

Background: Calreticulin retrotranslocated from the endoplasmic reticulum to the cytoplasm is post-translationally arginylated associates to stress granules following stress that decrease Ca2+.
Results: Arginylated calreticulin reaches the plasma membrane as a response to stress.
Conclusion: Arginylation confers to calreticulin a function as one of the preapoptotic signals of the cells.
Significance: Arginylated calreticulin is a novel factor involved in stress-induced apoptosis.
Keywords: Apoptosis, Calcium Signaling, Cell Surface Protein, Stress Granule, Stress Response, Arginylation, Calreticulin, Post-translational Modifications
Abstract
Post-translational modifications of proteins are important for the regulation of cell fate and functions; one of these post-translational modifications is arginylation. We have previously established that calreticulin (CRT), an endoplasmic reticulum resident, is also one of the arginylated substrates found in the cytoplasm. In the present study, we describe that arginylated CRT (R-CRT) binds to the cell membrane and identified its role as a preapoptotic signal. We also show that cells lacking arginyl-tRNA protein transferase are less susceptible to apoptosis than wild type cells. Under these conditions R-CRT is present on the cell membrane but at early stages is differently localized in stress granules. Moreover, cells induced to undergo apoptosis by arsenite show increased R-CRT on their cell surface. Exogenously applied R-CRT binds to the cell membrane and is able to both increase the number of cells undergoing apoptosis in wild type cells and overcome apoptosis resistance in cells lacking arginyl-tRNA protein transferase that express R-CRT on the cell surface. Thus, these results demonstrate the importance of surface R-CRT in the apoptotic response of cells, implying that post-translational arginylation of CRT can regulate its intracellular localization, cell function, and survival.
Introduction
The multifaceted mechanism of post-translational modification of proteins is important for the regulation of cell functions. We have previously demonstrated the post-translational incorporation of arginine into calreticulin (CRT)3 in the cytoplasm after its retrotranslocation from the endoplasmic reticulum (ER) (1). We also established that arginylated CRT (R-CRT) is recruited to stress granules (SGs) under conditions that promote a decrease in cytoplasmic Ca2+ levels and SG formation. These conditions were heat shock, thapsigargin combined with EGTA, and arsenite treatment. Furthermore, arginylation is essential for CRT association to SGs (2). These observations led us to suggest that arginylation confers different fates to CRT, but the precise characteristics of this isospecies and its differential compartmentalization within cells have not been entirely clarified.
CRT resides mainly in the ER and is a Ca2+-binding protein that function as a Ca2+ buffer and molecular chaperone, although it has also been linked to a number of processes occurring outside the ER lumen (3–17), which implies other subcellular localizations for CRT other than the ER. However, the subcellular origin, biochemical features, and the precise molecular mechanism responsible for targeting CRT to the plasma membrane is not fully understood.
SGs are formed in response to several stress stimuli and contain, among other components, proapoptotic factors. If the stress stimulus exceeds the cell survival threshold, it activates mechanisms leading to apoptosis. Reflecting the appearance of R-CRT associated with cytoplasmic SGs under stress conditions, we decided to explore R-CRT fate when apoptotic pathways are activated. We demonstrate that cells lacking the arginylation system are less susceptible to apoptosis than WT cells. It is hypothesized that this resistance is due to an increase in the amount of R-CRT present on the cell membrane.
EXPERIMENTAL PROCEDURES
Chemical Reagents
Chemical reagents and protease inhibitors were purchased from Sigma-Aldrich. Sodium arsenite was from Mallinckrodt.
Cell Cultures
All of the cell lines were grown in 100-mm-diameter Petri dishes with Dulbecco's modified Eagle's medium (high glucose; Invitrogen), containing 4.5 g/liter glucose, 4 mm l-glutamine, and 25 mmol/liter HEPES, and supplemented with 1 mm sodium pyruvate, 0.25 μg/ml amphotericin B (Sigma-Aldrich), 200 units/ml penicillin, 100 μg/ml streptomycin (Invitrogen), and 10% heat-inactivated fetal bovine serum (Invitrogen). For some experiments, cells were incubated for 3 h in starvation media, Earle's balanced salt solution.
ATE1−/− and ATE1+/+ mouse EF (embryonic fibroblast) cell lines were kindly provided by Dr Anna Kashina (Department of Animal Biology/Biochemistry, University of Pennsylvania, Philadelphia, PA). CRT−/− and CRT+/+ mouse EF cell lines were a gift from Dr Marek Michalak (Department of Biochemistry, University of Alberta, Edmonton, Canada).
To induce stress conditions, the cells were incubated for the indicated times with 0.5 mm sodium arsenite. We chose this stressor because it allows studies of cellular changes at different times on the same experiment. Recombinant R-CRT-FLAG was added to culture media overnight at 50 μg/ml.
Expression and Purification of pHUE-R-CRT-FLAG Protein
The coding sequence of mature CRT (CRT without signal peptide) was amplified by PCR from pCMV-Sport6-hCRT (cDNA provided by Professor H. L. Monaco, Biocrystallography Laboratory, University of Verona, Verona, Italy) using a forward primer containing an SacII restriction site (underlined), an arginine sequence (in bold), and the CRT sequence (in italics) (5′-ATCCGCCGC↓GGTGGACGTGAGCCTGCCGTCTACTTCAAGG-3′) and a reverse primer containing a KpnI restriction site (underlined), a FLAG sequence (in bold), and the CRT sequence (italics) (5′-ATCGGGGTAC↓CCTAGTCGTCGTCGTCTTTGTAGTCCAGCTCGTCCTTGGCCTGG 3′). The PCR product was digested with SacII and KpnI endonucleases and cloned into the histidine-tagged ubiquitin expression vector (pHUE) (provided by Professor Rohan T. Baker, Molecular Genetics Group, The John Curtin School of Medical Research, The Australian National University), and the construct was confirmed by DNA sequencing. R-CRT-FLAG protein was express and purified as described in Ref. 18.
Biotinylation and Streptavidin Precipitation
Biotinylation and recovery of cell surface proteins were performed on intact cell monolayers using EZ-link Sulfo-NHS-SS Biotin (Pierce) and isolated using streptavidin-agarose beads (Sigma-Aldrich). The cells were placed on ice and washed three times with PBS. The cells were then incubated with EZ-link Sulfo-NHS-SS-Biotin at a final concentration of 0.5 mg/ml into PBS for 60 min at 4 °C, followed by glycine (100 mm) in PBS to quench unbound labeling reagent, before being washed twice with PBS to completely remove any remaining quenching buffer. Biotinylated cells were scraped off the plates in lysis buffer (20 mm Tris-HCl, pH 7.5, 1 mm EDTA, 1% w/v Triton X-100, 150 mm NaCl, 10 mm glycine, 3 mg/ml leupeptin, 1 mm phenylmethylsulfonyl fluoride, 3 mg/ml aprotinin) and agitated on a shaker for 60 min at 4 °C. The cell lysate was centrifuged for 10 min at 14,000 × g, and the resulting supernatant was incubated with streptavidin-agarose beads, suspended in lysis buffer, and mixed at 4 °C overnight The beads were recovered by centrifugation (5,000 × g for 15 s) and aliquots of supernatants were taken to represent the unbound, intracellular pool of proteins. Biotinylated proteins were eluted from the beads by heating to 100 °C for 5 min in SDS-PAGE sample buffer before loading onto a 10% SDS-PAGE gel.
Electrophoresis and Immunodetection of Proteins
Proteins from cell lysates and biotinylated material were separated using SDS/PAGE (27) and transferred onto nitrocellulose membrane (28). As primary antibodies, we used mouse anti-CRT mAb (monoclonal antibody; 1:2500) (BD Biosciences), and rabbit anti-R-CRT pAb (polyclonal antibody; dilution 1:100) (custom made by Eurogentec, Seraing, Belgium), which specifically detects the arginylated form of CRT (1). As secondary antibodies, IRDye 800CW goat anti-mouse antibody or IRDye 800CW goat anti-rabbit antibody (1:30000) (LI-CORBiosciences) were used and visualized by the Odyssey infrared imaging system (LI-COR Biosciences). Total biotinylated proteins were revealed using IRDye 800CW Streptavidin. The samples were normalized to equal amounts of tubulin.
To assess autophagy, ATE1+/+ cells were incubated in starvation conditions or in complete medium treated with 0.5 mm of sodium arsenite and then lysed with radioimmune precipitation assay buffer. Protein samples from total cell lysates were run on a 12% polyacrylamide gel and transfected to nitrocellulose membranes. As primary antibodies, we used rabbit anti-LC3 pAb (polyclonal antibody; dilution 1:800) (Sigma), rabbit anti-Beclin-1 pAb (polyclonal antibody; dilution 1:1000) (Cell Signaling), and mouse anti-β-actin mAb (monoclonal antibody; 1:10000) (Sigma). As secondary antibodies, IRDye 800CW goat anti-rabbit antibody or IRDye 800CW goat anti-mouse antibody (1:30000) (LI-COR Biosciences) was used and visualized by the Odyssey infrared imaging system (LICOR Biosciences). The bands intensity was quantify with ImageJ program normalized with amounts of β-actin.
Assessment of Apoptosis
Redistribution of plasma membrane phosphatidylserine (PS) is a marker of apoptosis and was assessed by annexin V phycoerythrin (BD Biosciences) according to the manufacturer's protocol. Briefly, 1 × 106 cells/sample were collected, washed in PBS, pelleted, and resuspended in incubation buffer (10 mm HEPES/NaOH, pH 7.4, 140 mm NaCl, 2.5 mm CaCl2) containing 1% annexin V and 1% 7-Amino-actinomycin D or propidium iodide, to identify dead cells. The samples were kept in the dark and incubated for 15 min prior to analysis by flow cytometry on a FACSCantoII cytometer (BD Biosciences) using BD FACSDiva software (BD Biosciences).
Flow Cytometric Analysis of Cell Surface Proteins
Flow cytometry was used to detect R-CRT and annexin V exposure to the cell surface induced by sodium arsenite. Briefly, 4 × 105 cells were plated in 6-well plates, and the next day the cells were treated with 0.5 mm sodium arsenite for the indicated times. Both adherent and detached cells were harvest with EDTA 2 mm, washed twice with cold PBS, and incubated for 1 h at 4 °C with primary antibody, rabbit anti-R-CRT pAb (1:100) in cold PBS, followed by washing and incubation with goat anti-rabbit IgG conjugated to Cy5 (1:800) (Molecular Probes). Each sample was then analyzed by FACSCantoII cytometer (BD Biosciences) to identify cell surface R-CRT and annexin V (as described above). Isotype matched IgG antibodies were used as a control. A total of 50,000 cells were counted for each point. The data were recorded on a logarithmic scale and analyzed by BD FACSDiva software (BD Biosciences). The results are shown as representative frequency histograms or double fluorescence plots showing the percentage of cells positive for annexin V binding alone, percentage positive for R-CRT binding alone, and percentage positive for both R-CRT and annexin V binding, and after quantification, the percentage of double positive cells of four independent experiments were represented in line charts. As a control of integrity of nonpermeabilized cells after arsenite treatment, neglected immunostaining of the cytoplasmic enzyme tubulin tyrosine ligase was detected.
Immunofluorescence
For surface staining, the cells were grown on glass coverslips to 60% confluence. After arsenite treatment, the cells were placed on ice, washed twice with PBS and incubated at 4 °C with the primary antibody rabbit anti-R-CRT pAb (1:100) or mouse anti-FLAG M2 mAb (1:2000) for 1 h. The cells were then washed three times in PBS and incubated for 45 min with the secondary antibody Alexa Fluor 543-conjugated goat anti-rabbit IgG (1:1000) (Molecular Probes) (1:1000) in PBS. The cells were washed with PBS fixed with 3% (w/v) formaldehyde in PBS containing 4% w/v sucrose for 30 min at room temperature and mounted on slides with the mounting medium including DAPI from Vectashield.
Intracellular Staining
For intracellular staining, after arsenite treatment, the cells were washed with PBS, fixed with 4% formaldehyde for 15 min, permeabilized with 0.1% Triton X-100 for 10 min, and rinsed three times with PBS, and nonspecific binding sites were blocked with 10% FBS in PBS for 30 min. Rabbit anti-R-CRT pAb (1:100) and goat anti-TIA-1 (T-cell intracytoplasmic antigen 1) pAb (1:100) (Santa Cruz Biotechnology) primary antibodies were added for 1 h. Subsequently, the cells were washed three times with PBS and incubated for 30 min with Alexa Fluor 543-conjugated goat anti-rabbit IgG and Alexa Fluor 488-conjugated donkey anti-goat IgG (1:1000) (Molecular Probes) secondary antibodies respectively. All of the antibodies were diluted in PBS containing 5% (w/v) BSA. The specimens were imaged by Zeiss LSM 510 confocal system as described in Ref. 1.
In Vivo Measurement of Intracellular Ca2+
The intracellular Ca2+ concentration was quantified using the fluorescent Ca2+ dye indicator Fluo-3/AM (Fluo-3 acetoxymethyl ester; Molecular Probes). Briefly, ATE1+/+ and ATE1−/− cells were loaded with Fluo-3/AM (5 μm) and Pluronic F-127 acid (0.2%, w/v) at 37°C for 30 min. Ca2+ was measured by exciting the indicator at the range of 450–500 nm for 3–5 min. The cells were then treated with arsenite, and the fluorescence intensity was measured every 30 s for 60 min. The cells were imaged by confocal microscopy. The Ca2+ fluorescence intensity ratio (Ft/Fo) was plotted as a function of time. Confocal images were captured with an Olympus FV1000 (Olympus, Japan) laser confocal microscope, using a UPLFLN 40×, NA 1.3 Plan-Apochromat objective. Pinholes were set for a nominal axial resolution of less than 0.6 μm. Excitation on the Olympus FV1000 laser confocal microscope was produced with a 25-milliwatt argon laser emitting at 488 nm. Emissions were collected using a 505–530-nm band pass filter for Alexa Fluor® 488. To quantify the fluorescence intensity, we employed an image analysis system using Olympus FV1000 FLUOVIEW 1.7 software.
RESULTS
R-CRT Is Present on Cell Surface
There are numerous reports showing the presence of CRT on the surface of fibroblasts, lymphocytes, neutrophils, neurons, and B16 melanoma cells (19–23). Nevertheless, it remains unknown which pool of CRT translocates to the cell surface, and it has been speculated that only a fraction of CRT may reach the cell membrane. The identification of R-CRT not associated to the ER (1, 2) suggests that CRT could be localized on the cell plasma membranes after it is arginylated.
To study the presence of R-CRT on the cell surface, we use ATE1+/+ and ATE1−/− cells. Cells lacking arginyl-tRNA transferase (ATE1−/− cells) have no capacity to post-translationally arginylate proteins. Cell surface proteins present from both cells types (ATE1+/+ and ATE1−/−) were labeled using a membrane-impermeable form of biotin (EZ-Link Sulfo-NHS-biotin) and precipitated by streptavidin-agarose beads. Equal amounts of biotin-labeled plasma membrane proteins and total cell lysates were then analyzed by SDS-PAGE and Western blot (WB). Immunoblotting with anti-R-CRT pAb identified a single 60-kDa band indicative of R-CRT on the plasma membrane of ATE1+/+ but not on that of ATE1−/− cells (Fig. 1A, upper panel). Both cell lines expressed the same levels of CRT, as determined by anti-CRT mAb (Fig. 1A, lower panel).
FIGURE 1.

R-CRT is present on the cell surface. A, surface proteins present on ATE1+/+ (right lane) or ATE1−/− (left lane) were labeled using a membrane-impermeable form of biotin and precipitated by streptavidin-agarose beads. Biotin-labeled plasma membrane proteins and total cell lysates were subjected to SDS-PAGE and analyzed using WB against R-CRT (upper panel) and CRT (lower panel), respectively. B, Triton-permeabilized (panel a) or nonpermeabilized (panel b) ATE1+/+ cells were analyzed by immunofluorescence using anti-R-CRT pAb to visualize R-CRT. Note the subcellular localization of R-CRT in both the cytosol and on the cell membrane. Anti-CRT mAb was also used to detect CRT on the cell surface of ATE1 −/− cells (panel c). C, ATE1+/+ cells were immunostained with anti-R-CRT pAb, and fluorescence associated with plasma membrane was analyzed by flow cytometry. Unstained cells and cells incubated with the secondary antibody (Cy5) alone (isotype control) were used to determine the background fluorescence signal. The positive signal was determined as fluorescence above that seen with the secondary antibody alone. 20% of the cells express R-CRT on the cell surface. The results shown are from one experiment representative of three independent experiments.
To further support the above findings, the localization of R-CRT in permeabilized and nonpermeabilized ATE1+/+ cells was examined by immunofluorescence with an anti-R-CRT pAb. The staining patterns for R-CRT in both conditions are distinct (Fig. 1B). In permeabilized ATE1+/+ cells, R-CRT exhibits an expected (1), cytosolic-like immunostaining with occasional cluster distribution (Fig. 1B, panel a). In contrast, in nonpermeabilized cells R-CRT staining shows a diffuse to small punctuates distribution over the cell surface (Fig. 1B, panel b). The presence of CRT in the surface of ATE1−/− cells is noticeable as shown by immunofluorescence with anti-CRT mAb. (Fig. 1B, panel c). As a control, these staining patterns were not detected in cells in which the primary antibodies had been omitted (not shown).
To further confirm the cell surface localization of R-CRT, intact ATE1+/+ cells were stained for R-CRT using anti-R-CRT pAb followed by Cy5-conjugated secondary antibody and then analyzed by flow cytometry for surface-bound immunofluorescence (Fig. 1C). Under these conditions, 20% of the cells were positive for cell surface R-CRT (Fig. 1C, right panel). Together these results provide evidence that CRT, after its arginylation, reaches the plasma membrane, which implies that it came from the cytoplasmic pool. Finally, the addition of recombinant R-CRT-FLAG to CRT−/− and ATE1−/− cells shows surface R-CRT by immunocytochemistry using anti-R-CRT and anti-FLAG antibodies in nonpermeabilized cells (Fig. 2).
FIGURE 2.

Exogenous R-CRT binds to the cell surface. ATE1−/− and CRT−/− cells were incubated overnight with 50 μg/ml of recombinant R-CRT-FLAG protein and then analyzed by immunofluorescence in nonpermeabilized cells with anti-R-CRT pAb and anti-FLAG mAb. Note that exogenous R-CRT was observed on the cell surface of ATE1−/− and CRT−/− cells using a confocal microscope.
Cells Lacking Arginyl-tRNA Protein Transferase Are Less Susceptible to Apoptosis Compared with WT Cells
It has been described that the ER-Ca2+ release triggers apoptosis by activation of transcriptional cascades and a direct regulation of proapoptotic proteins as CRT (24–26). We have described an arginylated isoform of CRT, which is increased and up-regulated after arsenite treatment by the decrease in intracellular Ca2+ concentrations (2). In this paper we show that under oxidative stress, CRT-deficient cells (CRT−/−) are consistently more resistant to apoptosis compared with CRT+/+ cells after arsenite treatment up to 120 min (Fig. 3A). The resistance observed is also found under other stress conditions (27). Because of these differences, our aim was to analyze the sensitivity to apoptosis of cells lacking Ate1 enzyme (ATE1−/− cells). By flow cytometry analysis, we compared annexin V and propidium iodide binding in ATE1−/− and ATE1+/+ cells subjected to stress induction by arsenite treatment at different times (Fig. 3, B and C). We show that ATE1−/− cells are statistically significantly more resistant to apoptosis than ATE1+/+ cells at 60 and 120 min of arsenite treatment. At 30 min no difference was observed.
FIGURE 3.

Induction of apoptosis by arsenite treatment in CRT- and ATE1-deficient cells. Quantitative analyses of annexin V (AnnV) and propidium iodide (PI) staining at different times (0, 30, 60, and 120 min) of arsenite treatment (0.5 mm) are shown in A for CRT+/+ and CRT−/− cells and in B for ATE1+/+ and ATE1−/− cells. 100% values correspond to 50,000 cells. The data are the means of three independent experiments. Significance was calculated using an ANOVA test. *, p < 0.05, **, p < 0.02; ***, p < 0.01. The results indicated a reduction of apoptosis in CRT−/− and ATE1−/− cells compared with CRT+/+ and ATE1+/+ cells, respectively. C, representative flow cytometry dot plots of ATE1+/+ and ATE1−/− cells that were treated as in B, where the percentages of annexin V-positive and propidium iodide-negative cells and annexin V/propidium iodide double positive cells are indicated (Q1, Q2).
These results reveal that in the absence of the substrate (CRT) or the enzyme Ate1, the susceptibility of cells to apoptosis is decreased. The observed effect in CRT−/− cells could be due to the absence of R-CRT or both CRT and R-CRT.
We next determined the dynamics of the subcellular localization of R-CRT upon arsenite treatment. Because ∼60% R-CRT is recruited to SGs after 15 min of treatment with arsenite (Fig. 4A, panel b) (2), we explored R-CRT distribution in ATE1+/+ cells up to 90 min of arsenite treatment by using immunofluorescence with anti-R-CRT and anti-TIA-1 (SGs marker) antibodies. We found R-CRT associated with SGs up to 30 min of treatment (Fig. 4A, panels b and c). However, after 60 min, SGs were disassembled, and R-CRT was reduced to levels comparable with control conditions (Fig. 4, A, panels d and e; B; and C). These results suggest that when the stress exceeds the cell survival threshold, SGs are disassembled, and R-CRT is released from these aggregates.
FIGURE 4.

R-CRT association with SGs during arsenite treatment. A, ATE1+/+ cells at different times (0, 30, 60, and 90 min) of arsenite treatment (0.5 mm) were analyzed by double immunofluorescence using anti-R-CRT pAb and anti-TIA-1 pAb. Yellow pseudocolors in the merged images and in the enlarged regions of interest (insets) represent co-localization of R-CRT and TIA-1 as seen by confocal microscopy. R-CRT is increased and associated with SGs up to 30 min of treatment as compared with untreated cells. After 60 min, R-CRT is reduced to control levels and does not co-localize with SGs that are disassembled. B and C, the percentage of cells containing SGs (B) and the amount of cytoplasmic R-CRT expressed as relative fluorescence intensity (IF) (C) are represented at the different times. The values are the means ± S.D. from at least 60 cells analyzed in two independent experiments.
There is a possibility that the autophagy degradative pathway could be activated upon the stress condition, which would explain the reduced levels of R-CRT observed after arsenite treatment. To address this issue, WB analyses using LC3 and Beclin-1 antibodies (markers of autophagy that had been shown to correlate with the number of autophagosomes (28, 29)) were examined in total cell lysates of ATE1+/+ cells treated or not with arsenite for different times (15, 457 and 90 min). As a positive control of autophagy, these cells were incubated in starvation medium for 3 h (Fig. 5). In cells incubated in starvation conditions (Fig. 5, first lane), the amount of both proteins are higher than those observed in ATE1+/+ cells treated or not with arsenite in complete medium (Fig. 5, second through fifth lanes). Moreover LC3II and Beclin-1 levels do not vary significantly along our experimental assay conditions. Thus, the autophagy remains at basal levels during arsenite stress up to 90 min of treatment as compared with the untreated condition.
FIGURE 5.

Autophagy pathway is not activated upon arsenite treatment. ATE1+/+ cells were incubated in starvation (Stv.) medium for 3 h or in complete medium in the absence or in the presence of 0.5 mm arsenite for the indicated times (15, 45, and 90 min). Then cells were lysed with radioimmune precipitation assay buffer, and the samples were subjected to WB analysis using LC3 and Beclin-1 antibodies. Band intensity was quantified using the ImageJ program, and the LC3II/LC3I ratio and Beclin-1 amount was calculated in each condition normalized with β-actin. Statistical significance was calculated with ANOVA. *, p < 0.05; **, p < 0.02 indicate significant changes between the levels of proteins in cells incubated in starvation medium with respect to those expressed in cells treated or not with arsenite.
The results presented clearly indicate that arsenite stress does not induce an autophagic response. In addition, this evidence also suggests that autophagy is not the responsible for the apoptotic manifestation observed in these cells upon arsenite treatment as shown in Fig. 3B. Taken together these results further indicate that the SGs could provide a mechanism to delay an apoptotic signal and the R-CRT course to other cellular compartments such as the cell membrane.
R-CRT Increases on Cell Surface during Apoptosis Induction
It has been described that CRT translocates to the cell membrane after apoptosis induction (17, 30–32). Considering our findings of R-CRT on the cell surface (Fig. 1), we then examined R-CRT on the cell surface after stressing ATE1+/+ cells with arsenite for 45 and 90 min, the period in which we showed SG are disassembled (Fig. 4). R-CRT levels on intact ATE1+/+ cells were assessed by flow cytometry using anti-R-CRT pAb (Fig. 6). The results show that the overall amount of R-CRT on the cell surface increased during apoptosis induction, from 17% in untreated cells, up to 21 and 31% at 45 or 90 min arsenite treatment, respectively. Histograms represent the percentage of cells positive for R-CRT on the plasma membrane gated within the marker (M1) under arsenite treatment (Fig. 6, c and d) or untreated conditions (Fig. 6b). The background fluorescence signal of isotope control cells is shown in Fig. 6a.
FIGURE 6.

R-CRT at the cell membrane increases during apoptosis induction. Flow cytometry analysis of surface R-CRT in ATE1+/+ untreated cells (b) or treated for 45 min (c) or 90 min (d) with arsenite 0.5 mm. ATE1+/+ cells were incubated with anti-R-CRT pAb antibody followed by incubation with Cy5-conjugated goat anti-rabbit IgG. The same analysis was carried out in cells incubated with recombinant R-CRT-FLAG (50 μg/ml) (e–h). Histograms show the percentage of cells positive for surface R-CRT gated within a marker (M1) and (a and e) show the background fluoresce signal of cells without the primary antibody (isotype control). Dot plots display mode of forward scatter (FCS) versus side scatter (SSC), and the ellipse gate shows the population of cells positive for surface R-CRT on each condition. These cells appear to be smaller and more granular, indicating that they could be apoptotic cells. The results shown are representative of three independent experiments, 100% values correspond to 50,000 cells.
The addition of recombinant R-CRT-FLAG to ATE1+/+ cells enlarged the cell population having R-CRT on the cell surface in both untreated and arsenite-treated cells (Fig. 6, f–h). Cells positive for R-CRT were 31, 37, and 41% at the different time. Dot plots display mode of forward scatter versus side scatter; the population of cells positive for surface R-CRT is gated by an ellipse that corresponds to the cell population of greater granularity and the smaller area, presumably they are apoptotic cells.
Ca2+ Level Are Not Only Cause for Different Response to Apoptosis Induction in ATE1+/+ and ATE1−/− Cells
It has been described that the decrease of ER-Ca2+ levels increases the plasma membrane exposure of CRT (26). Moreover, we have determined that low cytosolic Ca2+ favors CRT retrotranslocation from the ER, its arginylation in the cytoplasm, and its association to SGs in vivo (2). Taking into account this evidence plus that under arsenite treatment ATE1−/− are less prone than ATE1+/+ cells to apoptosis induction (Fig. 3B), we analyzed whether this different response could be related to differences in the homeostatic mechanism of ER-Ca2+. The time course of cytosolic Ca2+ levels in ATE1−/− cells during arsenite treatment was measured by Fluo-3/AM (Fig. 7). Although a gradual decrease in Ca2+ level was observed in both cell lines, ATE1−/− cells display a 3-fold decrease of Ca2+ values with respect to ATE1+/+ cells, which showed a 1.5-fold decrease. This result indicates that in ATE1−/− cells there is a more pronounced decrease of Ca2+ that could promote an increased translocation of CRT to the membrane. Thereafter, we analyzed, at different times, the exposure of CRT in ATE1−/− and ATE1+/+ cell lines during arsenite treatment. We observed that in both cell lines, CRT is translocated to the plasma membrane and that the amount of CRT increases during the treatment (Fig. 8). However, ATE1−/− cells exhibit less CRT at the cell membrane compared with ATE1+/+ cells under the same experimental conditions. It should be emphasized that the amount of CRT in ATE1+/+ cells could result from the contribution of both the non-arginylated and the arginylated isospecies. Taken together, these results indicate that the decrease in Ca2+ level is not the only factor involved in the incorporation of CRT at the plasma membrane, and they further suggest that arginylation also affects the apoptotic response.
FIGURE 7.
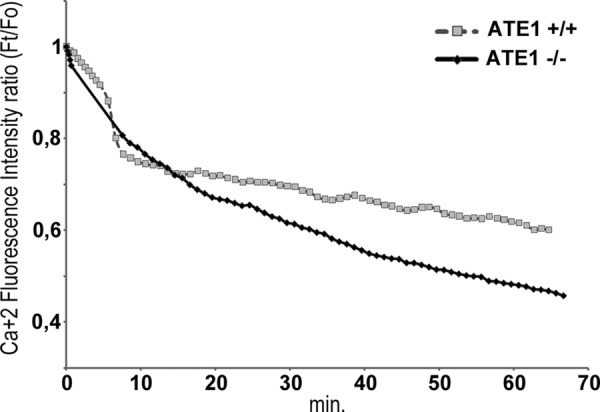
Measurements of intracellular Ca2+ levels in ATE1 cells subjected to arsenite stress conditions. ATE1+/+ and ATE1−/− cells were loaded with Fluo-3/AM (5 μm). The cells were then treated with arsenite, and the fluorescence intensity was measured every 30 s for 60 min. Ca2+ fluorescence intensity ratio (Ft/Fo) was plotted as a function of time. Cytosolic Ca2+ levels decrease during arsenite treatment in both ATE1+/+ cells (1.5-fold decrease) and even more in ATE1−/− cells (3-fold decrease). The data are the means ± S.D. of three independent experiment. Controls of the stability of Ca2+ signal during the time frame of this experiment were performed in ATE1+/+ and ATE1−/− cells (data not shown).
FIGURE 8.
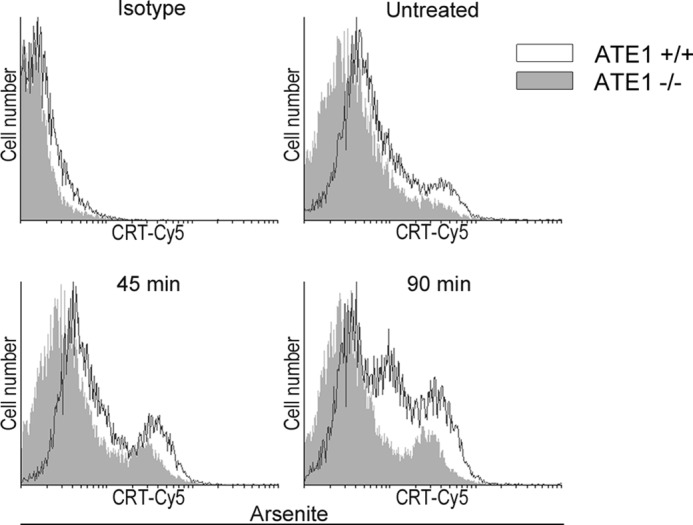
Presence of R-CRT on the plasma membrane of ATE1−/− compared with ATE1+/+ cells. ATE1+/+ and ATE1−/− cells not treated or treated for 45 or 90 min with arsenite were incubated with anti-CRT pAb antibody followed by incubation with Cy5-conjugated goat anti-rabbit IgG. Fluorescence was detected by flow cytometry. Histograms show CRT detected on the plasma membrane of ATE1−/− (solid gray) and ATE1+/+ (black line) cells. ATE1−/− cells exhibit less total CRT on the cell membrane compared with ATE1+/+ cells under the same experimental conditions.
Cells Exposing R-CRT on Cell Membrane Undergo Apoptosis
So far we have provided evidence indicating that R-CRT is found on the cell surface of apoptotic-induced cells. To confirm that R-CRT is predominantly present on the surface of apoptotic cells, detection of R-CRT and PS on intact ATE1+/+ cells was assessed by flow cytometry using an anti-R-CRT pAb and annexin V. The Fig. 9A show cells with annexin V labeling alone (upper left quadrant), cells that expose both R-CRT and annexin V (upper right quadrant), and cells that expose R-CRT alone (lower right quadrant). Clearly the same populations of cells exposing R-CRT on their cell surface are undergoing apoptosis (double positive cells). These cells at the different times during apoptosis induction were 11, 30, and 39%, as shown in the double plot (Fig. 9A) and in Table 1. It is important to note that only 1–2% cells exposing R-CRT on the membrane are nonapoptotic cells. Furthermore, the addition of recombinant R-CRT-FLAG to ATE1+/+ cells increased the amount of cells with surface R-CRT and PS compared with ATE1+/+ cells alone (Fig. 9B); the quantification of the percentage of cells under each condition is shown in Table 1. The sole addition of R-CRT-FLAG lead cells to apoptotic degrees comparable with those found after 90 min of stimulus with arsenite. A comparison between ATE1+/+ cells with or without R-CRT-FLAG shows the correlation between the apoptotic response and the presence of R-CRT on the cell membrane (Fig. 9C). The response elicited by the cells with added R-CRT-FLAG does not significantly vary at the different times of treatment. Finally it is worth mentioning that the percentage of apoptotic cells that are positive for surface R-CRT is augmented by a longer exposition to the stress agent and also by the addition of exogenous R-CRT-FLAG (Table 1).
FIGURE 9.

Cells exposing R-CRT on the cell membrane undergo apoptosis. A and B, ATE1+/+ cells (A) or ATE1+/+ plus R-CRT-FLAG (B) were left untreated or induced to undergo apoptosis with arsenite 0.5 mm for 45 and 90 min. Cells incubated with anti-R-CRT pAb followed by incubation with Cy5-conjugated goat anti-rabbit IgG and annexin V (AnnV) staining were analyzed by flow cytometry. The representative double fluorescence plot shows the percentage of cells positive for annexin V binding alone (upper left quadrant), percentage positive for R-CRT binding alone (lower right quadrant), and percentage positive for both R-CRT and annexin V binding (upper right quadrant, double positive). C, percentages of double positive ATE1+/+ cells (solid line) and ATE1+/+ plus R-CRT-FLAG cells (dashed line) were plotted as a function of time after arsenite treatment. The data are the means ± S.D. of four independent experiments. 100% values correspond to 50,000 cells for each point. Statistically significant at p < 0.01 (***) as determined by ANOVA test, indicating a significant increase in the amount of cells with surface R-CRT and PS when exogenous R-CRT was added to ATE1+/+ cells. D, percentage of annexin V-positive cells in ATE1−/− (solid line) and ATE1−/− plus R-CRT-FLAG cells (dashed line) were plotted as a function of time with arsenite treatment. Statistically significant at p < 0.02 (**) or p < 0.01 (***) as determined by ANOVA test, indicating an increase in the amount of PS when exogenous R-CRT was added to ATE1−/− cells. The data are the means ± S.D. of four independent experiments. 100% values correspond to 50,000 cells for each point.
TABLE 1.
Quantification of cell surface R-CRT and PS exposure of ATE1+/+ cells
ATE1+/+ cells without or with exogenous R-CRT-FLAG were analyzed by flow cytometry for surface exposure of R-CRT (with anti-R-CRT pAb) and PS (by annexin V binding) at 0, 45, or 90 min after arsenite treatment as described under “Experimental Procedures.” The different cell populations are shown as the percentages of cells positive for annexin V binding alone (AnnV cells) or R-CRT surface protein alone (R-CRT cells), cells double positive for annexin V and R-CRT (AnnV-R-CRT cells), total annexin V-positive cells (Total apoptotic cells), and total apoptotic cells that have surface R-CRT (Apoptotic cells/R-CRT). The data are the means ± S.D. of four independent experiments.
| ATE1+/+ |
ATE1+/+ plus R-CRT-FLAG |
|||||
|---|---|---|---|---|---|---|
| 0 min | 45 min | 90 min | 0 min | 45 min | 90 min | |
| % | % | |||||
| AnnV cells | 21 ± 3 | 21.5 ± 9 | 31 ± 13 | 12.7 ± 1.5 | 13.3 ± 2.5 | 13.6 ± 3 |
| R-CRT cells | 2 ± 1 | 3.5 ± 1.5 | 3 ± 1 | 2 ± 0 | 3 ± 1.4 | 2 ± 1.4 |
| AnnV-R-CRT cells | 10.1 ± 1.9 | 23 ± 9.9 | 30 ± 12 | 36.7 ± 4.9 | 42.3 ± 6.4 | 46 ± 3.6 |
| Total apoptotic cells | 29 ± 2.5 | 37.5 ± 11 | 64 ± 11 | 49.3 ± 4.7 | 55.7 ± 7.1 | 59.7 ± 3.2 |
| Apoptotic cells/R-CRT | 40 ± 3 | 67 ± 0.5 | 64 ± 14 | 74.2 ± 4.0 | 75.9 ± 4.1 | 79.4 ± 3.7 |
Addition of R-CRT-FLAG Increases Sensitivity to Apoptosis in ATE1−/− Cells
To conclusively define the role of R-CRT as a proapoptotic protein, we performed flow cytometry analyses with and without exogenous R-CRT-FLAG in ATE1−/− cells subjected to arsenite for 45 or 90 min. Although ATE1−/− cells expose CRT on the cell membrane (Figs. 1B and 8), with this experimental approach we were able to determine the consequence of CRT arginylation per se, in relation to the apoptotic susceptibility of these cells. ATE1−/− cells have 7% annexin V-positive cells, which increase to ∼20% between 45 and 90 min of arsenite treatment (Fig. 9D, solid line). When R-CRT-FLAG was added to ATE1−/− cells, a higher percentage of apoptotic cells was observed (20, 28, and 29% at 0, 45, and 90 min of arsenite treatment) (Fig. 9D, dashed line). Although the addition of R-CRT-FLAG increases cell sensitivity to apoptosis in ATE1−/− cells, the sole addition of this CRT isoform increases near 3-fold the population of apoptotic cells even in the absence of stress.
Moreover, the addition of R-CRT-FLAG to ATE1−/− cells restored the apoptotic response and R-CRT levels to degrees similar to those elicited in ATE1+/+ cells (Fig. 9). Because ATE1−/− cells do not have endogenous R-CRT, roughly the same levels of R-CRT are detected (∼20%) at all the time points, as a consequence of the addition of recombinant R-CRT-FLAG. These levels are similar to those observed in ATE1+/+ cells (Tables 1 and 2). Additionally it is important to note that of the total apoptotic cells, 70% contain R-CRT.
TABLE 2.
Quantification of cell surface R-CRT and PS exposure of ATE1−/− cells
ATE1−/− cells without or with exogenous R-CRT-FLAG were analyzed by flow cytometry for surface exposure of R-CRT (with anti-R-CRT pAb) and PS (by annexin V binding) at 0, 45, or 90 min after arsenite treatment as described under “Experimental Procedures.” The different cell populations are shown as the percentages of cells positive for annexin V binding alone (AnnV cells) or R-CRT surface protein alone (R-CRT cells), cells double positive for annexin V and R-CRT (AnnV-R-CRT cells), total annexin V-positive cells (Total apoptotic cells), and total apoptotic cells that have surface R-CRT (Apoptotic cells/R-CRT). The data are the means ± S.D. of four independent experiments.
| ATE1−/− |
ATE1−/− plus R-CRT-FLAG |
|||||
|---|---|---|---|---|---|---|
| 0 min | 45 min | 90 min | 0 min | 45 min | 90 min | |
| % | % | |||||
| AnnV cells | 7 ± 1.4 | 21.5 ± 3.5 | 23 ± 2.8 | 2 ± 1.4 | 8.5 ± 0.7 | 8.5 ± 3.5 |
| R-CRT cells | ||||||
| AnnV-R-CRT cells | 19 ± 2.8 | 19.5 ± 2.1 | 20.5 ± 0.7 | |||
| Total apoptotic cells | 7 ± 1.4 | 21.5 ± 3.5 | 23 ± 2.8 | 20 ± 1.4 | 28 ± 2.8 | 29 ± 2.8 |
| Apoptotic cells/R-CRT | 90 ± 7.8 | 70 ± 0.5 | 71 ± 9.3 | |||
DISCUSSION
In the present paper we demonstrate that R-CRT reaches the plasma membrane where it acts as a proapoptotic protein. It has been well established that CRT is translocated to the plasma membrane (17, 30–32), but the novelty of this paper is that not only CRT but also its arginylated form is incorporated in the plasma membrane, probably after passing through SGs. Kroemer and co-workers (33) had demonstrated that CRT is directed to the plasma membrane by exocytosis trafficking through the Golgi. In all cases the process of translocation is triggered by a Ca2+ decrease in the ER (26) or in the cytosol (2). However, Ca2+ changes might not be the only factor implicated in CRT incorporation to the cell surface. In our experiments R-CRT, coincident with the appearance of PS, associates to the plasma membrane 60 min after arsenite treatment (Figs. 3 and 9). Perhaps the arginylation of CRT orchestrate the mechanism by which CRT is incorporated on the plasma membrane. The 60-min delay in the increase of cell membrane incorporation that we see (Figs. 4 and 6) could be explained by the trapping of R-CRT in SGs up to 30 min. It is of interest that R-CRT does not increase on the plasma membrane under UV treatment (not shown); condition in which we described that R-CRT does not associate to SGs, and Ca2+ levels are unchanged (2). This suggests that arginylation of CRT and its passage through SGs might be a necessary step for the trafficking of this isospecies to the plasma membrane.
The above results are a demonstration that in vivo arginylation is important for the regulation of proteins because it can confer different fates or functions to a protein (1, 34, 35) and in cases determine cell destiny as happens with the arginylation of CRT. CRT is a multifunctional multicompartmental protein (36), is mainly located in the ER, but also has been implicated in a variety of processes that occur outside this organelle such as in the cytoplasm (3–6), in the nucleus (7, 8), on the cell plasma membrane, and in other extracellular compartments (9–17). CRT coordinates a number of cellular events from the cell surface where it has been suggested to participate in: wound healing (9, 10), thrombospondin signaling (13–16), antigen presentation and complement activation (11, 12), clearance of apoptotic cells (17), and immunogenicity of cancer cell death (37, 38). It is not surprising that post-translational modification of CRT plays a key role in its regulation, under several conditions, including stress. An indication of this postulate is the observation that cells lacking Ate1 enzyme responsible for post-translational arginylation (ATE1−/−), are less susceptible to apoptosis under arsenite treatment than WT (ATE1+/+) cells (Fig. 3B). This result does not exclude the possibility that other arginylated proteins besides CRT may be responsible for the additional phenotype in ATE1 −/− cells. It has been described that the restitution of a single arginylated protein does not restore completely a WT phenotype. This is the case with the arginylated B-actin involved in the control of the actin polymerization and lamella formation in motile cells. Nevertheless, in this paper we show that the WT apoptotic phenotype can be mainly restored by the addition of R-CRT-FLAG to ATE1−/− cells (Fig. 9D).
It has been well demonstrated that preapoptotic translocation of CRT to the cell surface is critical for immunogenic cell death, i.e., upon treatment with anthracyclins (33, 39, 40). Cell surface CRT in association with PS provides the obligate recognition signal for the removal of both professional (macrophages and neutrophils) and nonprofessional phagocytes (fibroblasts). In this paper we found that R-CRT exposure to the cell surface is increased during apoptosis elicited by arsenite treatment (Fig. 6). A positive correlation between PS and R-CRT exposure was also found (Fig. 9A). These findings could imply that R-CRT acts as a proapoptotic protein when located on the plasma membrane as we see under stress conditions. It has been demonstrated that CRT flips together with PS after cell stress and apoptosis in a Ca2+-dependent manner (32), controlling the local release of Ca2+, which is involved in the regulation of the amount and localization of PS in healthy cells. Additionally, upon anthracyclins or any other agent that promotes lower Ca2+ levels in the ER, CRT translocates to the cell surface (26). It is well known that arsenite provokes ER stress that leads to down-regulation of Ca2+ signaling, resulting in apoptosis (41). Certainly the perturbation of Ca2+ homeostasis plays a role in CRT trafficking to the cell membrane, most probably associated with the conformation that CRT adopts outside the ER (42, 43). However, in the present paper we observe no differences in Ca2+ homeostatic mechanism in ATE1−/− cells compared with WT cells (Fig. 7), indicating that the difference in susceptibility to apoptosis observed in ATE1−/− cells is not only due to an impairment of CRT to reach the membrane by this mechanism (Figs. 1B and 8). Because it was previously described that Ca2+ homeostasis and arginylation regulate the association of R-CRT to SGs in an early stress response (2), here we show that these two factors are also involved in CRT fate after longer stress periods, when the protein reaches the plasma membrane and the cells display preapoptotic signals.
When cells are confronted with chemical or physical stress, they can react in two opposite ways. On one hand they can activate the suicidal biochemical machinery leading to apoptotic demise. On the other hand, they can activate defense mechanisms designed to adapt to stressful conditions, to repair damage and to resume normal cellular functions by inducing cells to assembly SGs (44–46). The choice between these two responses is dictated by the intensity of stress, as well as cell-intrinsic parameters, for instance those that set the apoptotic threshold. It is of interest to mention that the formation of SGs upon exposure to hypoxia (47) or oxidative stress (48) leads to tumor cell resistance to apoptosis, by a mechanism that involves the sequestration and inactivation of proapoptotic proteins in SGs (49). It is an intriguing possibility that arginylation may participate in this cancer process.
It should be noted that the association of CRT to SGs is regulated by post-translational arginylation because R-CRT but not CRT is recruited to these aggregates under stress conditions (2). Herein we found that R-CRT is recruited to SGs up to 30 min of arsenite treatment, whereas when the stress persists, SGs are disassembled; R-CRT is released from these structures (Fig. 4) and perhaps translocated to the cell membrane (Fig. 6) where it acts as a preapoptotic determinant. Thus it can be speculated that R-CRT, when associated to SGs, prevents cells from apoptosis. The underlying mechanism appears to involve the sequestration and inactivation of this proapoptotic factor in SGs. However, we cannot discard the possibility that R-CRT, when present in SGs, regulate particular mRNA's encoding key antiapoptotic factors, thus either preventing its degradation necessary for the survival response or sequestering mRNA of proapoptotic proteins, inhibiting its translation.
This is not the only case where a post-translational modification is involved in the assembly and remodeling of SGs (50). Other examples include the histone deacetylase 6 activity in SG formation (47), the inhibition of the ubiquitin proteasome system in SG disassembly (51), and the demonstration that SGs contain methylarginine proteins (52). Interestingly the trigger for SGs formation under arsenite treatment involves the phosphorylation of eukaryotic initiation factor 2α, a fundamental regulatory mechanism that controls global rates of protein synthesis (53, 54), which is also critical for CRT exposure (55).
In conclusion, under stress conditions where cytosolic Ca2+ levels are decreased, the SGs, in addition to being an initial platform for R-CRT, are a major step for the storage of this protein before its trafficking to the plasma membrane. If the exerted stress persists, the protein will be released from these aggregates and eventually become an important factor implicated in programmed cell death. Consequently R-CRT is emerging as a novel protein that has an impact on cell death and whose regulation is influenced by its subcellular compartmentalization. It can be envisaged that a significant role in many physiological and pathological processes will be described in the near future for R-CRT.
Acknowledgments
We are grateful to Drs. G. Pilar, B. Caputto, and H. Maccioni for invaluable and helpful discussions; G. Schachner, S. Deza, C. Mas, C. Sampedro, P. Abadie, and P. Crespo for excellent technical assistance; and M. Sambrooks for editing work.
This work was supported by Grant BID 1728/OC-AR PICT 1661 and grants from the Agencia Nacional de Promocion Científica y Technologica, Consejo Nacional de Investigaciones Científicas y Técnicas, and Secretaria de Ciencia y Tecnica, Universidad Nacional de Córdoba.
- CRT
- calreticulin
- R-CRT
- arginylated calreticulin
- SG
- stress granule
- ER
- endoplasmic reticulum
- WB
- Western blot
- PS
- phosphatidylserine
- pAb
- polyclonal antibody
- ATE1
- arginyltRNA protein transferase.
REFERENCES
- 1. Decca M. B., Carpio M. A., Bosc C., Galiano M. R., Job D., Andrieux A., Hallak M. E. (2007) Post-translational arginylation of calreticulin. A new isospecies of calreticulin component of stress granules. J. Biol. Chem. 282, 8237–8245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carpio M. A., López Sambrooks C., Durand E. S., Hallak M. E. (2010) The arginylation-dependent association of calreticulin with stress granules is regulated by calcium. Biochem. J. 429, 63–72 [DOI] [PubMed] [Google Scholar]
- 3. Coppolino M. G., Woodside M. J., Demaurex N., Grinstein S., St-Arnaud R., Dedhar S. (1997) Calreticulin is essential for integrin-mediated calcium signalling and cell adhesion. Nature 386, 843–847 [DOI] [PubMed] [Google Scholar]
- 4. Timchenko L. T., Iakova P., Welm A. L., Cai Z. J., Timchenko N. A. (2002) Calreticulin interacts with C/EBPα and C/EBPβ mRNAs and represses translation of C/EBP proteins. Mol. Cell Biol. 22, 7242–7257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singh N. K., Atreya C. D., Nakhasi H. L. (1994) Identification of calreticulin as a rubella virus RNA binding protein. Proc. Natl. Acad. Sci. U.S.A. 91, 12770–12774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Iakova P., Wang G. L., Timchenko L., Michalak M., Pereira-Smith O. M., Smith J. R., Timchenko N. A. (2004) Competition of CUGBP1 and calreticulin for the regulation of p21 translation determines cell fate. EMBO J. 23, 406–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Holaska J. M., Black B. E., Love D. C., Hanover J. A., Leszyk J., Paschal B. M. (2001) Calreticulin is a receptor for nuclear export. J. Cell Biol. 152, 127–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Olkku A., Mahonen A. (2009) Calreticulin mediated glucocorticoid receptor export is involved in β-catenin translocation and Wnt signalling inhibition in human osteoblastic cells. Bone 44, 555–565 [DOI] [PubMed] [Google Scholar]
- 9. Gold L. I., Rahman M., Blechman K. M., Greives M. R., Churgin S., Michaels J., Callaghan M. J., Cardwell N. L., Pollins A. C., Michalak M., Siebert J. W., Levine J. P., Gurtner G. C., Nanney L. B., Galiano R. D., Cadacio C. L. (2006) Overview of the role for calreticulin in the enhancement of wound healing through multiple biological effects. J. Investig. Dermatol. Symp. Proc. 11, 57–65 [DOI] [PubMed] [Google Scholar]
- 10. Nanney L. B., Woodrell C. D., Greives M. R., Cardwell N. L., Pollins A. C., Bancroft T. A., Chesser A., Michalak M., Rahman M., Siebert J. W., Gold L. I. (2008) Calreticulin enhances porcine wound repair by diverse biological effects. Am. J. Pathol. 173, 610–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gao B., Adhikari R., Howarth M., Nakamura K., Gold M. C., Hill A. B., Knee R., Michalak M., Elliott T. (2002) Assembly and antigen-presenting function of MHC class I molecules in cells lacking the ER chaperone calreticulin. Immunity 16, 99–109 [DOI] [PubMed] [Google Scholar]
- 12. Donnelly S., Roake W., Brown S., Young P., Naik H., Wordsworth P., Isenberg D. A., Reid K. B., Eggleton P. (2006) Impaired recognition of apoptotic neutrophils by the C1q/calreticulin and CD91 pathway in systemic lupus erythematosus. Arthritis Rheum. 54, 1543–1556 [DOI] [PubMed] [Google Scholar]
- 13. Goicoechea S., Pallero M. A., Eggleton P., Michalak M., Murphy-Ullrich J. E. (2002) The anti-adhesive activity of thrombospondin is mediated by the N-terminal domain of cell surface calreticulin. J. Biol. Chem. 277, 37219–37228 [DOI] [PubMed] [Google Scholar]
- 14. Orr A. W., Pedraza C. E., Pallero M. A., Elzie C. A., Goicoechea S., Strickland D. K., Murphy-Ullrich J. E. (2003) Low density lipoprotein receptor-related protein is a calreticulin coreceptor that signals focal adhesion disassembly. J. Cell Biol. 161, 1179–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Orr A. W., Elzie C. A., Kucik D. F., Murphy-Ullrich J. E. (2003) Thrombospondin signaling through the calreticulin/LDL receptor-related protein co-complex stimulates random and directed cell migration. J. Cell Sci. 116, 2917–2927 [DOI] [PubMed] [Google Scholar]
- 16. Orr A. W., Pallero M. A., Murphy-Ullrich J. E. (2002) Thrombospondin stimulates focal adhesion disassembly through Gi- and phosphoinositide 3-kinase-dependent ERK activation. J. Biol. Chem. 277, 20453–20460 [DOI] [PubMed] [Google Scholar]
- 17. Gardai S. J., McPhillips K. A., Frasch S. C., Janssen W. J., Starefeldt A., Murphy-Ullrich J. E., Bratton D. L., Oldenborg P. A., Michalak M., Henson P. M. (2005) Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 123, 321–334 [DOI] [PubMed] [Google Scholar]
- 18. Catanzariti A. M., Soboleva T. A., Jans D. A., Board P. G., Baker R. T. (2004) An efficient system for high-level expression and easy purification of authentic recombinant proteins. Protein Sci. 13, 1331–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gray A. J., Park P. W., Broekelmann T. J., Laurent G. J., Reeves J. T., Stenmark K. R., Mecham R. P. (1995) The mitogenic effects of the B β chain of fibrinogen are mediated through cell surface calreticulin. J. Biol. Chem. 270, 26602–26606 [DOI] [PubMed] [Google Scholar]
- 20. Arosa F. A., de Jesus O., Porto G., Carmo A. M., de Sousa M. (1999) Calreticulin is expressed on the cell surface of activated human peripheral blood T lymphocytes in association with major histocompatibility complex class I molecules. J. Biol. Chem. 274, 16917–16922 [DOI] [PubMed] [Google Scholar]
- 21. Eggleton P., Lieu T. S., Zappi E. G., Sastry K., Coburn J., Zaner K. S., Sontheimer R. D., Capra J. D., Ghebrehiwet B., Tauber A. I. (1994) Calreticulin is released from activated neutrophils and binds to C1q and mannan-binding protein. Clin. Immunol. Immunopathol. 72, 405–409 [DOI] [PubMed] [Google Scholar]
- 22. Xiao G., Chung T. F., Fine R. E., Johnson R. J. (1999) Calreticulin is transported to the surface of NG108–15 cells where it forms surface patches and is partially degraded in an acidic compartment. J. Neurosci. Res. 58, 652–662 [DOI] [PubMed] [Google Scholar]
- 23. Zhu Q., Zelinka P., White T., Tanzer M. L. (1997) Calreticulin-integrin bidirectional signaling complex. Biochem. Biophys. Res. Commun. 232, 354–358 [DOI] [PubMed] [Google Scholar]
- 24. Lim S., Chang W., Lee B. K., Song H., Hong J. H., Lee S., Song B. W., Kim H. J., Cha M. J., Jang Y., Chung N., Choi S. Y., Hwang K. C. (2008) Enhanced calreticulin expression promotes calcium-dependent apoptosis in postnatal cardiomyocytes. Mol. Cells 25, 390–396 [PubMed] [Google Scholar]
- 25. Rong Y., Distelhorst C. W. (2008) Bcl-2 protein family members. Versatile regulators of calcium signaling in cell survival and apoptosis. Annu. Rev. Physiol. 70, 73–91 [DOI] [PubMed] [Google Scholar]
- 26. Tufi R., Panaretakis T., Bianchi K., Criollo A., Fazi B., Di Sano F., Tesniere A., Kepp O., Paterlini-Brechot P., Zitvogel L., Piacentini M., Szabadkai G., Kroemer G. (2008) Reduction of endoplasmic reticulum Ca2+ levels favors plasma membrane surface exposure of calreticulin. Cell Death Differ. 15, 274–282 [DOI] [PubMed] [Google Scholar]
- 27. Nakamura K., Bossy-Wetzel E., Burns K., Fadel M. P., Lozyk M., Goping I. S., Opas M., Bleackley R. C., Green D. R., Michalak M. (2000) Changes in endoplasmic reticulum luminal environment affect cell sensitivity to apoptosis. J. Cell Biol. 150, 731–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mizushima N., Yoshimori T. (2007) How to interpret LC3 immunoblotting. Autophagy 3, 542–545 [DOI] [PubMed] [Google Scholar]
- 29. Liang X. H., Jackson S., Seaman M., Brown K., Kempkes B., Hibshoosh H., Levine B. (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402, 672–676 [DOI] [PubMed] [Google Scholar]
- 30. Chen D., Texada D. E., Duggan C., Liang C., Reden T. B., Kooragayala L. M., Langford M. P. (2005) Surface calreticulin mediates muramyl dipeptide-induced apoptosis in RK13 cells. J. Biol. Chem. 280, 22425–22436 [DOI] [PubMed] [Google Scholar]
- 31. Obeid M., Tesniere A., Ghiringhelli F., Fimia G. M., Apetoh L., Perfettini J. L., Castedo M., Mignot G., Panaretakis T., Casares N., Métivier D., Larochette N., van Endert P., Ciccosanti F., Piacentini M., Zitvogel L., Kroemer G. (2007) Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 13, 54–61 [DOI] [PubMed] [Google Scholar]
- 32. Tarr J. M., Young P. J., Morse R., Shaw D. J., Haigh R., Petrov P. G., Johnson S. J., Winyard P. G., Eggleton P. (2010) A mechanism of release of calreticulin from cells during apoptosis. J. Mol. Biol. 401, 799–812 [DOI] [PubMed] [Google Scholar]
- 33. Panaretakis T., Kepp O., Brockmeier U., Tesniere A., Bjorklund A. C., Chapman D. C., Durchschlag M., Joza N., Pierron G., van Endert P., Yuan J., Zitvogel L., Madeo F., Williams D. B., Kroemer G. (2009) Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 28, 578–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hamilton M. H., Cook L. A., McRackan T. R., Schey K. L., Hildebrandt J. D. (2003) Gamma 2 subunit of G protein heterotrimer is an N-end rule ubiquitylation substrate. Proc. Natl. Acad. Sci. U.S.A. 100, 5081–5086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Karakozova M., Kozak M., Wong C. C., Bailey A. O., Yates J. R., 3rd, Mogilner A., Zebroski H., Kashina A. (2006) Arginylation of β-actin regulates actin cytoskeleton and cell motility. Science 313, 192–196 [DOI] [PubMed] [Google Scholar]
- 36. Michalak M., Groenendyk J., Szabo E., Gold L. I., Opas M. (2009) Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 417, 651–666 [DOI] [PubMed] [Google Scholar]
- 37. Obeid M., Panaretakis T., Joza N., Tufi R., Tesniere A., van Endert P., Zitvogel L., Kroemer G. (2007) Calreticulin exposure is required for the immunogenicity of γ-irradiation and UVC light-induced apoptosis. Cell Death Differ. 14, 1848–1850 [DOI] [PubMed] [Google Scholar]
- 38. Gardai S. J., Xiao Y. Q., Dickinson M., Nick J. A., Voelker D. R., Greene K. E., Henson P. M. (2003) By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 115, 13–23 [DOI] [PubMed] [Google Scholar]
- 39. Obeid M., Panaretakis T., Tesniere A., Joza N., Tufi R., Apetoh L., Ghiringhelli F., Zitvogel L., Kroemer G. (2007) Leveraging the immune system during chemotherapy. Moving calreticulin to the cell surface converts apoptotic death from “silent” to immunogenic. Cancer Res. 67, 7941–7944 [DOI] [PubMed] [Google Scholar]
- 40. Obeid M., Tesniere A., Panaretakis T., Tufi R., Joza N., van Endert P., Ghiringhelli F., Apetoh L., Chaput N., Flament C., Ullrich E., de Botton S., Zitvogel L., Kroemer G. (2007) Ecto-calreticulin in immunogenic chemotherapy. Immunol. Rev. 220, 22–34 [DOI] [PubMed] [Google Scholar]
- 41. Zhang X. W., Yan X. J., Zhou Z. R., Yang F. F., Wu Z. Y., Sun H. B., Liang W. X., Song A. X., Lallemand-Breitenbach V., Jeanne M., Zhang Q. Y., Yang H. Y., Huang Q. H., Zhou G. B., Tong J. H., Zhang Y., Wu J. H., Hu H. Y., de Thé H., Chen S. J., Chen Z. (2010) Arsenic trioxide controls the fate of the PML-RARα oncoprotein by directly binding PML. Science 328, 240–243 [DOI] [PubMed] [Google Scholar]
- 42. Afshar N., Black B. E., Paschal B. M. (2005) Retrotranslocation of the chaperone calreticulin from the endoplasmic reticulum lumen to the cytosol. Mol. Cell Biol. 25, 8844–8853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Corbett E. F., Michalak K. M., Oikawa K., Johnson S., Campbell I. D., Eggleton P., Kay C., Michalak M. (2000) The conformation of calreticulin is influenced by the endoplasmic reticulum luminal environment. J. Biol. Chem. 275, 27177–27185 [DOI] [PubMed] [Google Scholar]
- 44. Buchan J. R., Parker R. (2009) Eukaryotic stress granules. The ins and outs of translation. Mol. Cell 36, 932–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Anderson P., Kedersha N. (2008) Stress granules. The Tao of RNA triage. Trends Biochem. Sci. 33, 141–150 [DOI] [PubMed] [Google Scholar]
- 46. Thomas M. G., Loschi M., Desbats M. A., Boccaccio G. L. (2011) RNA granules. The good, the bad and the ugly. Cell Signal. 23, 324–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Arimoto K., Fukuda H., Imajoh-Ohmi S., Saito H., Takekawa M. (2008) Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat. Cell Biol. 10, 1324–1332 [DOI] [PubMed] [Google Scholar]
- 48. Kwon S., Zhang Y., Matthias P. (2007) The deacetylase HDAC6 is a novel critical component of stress granules involved in the stress response. Genes Dev. 21, 3381–3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gareau C., Fournier M. J., Filion C., Coudert L., Martel D., Labelle Y., Mazroui R. (2011) p21(WAF1/CIP1) upregulation through the stress granule-associated protein CUGBP1 confers resistance to bortezomib-mediated apoptosis. PLoS One 6, e20254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xie W., Denman R. B. (2011) Protein methylation and stress granules. Posttranslational remodeler or innocent bystander? Mol. Biol. Int. 2011, 137459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mazroui R., Di Marco S., Kaufman R. J., Gallouzi I. E. (2007) Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol. Biol. Cell 18, 2603–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dolzhanskaya N., Merz G., Aletta J. M., Denman R. B. (2006) Methylation regulates the intracellular protein-protein and protein-RNA interactions of FMRP. J. Cell Sci. 119, 1933–1946 [DOI] [PubMed] [Google Scholar]
- 53. Anderson P., Kedersha N. (2002) Stressful initiations. J. Cell Sci. 115, 3227–3234 [DOI] [PubMed] [Google Scholar]
- 54. Mokas S., Mills J. R., Garreau C., Fournier M. J., Robert F., Arya P., Kaufman R. J., Pelletier J., Mazroui R. (2009) Uncoupling stress granule assembly and translation initiation inhibition. Mol. Biol. Cell 20, 2673–2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kepp O., Galluzzi L., Giordanetto F., Tesniere A., Vitale I., Martins I., Schlemmer F., Adjemian S., Zitvogel L., Kroemer G. (2009) Disruption of the PP1/GADD34 complex induces calreticulin exposure. Cell Cycle 8, 3971–3977 [DOI] [PubMed] [Google Scholar]