosuppressive drugs FK506, rapamycin, or cyclosporin A at the indicated concentrations for 24 h at 37 °C. B and C, immunoblot analysis of ΔF508-CFTR (B) or WT-CFTR (C) following the treatment of the respective CFBE41o− cells with the indicated concentrations of the FKBP8-specific inhibitor DM-CHX. Quantitative analysis of the band B (B) and C (C) glycoforms of ΔF508-CFTR or WT-CFTR are shown on the right. Immunoblot for FKBP8 is also shown. Data in B and C are shown as fold-change relative to control condition and as a mean ± S.E., n = 3. p values were determined by two-tailed t test using Hepes treatment as the reference point; asterisks indicate p < 0.05.
In contrast to our observations, Banasavadi-Siddegowda et al. (54) reported a destabilizing effect of DM-CHX on the maturation of WT-CFTR to band C. However, these investigators used a high dose of the inhibitor (200 μm) for a brief 2-h treatment followed by pulse-chase analysis. This dose reduces protein synthesis by >20% (64), and the destabilizing effect seen could be the result of indirect effects on global protein synthesis over the 2-h time frame. Additionally, Banasavadi-Siddegowda et al. (54) only monitored pulse-chase data for WT-CFTR maturation and therefore limited their analysis to the effect of the compound on de novo synthesized CFTR. In contrast, our treatment scheme involved much lower doses of DM-CHX, which do not affect global protein synthesis (64), for a 24-h dosing regiment. Moreover, we monitored the steady-state pool of CFTR, which accounts for both de novo synthesized and existing pools of CFTR.
Effect of FKBP8 on Recruitment of Heat Shock Proteins by CFTR
We previously demonstrated that the ΔF508-CFTR interactome (18) contains significantly more core chaperone components, including Hsp70 and Hsp90, when compared with that seen in the WT-CFTR interactome (18). Furthermore, we observed a decrease in the association of these core PN components under conditions in which CFTR trafficking occurs at 30 °C (65).6 The latter observation suggests that a reduced recovery of core chaperones is indicative of improved folding, stability, and/or trafficking, whereas their persistent association correlates with reduced folding and stability. We therefore examined the effect of FKBP8 silencing on the recovery of core chaperones bound to either WT- or ΔF508-CFTR to determine the impact on the fate of both WT and mutant CFTR function.
Following knockdown of FKBP8, we observed an increase in the chaperone/CFTR ratio for Hsp70 and Hsp90 with both ΔF508- and WT-CFTR at physiological temperature, a result that was coupled to the destabilization of CFTR (Fig. 7, A and B). There are two possible interpretations for this result. One possibility is that FKBP8 presented a hurdle for CFTR progression to the Hsp70/Hsp90 containing a folding intermediate and that its silencing alleviated this road block. Alternatively, the ability of CFTR to progress beyond the Hsp70/90 checkpoint is linked to the PPIase activity of FKBP8, a step that is already severely compromised by the F508 deletion in NBD1. Given that Hsp70 has been implicated in the co-translational folding of CFTR and that Hsp90 is critical for stabilizing an early folding intermediate during CFTR biogenesis (9, 17–25, 30, 65), it is more likely that FKBP8 is acting at a late step in CFTR biogenesis.
FIGURE 7.
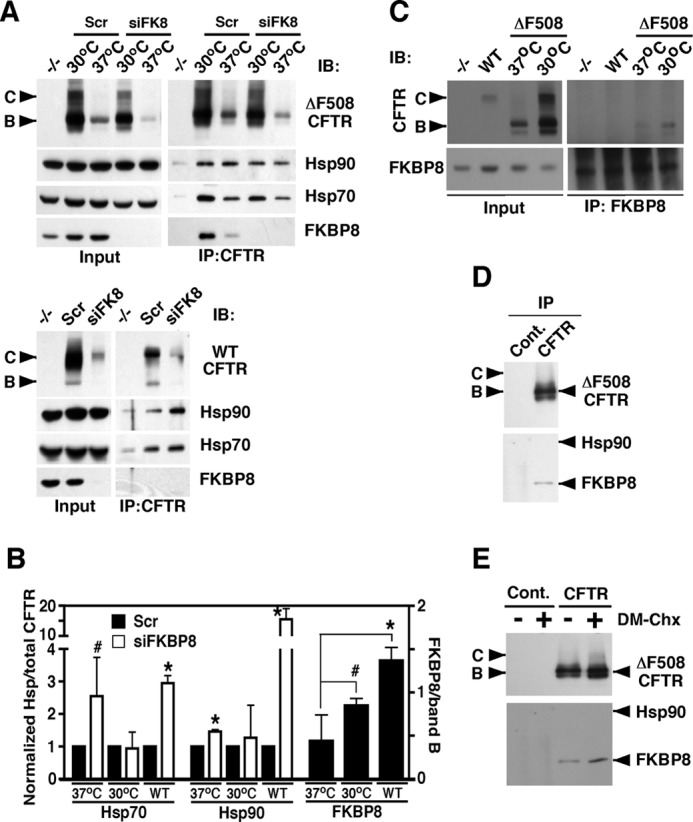
FKBP8 facilitates CFTR biogenesis. A, immunoblot (IB) analysis of CFTR, Hsp70, Hsp90, and FKBP8 following immunoprecipitation (IP) of ΔF508-CFTR (top) or WT-CFTR (bottom) and treatment of the respective CFBE cells with siFKBP8 or scramble control siRNA. Cells were treated at either 37 or 30 °C as indicated. Controls cells lacking CFTR are indicated by −/−. B, quantitative analysis of the recovery of Hsp70, Hsp90, and FKBP8 by co-immunoprecipitation of CFTR following treatment with siFKBP8 or scramble control for the indicated CFTR isoform. The 37 and 30 °C labels indicate ΔF508-CFTR following culture of the CFBE41o− cells at the indicated temperature. Data for Hsp70 and -90 are shown as a ratio to total CFTR. Data were then normalized to the scramble condition that was set to 1. FKBP8 data are shown as a ratio to band B (B). All data are shown as mean ± S.E. p values were determined using a two-tailed t test where asterisks indicate p ≤ 0.05 and pound signs indicate p ≤ 0.10. C, immunoblot analysis of CFTR and FKBP8 following immunoprecipitation of FKBP8 from CFBE cells expressing no CFTR (−/−), wild-type CFTR (WT), or ΔF508-CFTR at 37 or 30 °C. The left panels show the input for the experiment, and the right panels represent the immunoprecipitated material. D, immunoblot analysis of CFTR, Hsp90, and FKBP8 following immunoprecipitation of ΔF508-CFTR from CFBE41o− cells cultured at 37 °C using more stringent washing conditions to remove Hsp90/70 than shown in A. E, immunoblot analysis of CFTR, Hsp90, and FKBP8 following immunoprecipitation of ΔF508-CFTR from CFBE41o− cells treated with the FKBP8 inhibitor DM-CHX or Hepes control (“−”) at 37 °C. The samples were washed as in D. Cont, control.
To determine how FKBP8 may function in promoting CFTR folding, we monitored its level of recovery with CFTR by immunoprecipitation. Given that FKBP8 is an ER membrane protein and that only band B CFTR is recovered in an FKBP8 immunoprecipitation (Fig. 7C), we normalized its recovery to the band B ER glycoform, as opposed to total CFTR, which also reflects post-ER trafficking events. We observed that the ability of CFTR to bind FKBP8 correlated directly with its capacity to exit the ER (Fig. 7B). This observation, in combination with the loss of chaperone proteins under permissive trafficking conditions of 30 °C for ΔF508, supports our hypothesis that FKBP8 is a late acting PN component. This interpretation is also consistent with the fact that we observed a more persistent association of FKBP8 with ΔF508-CFTR following stringent washing conditions that removed Hsp90 (Fig. 7D). Furthermore, we observed an increase in FKBP8 binding to ΔF508-CFTR following treatment with the FKBP8 inhibitor, DM-CHX (Fig. 7E). Together, these results suggest that FKBP8 acts at a late stage in the chaperone bucket brigade during CFTR biogenesis. Furthermore, we suggest that FKBP8 uncouples the ERAD machinery from CFTR folding intermediates and promotes CFTR association with the ER export machinery.
FKBP8 Is Required for Aha1-mediated Correction of ΔF508-CFTR
To further test our hypothesis that FKBP8 is a late acting CFTR chaperone, functioning downstream of the Hsp90-dependent folding step, we monitored the effect of simultaneously silencing FKBP8 and the Hsp90 co-chaperone Aha1. We previously showed that Aha1 increases the stability and trafficking of both ΔF508- and WT-CFTR (Fig. 8, A–F). Increased trafficking in response to Aha1 silencing is characterized by an increase in the trafficking index (C/B ratio) (Fig. 8, B, D and F) (65). Consistent with the data in Fig. 2, the silencing of FKBP8 alone resulted in the destabilization of both ΔF508- and WT-CFTR (Fig. 8, A–F). When we silenced both FKBP8 and Aha1, we observed a minimal effect on the stability of the ER pool (band B) of ΔF508-CFTR at either 37 or 30 °C (Fig. 8, A–D) and WT-CFTR (Fig. 8, E and F). However, the ability of the stabilized ΔF508- and WT-CFTR pool to exit the ER was dramatically reduced in the absence of FKBP8, resulting in a depleted band C pool (Fig. 8, A–F) and a reduction of the C/B ratio (Fig. 8, B, D and F) (65). If FKBP8 were acting upstream of Aha1, then the destabilizing effect of FKBP8 silencing would provide a reduced pool of CFTR that could be rescued by Aha1 silencing, resulting in a reduction in band B levels, but the trafficking index should remain the same. However, because we see similar stability of the ER pool of CFTR in the presence or absence of FKBP8, we suggest that CFTR encounters the stabilizing effect of reduced Aha1 levels before it requires FKBP8. These data, coupled with the chaperone interaction data above, help support the temporal placement of FKBP8 at a step in CFTR biogenesis that is downstream of the Hsp90-dependent folding step.
FIGURE 8.

FKBP8 facilitates the Aha1-mediated correction of CFTR trafficking. Immunoblot analysis of CFTR, Hsp90, Hsp70, FKBP8, and Aha1 following siFKBP8, siAha1, or siFKBP8 + siAha1 treatment of CFBE41o− cells expressing ΔF508-CFTR at 37 °C (A), ΔF508-CFTR at 30 °C (C), or WT-CFTR at 37 °C (E) is shown. Quantitative analysis of CFTR band B (B) and C (C) glycoforms following siRNA-mediated silencing in CFBE41o− cells expressing ΔF508-CFTR at 37 °C (B), ΔF508-CFTR at 30 °C (D), or WT-CFTR at 37 °C (F). B and D, CFTR glycoform analysis is normalized to band B in the scramble control, and in F, the data are normalized to band C in the scramble control due to the low levels of band B in WT-CFTR lysates. B, D, and F, the data are shown as a fold increase relative to scramble control as a mean ± S.E., n = 4. p values were determined by two-tailed t test using the scramble control or siAha1 as the reference points. The asterisks indicate p < 0.05 relative to scramble control, and the pound signs indicate p < 0.05 relative to siAha1.
DISCUSSION
FKBP family members have been shown to exhibit both co-chaperoning and chaperoning activities. The prototypical member of the FK506-binding protein family, FKBP12, modulates the electrophysiological properties of both ryanodine receptors and inositol 1,4,5-triphosphate receptors and thus acts as a chaperone to modulate the properties of these calcium channels (68–72). Conversely, the role of FKBP52 as an Hsp90 co-chaperone in the functional cycling of steroid hormone receptors is well established (73–76). Although the similar domain organizations between FKBP52 and FKBP8 (Fig. 1) would suggest a common cellular function, a difference in their ability to interact with client proteins suggests otherwise. Whereas FKBP52 can only bind to the SHR receptor in the presence of Hsp90 (77–81), the binding of FKBP8 to a client, Bcl-2, is abolished upon Hsp90 recruitment (47). One possibility is that FKBP8 delivers client proteins to Hsp90, an event requiring client release upon binding of the TPR domain of FKBP8 to the C terminus of Hsp90. Alternatively, the functional pairing between FKBP8 and Hsp90 may work in the other direction; FKBP8 may be recruited to Hsp90 and accept the transfer of a nearly folded Hsp90 client(s) for final chaperoning of the prolyl bonds. The pre-binding of FKBP8 with Hsp90 in this case would serve to couple transfer of the Hsp90-client complex to FKBP8.
Our data reveal that the loss of FKBP8 results in the accumulation of CFTR in an Hsp70/90 folding intermediate complex (Fig. 7, A and B). Therefore, it is unlikely that FKBP8 acts upstream of the core chaperone machinery. Combined with the observed effects of the double silencing of FKBP8 and Aha1 (Fig. 8), we instead propose a model in which FKBP8 plays a late acting role in directing CFTR for ER export (Fig. 9). In this view, in a first step, CFTR associates with a functional Hsp90 dimer, which is regulated by the co-chaperone Aha1 (18). FKBP8 is subsequently recruited to this complex via its Hsp90-interacting TPR motif (Fig. 9, step 1), allowing for the binding partners to be in close proximity to one another to ensure proper client transfer. Following the release of the CFTR client from Hsp90, FKBP8, via its TPR/LZ domain, binds to CFTR and increases client stability (Fig. 9, step 2). This step is blocked upon FKBP8 silencing, leading to its accumulation in a “chaperone trap” (Fig. 7, A and B) and subsequent targeting for ERAD (Fig. 2) (19, 22, 66, 82). Conversely, the overexpression of FKBP8 results in increased stability and trafficking of ΔF508-CFTR (Figs. 3 and 4). The final chaperoning step for FKBP8 is the transfer of the client from its TPR/LZ domain to the FBD, which contains the PPIase activity (Fig. 9, step 3). This late step completes the protein fold, perhaps through the isomerization of prolyl bonds to allow the protein to adopt a more final, compact structure. This hypothesis is supported by recent evidence on the mechanism of action of a subclass of FKBP proteins that contain chaperone domains (8).
FIGURE 9.
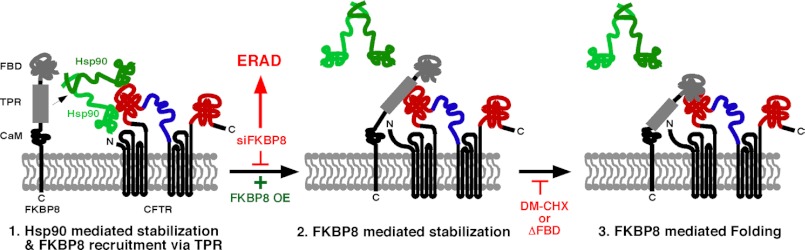
FKBP8 triggers CFTR release from the chaperone-bound intermediate in the ER. Step 1, Hsp90 chaperone is bound to CFTR in the ER and promotes its proper folding. FKBP8 is recruited and binds CFTR through its TPR domain to mediate Hsp90 release (step 2) and CFTR stabilization (step 3). After FKBP8 silencing, CFTR would be trapped in an Hsp90 complex and targeted for ERAD, and following FKBP8 overexpression (OE), CFTR would be stabilized. DM-CHX inhibits FKBP8 PPIase activity and transfer of CFTR from the TPR (chaperone domain) to FBD (catalytic domain), stabilizing CFTR in the ER, but not promoting maturation and trafficking to band C.
The substrate specificity of FKBPs containing only an FK506-binding domain, such as that seen in FKBP12, is dictated by the amino acid in the −1 position, with Leu-Pro being isomerized with 400-fold increased efficiency compared with Glu-Pro (8). However, when the chaperone domain of the immunophilin SlyD was grafted onto FKBP12, substrate specificity was eliminated, and the isomerization of a Xaa-Pro library exhibited similar rates for all 20 amino acids in the Xaa position (8). In this case, the non-native substrate was recognized by the chaperone domain, and the rate-limiting step in this enzymatic process was dictated by the rate of transfer of the substrate from the chaperone domain to the enzymatic domain (8). Indeed, FKBP8 harbors an N-terminal PPIase domain and a TPR/LZ domain that exhibits chaperone activity. The proposed intramolecular shuttling of the client from the TPR/LZ domain to the FBD domain (Fig. 9, step 3) would be inhibited by the addition of DM-CHX or by the deletion of the FBD domain, which blocks access to the catalytic pocket of the FKBP8 FBD (64). This would result in the accumulation of CFTR bound to the chaperone domain in FKBP8 (Fig. 9, step 2), and it accounts for the increased stability of CFTR upon addition of the PPIase inhibitor. This model also explains the difference seen between the siRNA-mediated knockdown of the protein and the use of a chemical inhibitor.
Overall, our data support a role for FKBP8 as a critical component that chaperones the maturation of CFTR in the ER. In the absence of this PN factor, CFTR accumulates in an early folding intermediate bound to the core chaperone components, resulting in reduced trafficking to the cell surface and targeting to ERAD of even the otherwise stable WT- and ΔF508-CFTR at 30 °C. TPR domain containing proteins, such as FKBP8, have long been viewed as accessory co-chaperones involved in modulating the activity of the core Hsp70 and Hsp90 proteins. In contrast, the activity of FKBP8 shown in this report and for additional client proteins, such as Bcl-2 (52) and the HERG potassium channel (53), reveals that the function of a co-chaperone may be more complex then previously thought. A related phenomenon has recently been observed for the Hsp90 co-chaperone, p23, which appears to possess Hsp90 independent chaperone functions (67). Furthermore, the temporal ordering of FKBP8 in the chaperone cycle provides new insight into the more general role of co-chaperones in mediating the folding of the large cohort of metastable clients, such as CFTR. We suggest that the identification of FKBP8 as both a co-chaperone of the Hsp90 system and as a chaperone functioning in late-acting events in the CFTR folding cascade provides a new molecular target for the development of pharmacological approaches to overcome CF disease associated with the ΔF508-CFTR mutant.
This work was supported, in whole or in part, by National Institutes of Health Grants HL079442 (to W. E. B.), GM42336 (to W. E. B.), DK785483 (to W. E. B.), and GM75061 (to J. L. B.). This work was also supported by Grant BRODSK08XX0 (to J. L. B.) from Cystic Fibrosis Foundation Therapeutics, by fellowships from the Cystic Fibrosis Foundation, and by funding from the Cystic Fibrosis Consortium.
Coppinger, J. A., Hutt, D. M., Razvi, A., Koulov, A. V., Pankow, S., Yates, J. R., III, and Balch, W. E. (2012) A chaperone trap contributes to the onset of cystic fibrosis. PLoS One, in press.
- PN
- proteostasis network
- CF
- cystic fibrosis
- CFTR
- cystic fibrosis transmembrane conductance regulator
- ER
- endoplasmic reticulum
- ERAD
- endoplasmic reticulum-associated degradation
- FBD
- FK506-binding domain
- PPIase
- peptidylprolyl isomerase
- Hsp
- heat shock protein
- FKBP
- FK506-binding protein
- TPR
- tetratricopeptide repeat
- LZ
- leucine zipper
- DM-CHX
- N-(N′,N′-dimethylcarboxamidomethyl)cycloheximide
- HBE
- human bronchial epithelial cell expressing WT CFTR
- CFBE
- cystic fibrosis bronchial epithelial cell expressing ΔF508-CFTR.
REFERENCES
- 1. Oliveberg M., Wolynes P. G. (2005) The experimental survey of protein-folding energy landscapes. Q. Rev. Biophys. 38, 245–288 [DOI] [PubMed] [Google Scholar]
- 2. Balch W. E., Morimoto R. I., Dillin A., Kelly J. W. (2008) Adapting proteostasis for disease intervention. Science 319, 916–919 [DOI] [PubMed] [Google Scholar]
- 3. Balch W. E., Roth D. M., Hutt D. M. (2011) Cold Spring Harbor Perspect. Biol. 3, p. ii:a004499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hutt D., Balch W. E. (2010) Cell biology. The proteome in balance. Science 329, 766–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hutt D. M., Powers E. T., Balch W. E. (2009) The proteostasis boundary in misfolding diseases of membrane traffic. FEBS Lett. 583, 2639–2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sawkar A. R., Schmitz M., Zimmer K. P., Reczek D., Edmunds T., Balch W. E., Kelly J. W. (2006) Chemical chaperones and permissive temperatures alter localization of Gaucher disease-associated glucocerebrosidase variants. ACS Chem. Biol. 1, 235–251 [DOI] [PubMed] [Google Scholar]
- 7. Powers E. T., Morimoto R. I., Dillin A., Kelly J. W., Balch W. E. (2009) Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 78, 959–991 [DOI] [PubMed] [Google Scholar]
- 8. Jakob R. P., Zoldák G., Aumüller T., Schmid F. X. (2009) Chaperone domains convert prolyl isomerases into generic catalysts of protein folding. Proc. Natl. Acad. Sci. U.S.A. 106, 20282–20287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Turnbull E. L., Rosser M. F., Cyr D. M. (2007) The role of the UPS in cystic fibrosis. BMC Biochem. 8, S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brodsky J. L., Wojcikiewicz R. J. (2009) Substrate-specific mediators of ER-associated degradation (ERAD). Curr. Opin. Cell Biol. 21, 516–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vembar S. S., Brodsky J. L. (2008) One step at a time. Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 9, 944–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Riordan J. R. (2008) CFTR function and prospects for therapy. Annu. Rev. Biochem. 77, 701–726 [DOI] [PubMed] [Google Scholar]
- 13. Amaral M. D. (2006) Therapy through chaperones. sense or antisense? Cystic fibrosis as a model disease. J. Inherit. Metab. Dis. 29, 477–487 [DOI] [PubMed] [Google Scholar]
- 14. Chang X. B., Mengos A., Hou Y. X., Cui L., Jensen T. J., Aleksandrov A., Riordan J. R., Gentzsch M. (2008) Role of N-linked oligosaccharides in the biosynthetic processing of the cystic fibrosis membrane conductance regulator. J. Cell Sci. 121, 2814–2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Du S. J., Li H., Bian Y., Zhong Y. (2008) Heat-shock protein 90α1 is required for organized myofibril assembly in skeletal muscles of zebrafish embryos. Proc. Natl. Acad. Sci. U.S.A. 105, 554–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rosser M. F., Grove D. E., Chen L., Cyr D. M. (2008) Assembly and misassembly of cystic fibrosis transmembrane conductance regulator. Folding defects caused by deletion of F508 occur before and after the calnexin-dependent association of membrane spanning domain (MSD) 1 and MSD2. Mol. Biol. Cell 19, 4570–4579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun F., Mi Z., Condliffe S. B., Bertrand C. A., Gong X., Lu X., Zhang R., Latoche J. D., Pilewski J. M., Robbins P. D., Frizzell R. A. (2008) Chaperone displacement from mutant cystic fibrosis transmembrane conductance regulator restores its function in human airway epithelia. FASEB J. 22, 3255–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang X., Venable J., LaPointe P., Hutt D. M., Koulov A. V., Coppinger J., Gurkan C., Kellner W., Matteson J., Plutner H., Riordan J. R., Kelly J. W., Yates J. R., 3rd, Balch W. E. (2006) Hsp90 cochaperone Aha1 down-regulation rescues misfolding of CFTR in cystic fibrosis. Cell 127, 803–815 [DOI] [PubMed] [Google Scholar]
- 19. Younger J. M., Chen L., Ren H. Y., Rosser M. F., Turnbull E. L., Fan C. Y., Patterson C., Cyr D. M. (2006) Sequential quality control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell 126, 571–582 [DOI] [PubMed] [Google Scholar]
- 20. Ahner A., Nakatsukasa K., Zhang H., Frizzell R. A., Brodsky J. L. (2007) Small heat-shock proteins select ΔF508-CFTR for endoplasmic reticulum-associated degradation. Mol. Biol. Cell 18, 806–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morishima Y., Wang A. M., Yu Z., Pratt W. B., Osawa Y., Lieberman A. P. (2008) CHIP deletion reveals functional redundancy of E3 ligases in promoting degradation of both signaling proteins and expanded glutamine proteins. Hum. Mol. Genet. 17, 3942–3952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morito D., Hirao K., Oda Y., Hosokawa N., Tokunaga F., Cyr D. M., Tanaka K., Iwai K., Nagata K. (2008) Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRΔF508. Mol. Biol. Cell 19, 1328–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schmid K., Haslbeck M., Buchner J., Somoza V. (2008) Induction of heat shock proteins and the proteasome system by casein-Nϵ-(carboxymethyl)lysine and Nϵ-(carboxymethyl)lysine in Caco-2 cells. Ann. N.Y. Acad. Sci. 1126, 257–261 [DOI] [PubMed] [Google Scholar]
- 24. Sun F., Zhang R., Gong X., Geng X., Drain P. F., Frizzell R. A. (2006) Derlin-1 promotes the efficient degradation of the cystic fibrosis transmembrane conductance regulator (CFTR) and CFTR folding mutants. J. Biol. Chem. 281, 36856–36863 [DOI] [PubMed] [Google Scholar]
- 25. Loo M. A., Jensen T. J., Cui L., Hou Y., Chang X. B., Riordan J. R. (1998) Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J. 17, 6879–6887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McClellan A. J., Xia Y., Deutschbauer A. M., Davis R. W., Gerstein M., Frydman J. (2007) Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell 131, 121–135 [DOI] [PubMed] [Google Scholar]
- 27. Pratt W. B., Morishima Y., Osawa Y. (2008) The Hsp90 chaperone machinery regulates signaling by modulating ligand binding clefts. J. Biol. Chem. 283, 22885–22889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wandinger S. K., Richter K., Buchner J. (2008) The Hsp90 chaperone machinery. J. Biol. Chem. 283, 18473–18477 [DOI] [PubMed] [Google Scholar]
- 29. Youker R. T., Walsh P., Beilharz T., Lithgow T., Brodsky J. L. (2004) Distinct roles for the Hsp40 and Hsp90 molecular chaperones during cystic fibrosis transmembrane conductance regulator degradation in yeast. Mol. Biol. Cell 15, 4787–4797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koulov A. V., Lapointe P., Lu B., Razvi A., Coppinger J., Dong M. Q., Matteson J., Laister R., Arrowsmith C., Yates J. R., 3rd, Balch W. E. (2010) Biological and structural basis for Aha1 regulation of Hsp90 ATPase activity in maintaining proteostasis in the human disease cystic fibrosis. Mol. Biol. Cell 21, 871–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Retzlaff M., Hagn F., Mitschke L., Hessling M., Gugel F., Kessler H., Richter K., Buchner J. (2010) Asymmetric activation of the hsp90 dimer by its cochaperone aha1. Mol. Cell 37, 344–354 [DOI] [PubMed] [Google Scholar]
- 32. Qu B. H., Strickland E., Thomas P. J. (1997) Cystic fibrosis. A disease of altered protein folding. J. Bioenerg. Biomembr. 29, 483–490 [DOI] [PubMed] [Google Scholar]
- 33. Hoelen H., Kleizen B., Schmidt A., Richardson J., Charitou P., Thomas P. J., Braakman I. (2010) The primary folding defect and rescue of ΔF508 CFTR emerge during translation of the mutant domain. PLoS One 5, e15458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Du K., Lukacs G. L. (2009) Cooperative assembly and misfolding of CFTR domains in vivo. Mol. Biol. Cell 20, 1903–1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. He L., Aleksandrov A. A., Serohijos A. W., Hegedus T., Aleksandrov L. A., Cui L., Dokholyan N. V., Riordan J. R. (2008) Multiple membrane-cytoplasmic domain contacts in the cystic fibrosis transmembrane conductance regulator (CFTR) mediate regulation of channel gating. J. Biol. Chem. 283, 26383–26390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aleksandrov A. A., Kota P., Aleksandrov L. A., He L., Jensen T., Cui L., Gentzsch M., Dokholyan N. V., Riordan J. R. (2010) Regulatory insertion removal restores maturation, stability, and function of ΔF508 CFTR. J. Mol. Biol. 401, 194–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deuerling E., Schulze-Specking A., Tomoyasu T., Mogk A., Bukau B. (1999) Trigger factor and DnaK cooperate in folding of newly synthesized proteins. Nature 400, 693–696 [DOI] [PubMed] [Google Scholar]
- 38. Teter S. A., Houry W. A., Ang D., Tradler T., Rockabrand D., Fischer G., Blum P., Georgopoulos C., Hartl F. U. (1999) Polypeptide flux through bacterial Hsp70. DnaK cooperates with trigger factor in chaperoning nascent chains. Cell 97, 755–765 [DOI] [PubMed] [Google Scholar]
- 39. Ng K. K., Weis W. I. (1998) Coupling of prolyl peptide bond isomerization and Ca2+ binding in a C-type mannose-binding protein. Biochemistry 37, 17977–17989 [DOI] [PubMed] [Google Scholar]
- 40. Schmid F. X., Frech C., Scholz C., Walter S. (1996) Catalyzed and assisted protein folding of ribonuclease T1. Biol. Chem. 377, 417–424 [PubMed] [Google Scholar]
- 41. Zhou X. Z., Kops O., Werner A., Lu P. J., Shen M., Stoller G., Küllertz G., Stark M., Fischer G., Lu K. P. (2000) Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and Tau proteins. Mol. Cell 6, 873–883 [DOI] [PubMed] [Google Scholar]
- 42. Fruman D. A., Bierer B. E., Benes J. E., Burakoff S. J., Austen K. F., Katz H. R. (1995) The complex of FK506-binding protein 12 and FK506 inhibits calcineurin phosphatase activity and IgE activation-induced cytokine transcripts, but not exocytosis, in mouse mast cells. J. Immunol. 154, 1846–1851 [PubMed] [Google Scholar]
- 43. Fruman D. A., Klee C. B., Bierer B. E., Burakoff S. J. (1992) Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc. Natl. Acad. Sci. U.S.A. 89, 3686–3690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu J., Farmer J. D., Jr., Lane W. S., Friedman J., Weissman I., Schreiber S. L. (1991) Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66, 807–815 [DOI] [PubMed] [Google Scholar]
- 45. Lam E., Martin M., Wiederrecht G. (1995) Isolation of a cDNA encoding a novel human FK506-binding protein homolog containing leucine zipper and tetratricopeptide repeat motifs. Gene 160, 297–302 [DOI] [PubMed] [Google Scholar]
- 46. Nielsen J. V., Mitchelmore C., Pedersen K. M., Kjaerulff K. M., Finsen B., Jensen N. A. (2004) Fkbp8. Novel isoforms, genomic organization, and characterization of a forebrain promoter in transgenic mice. Genomics 83, 181–192 [DOI] [PubMed] [Google Scholar]
- 47. Edlich F., Erdmann F., Jarczowski F., Moutty M. C., Weiwad M., Fischer G. (2007) The Bcl-2 regulator FKBP38-calmodulin-Ca2+ is inhibited by Hsp90. J. Biol. Chem. 282, 15341–15348 [DOI] [PubMed] [Google Scholar]
- 48. Davies T. H., Sánchez E. R. (2005) FKBP52. Int. J. Biochem. Cell Biol. 37, 42–47 [DOI] [PubMed] [Google Scholar]
- 49. Scheufler C., Brinker A., Bourenkov G., Pegoraro S., Moroder L., Bartunik H., Hartl F. U., Moarefi I. (2000) Structure of TPR domain-peptide complexes. Critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 101, 199–210 [DOI] [PubMed] [Google Scholar]
- 50. Sinars C. R., Cheung-Flynn J., Rimerman R. A., Scammell J. G., Smith D. F., Clardy J. (2003) Structure of the large FK506-binding protein FKBP51, an Hsp90-binding protein and a component of steroid receptor complexes. Proc. Natl. Acad. Sci. U.S.A. 100, 868–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen S., Sullivan W. P., Toft D. O., Smith D. F. (1998) Differential interactions of p23 and the TPR-containing proteins Hop, Cyp40, FKBP52, and FKBP51 with Hsp90 mutants. Cell Stress Chaperones 3, 118–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Choi B. H., Feng L., Yoon H. S. (2010) FKBP38 protects Bcl-2 from caspase-dependent degradation. J. Biol. Chem. 285, 9770–9779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Walker V. E., Atanasiu R., Lam H., Shrier A. (2007) Co-chaperone FKBP38 promotes HERG trafficking. J. Biol. Chem. 282, 23509–23516 [DOI] [PubMed] [Google Scholar]
- 54. Banasavadi-Siddegowda Y. K., Mai J., Fan Y., Bhattacharya S., Giovannucci D. R., Sanchez E. R., Fischer G., Wang X. (2011) FKBP38 peptidylprolyl isomerase promotes the folding of cystic fibrosis transmembrane conductance regulator in the endoplasmic reticulum. J. Biol. Chem. 286, 43071–43080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Adams A., Gottschling D. E., Kaiser C. A., Stearns T. (1997) Methods in Yeast Genetics, p. 178, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 56. Labbé S., Thiele D. J. (1999) Copper ion-inducible and -repressible promoter systems in yeast. Methods Enzymol. 306, 145–153 [DOI] [PubMed] [Google Scholar]
- 57. Zhang Y., Nijbroek G., Sullivan M. L., McCracken A. A., Watkins S. C., Michaelis S., Brodsky J. L. (2001) Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol. Biol. Cell 12, 1303–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Edlich F., Weiwad M., Erdmann F., Fanghänel J., Jarczowski F., Rahfeld J. U., Fischer G. (2005) Bcl-2 regulator FKBP38 is activated by Ca2+/calmodulin. EMBO J. 24, 2688–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kang C. B., Tai J., Chia J., Yoon H. S. (2005) The flexible loop of Bcl-2 is required for molecular interaction with immunosuppressant FK-506-binding protein 38 (FKBP38). FEBS Lett. 579, 1469–1476 [DOI] [PubMed] [Google Scholar]
- 60. Shirane M., Nakayama K. I. (2003) Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat. Cell Biol. 5, 28–37 [DOI] [PubMed] [Google Scholar]
- 61. Denning G. M., Anderson M. P., Amara J. F., Marshall J., Smith A. E., Welsh M. J. (1992) Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 358, 761–764 [DOI] [PubMed] [Google Scholar]
- 62. Huang P., Stroffekova K., Cuppoletti J., Mahanty S. K., Scarborough G. A. (1996) Functional expression of the cystic fibrosis transmembrane conductance regulator in yeast. Biochim. Biophys. Acta 1281, 80–90 [DOI] [PubMed] [Google Scholar]
- 63. Ho S., Clipstone N., Timmermann L., Northrop J., Graef I., Fiorentino D., Nourse J., Crabtree G. R. (1996) The mechanism of action of cyclosporin A and FK506. Clin. Immunol. Immunopathol. 80, S40–45 [DOI] [PubMed] [Google Scholar]
- 64. Edlich F., Weiwad M., Wildemann D., Jarczowski F., Kilka S., Moutty M. C., Jahreis G., Lücke C., Schmidt W., Striggow F., Fischer G. (2006) The specific FKBP38 inhibitor N-(N′,N′-dimethylcarboxamidomethyl)cycloheximide has potent neuroprotective and neurotrophic properties in brain ischemia. J. Biol. Chem. 281, 14961–14970 [DOI] [PubMed] [Google Scholar]
- 65. Wang X., Koulov A. V., Kellner W. A., Riordan J. R., Balch W. E. (2008) Chemical and biological folding contribute to temperature-sensitive ΔF508 CFTR trafficking. Traffic 9, 1878–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Grove D. E., Rosser M. F., Ren H. Y., Naren A. P., Cyr D. M. (2009) Mechanisms for rescue of correctable folding defects in CFTRΔF508. Mol. Biol. Cell 20, 4059–4069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Toogun O. A., Zeiger W., Freeman B. C. (2007) The p23 molecular chaperone promotes functional telomerase complexes through DNA dissociation. Proc. Natl. Acad. Sci. U.S.A. 104, 5765–5770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ahern G. P., Junankar P. R., Dulhunty A. F. (1994) Single channel activity of the ryanodine receptor calcium release channel is modulated by FK-506. FEBS Lett. 352, 369–374 [DOI] [PubMed] [Google Scholar]
- 69. Brillantes A. B., Ondrias K., Scott A., Kobrinsky E., Ondriasová E., Moschella M. C., Jayaraman T., Landers M., Ehrlich B. E., Marks A. R. (1994) Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell 77, 513–523 [DOI] [PubMed] [Google Scholar]
- 70. Cameron A. M., Steiner J. P., Sabatini D. M., Kaplin A. I., Walensky L. D., Snyder S. H. (1995) Immunophilin FK506-binding protein associated with inositol 1,4,5-trisphosphate receptor modulates calcium flux. Proc. Natl. Acad. Sci. U.S.A. 92, 1784–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chen S. R., Zhang L., MacLennan D. H. (1994) Asymmetrical blockade of the Ca2+ release channel (ryanodine receptor) by 12-kDa FK506-binding protein. Proc. Natl. Acad. Sci. U.S.A. 91, 11953–11957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Timerman A. P., Wiederrecht G., Marcy A., Fleischer S. (1995) Characterization of an exchange reaction between soluble FKBP-12 and the FKBP·ryanodine receptor complex. Modulation by FKBP mutants deficient in peptidylprolyl isomerase activity. J. Biol. Chem. 270, 2451–2459 [DOI] [PubMed] [Google Scholar]
- 73. Callebaut I., Renoir J. M., Lebeau M. C., Massol N., Burny A., Baulieu E. E., Mornon J. P. (1992) An immunophilin that binds Mr 90,000 heat shock protein. Main structural features of a mammalian p59 protein. Proc. Natl. Acad. Sci. U.S.A. 89, 6270–6274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lebeau M. C., Massol N., Herrick J., Faber L. E., Renoir J. M., Radanyi C., Baulieu E. E. (1992) P59, an hsp 90-binding protein. Cloning and sequencing of its cDNA and preparation of a peptide-directed polyclonal antibody. J. Biol. Chem. 267, 4281–4284 [PubMed] [Google Scholar]
- 75. Peattie D. A., Harding M. W., Fleming M. A., DeCenzo M. T., Lippke J. A., Livingston D. J., Benasutti M. (1992) Expression and characterization of human FKBP52, an immunophilin that associates with the 90-kDa heat shock protein and is a component of steroid receptor complexes. Proc. Natl. Acad. Sci. U.S.A. 89, 10974–10978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Renoir J. M., Pahl A., Keller U., Baulieu E. E. (1993) Immunological identification of a 50-kDa Mr FK506-binding immunophilin as a component of the non-DNA binding, hsp90- and hsp70-containing, hetero-oligomeric form of the chick oviduct progesterone receptor. C. R. Acad. Sci. III 316, 1410–1416 [PubMed] [Google Scholar]
- 77. Czar M. J., Owens-Grillo J. K., Dittmar K. D., Hutchison K. A., Zacharek A. M., Leach K. L., Deibel M. R., Jr., Pratt W. B. (1994) Characterization of the protein-protein interactions determining the heat shock protein (hsp90·hsp70·hsp56) heterocomplex. J. Biol. Chem. 269, 11155–11161 [PubMed] [Google Scholar]
- 78. Czar M. J., Owens-Grillo J. K., Yem A. W., Leach K. L., Deibel M. R., Jr., Welsh M. J., Pratt W. B. (1994) The hsp56 immunophilin component of untransformed steroid receptor complexes is localized both to microtubules in the cytoplasm and to the same nonrandom regions within the nucleus as the steroid receptor. Mol. Endocrinol. 8, 1731–1741 [DOI] [PubMed] [Google Scholar]
- 79. Pratt W. B., Toft D. O. (1997) Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 18, 306–360 [DOI] [PubMed] [Google Scholar]
- 80. Ratajczak T., Ward B. K., Minchin R. F. (2003) Immunophilin chaperones in steroid receptor signaling. Curr. Top. Med. Chem. 3, 1348–1357 [DOI] [PubMed] [Google Scholar]
- 81. Sivils J. C., Storer C. L., Galigniana M. D., Cox M. B. (2011) Regulation of steroid hormone receptor function by the 52-kDa FK506-binding protein (FKBP52). Curr. Opin. Pharmacol. 11, 314–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Grove D. E., Fan C. Y., Ren H. Y., Cyr D. M. (2011) The endoplasmic reticulum-associated Hsp40 DNAJB12 and Hsc70 cooperate to facilitate RMA1 E3-dependent degradation of nascent CFTRΔF508. Mol. Biol. Cell 22, 301–314 [DOI] [PMC free article] [PubMed] [Google Scholar]