

Background: Zinc inhibits human P2X2 with high potency.
Results: Site-directed mutations of P2X2 with a 100,000-fold range in the potency of zinc inhibition were made.
Conclusion: A cluster of three histidines at the interface between subunits controls zinc potency.
Significance: Zinc-resistant mutants offer the opportunity to test the biological significance of zinc modulation of P2X2.
Keywords: ATP, Ion Channels, Mutagenesis, Purinergic Receptor, Zinc, P2X2 Receptor, Cross-linking, Homology Model, Potentiation, Zinc Inhibition
Abstract
Human P2X2 receptors (hP2X2) are strongly inhibited by zinc over the range of 2–100 μm, whereas rat P2X2 receptors (rP2X2) are strongly potentiated over the same range, and then inhibited by zinc over 100 μm. However, the biological role of zinc modulation is unknown in either species. To identify candidate regions controlling zinc inhibition in hP2X2 a homology model based on the crystal structure of zebrafish P2X4.1 was made. In this model, His-204 and His-209 of one subunit were near His-330 of the adjacent subunit. Cross-linking studies confirmed that these residues are within 8 Å of each other. Simultaneous mutation of these three histidines to alanines decreased the zinc potency of hP2X2 nearly 100-fold. In rP2X2, one of these histidines is replaced by a lysine, and in a background in which zinc potentiation was eliminated, mutation of Lys-197 to histidine converted rP2X2 from low potency to high potency inhibition. We explored whether the zinc-binding site lies within the vestibules running down the central axis of the receptor. Elimination of all negatively charged residues from the upper vestibule had no effect on zinc inhibition. In contrast, mutation of several residues in the hP2X2 middle vestibule resulted in dramatic changes in the potency of zinc inhibition. In particular, the zinc potency of P206C could be reversibly shifted from extremely high (∼10 nm) to very low (>100 μm) by binding and unbinding MTSET. These results suggest that the cluster of histidines at the subunit interface controls access of zinc to its binding site.
Introduction
In humans, seven different genes (P2RX1–P2RX7) code for subunits of P2X receptors. The proteins these genes encode (P2X1–P2X7) form ATP-gated cation channels, by assembling into homomeric or heteromeric trimers (1). Different subunit combinations have distinct physiological and pharmacological properties. The characterization of the crystal structure of one member of this gene family (2) provides a framework to allow a more sophisticated exploration of the molecular basis of properties specific to particular subunit combinations.
It is well established that rodent and human P2X2 receptors expressed in Xenopus oocytes or transfected cells are modulated by extracellular zinc (reviewed in Ref. 3), but the actions of zinc on P2X2 receptors vary dramatically by species. Mouse and rat P2X2 receptors (rP2X2)2 show a biphasic response to zinc. When exposed to zinc concentrations in the range of 2–100 μm the response to ATP shows dramatic potentiation, whereas at higher zinc concentrations the ATP response is inhibited (4–6). In contrast, human P2X2 receptors (hP2X2) show no potentiation at any zinc concentration; rather over the same concentration range that potentiates the rat receptor (2–100 μm) the response to ATP is inhibited by zinc (7).
Substantial evidence indicates that zinc is co-released with neurotransmitters from many synaptic terminals (8) and upon release can bind to receptors and modulate neuronal excitability of several types of neurotransmitter receptors (9). Some regions of high P2X2 expression also have a high density of zinc containing nerve terminals (for example, the cerebral cortex and hippocampus) but evidence for zinc modulation of P2X2 receptors in vivo or in brain slices is lacking. Indeed, the role of P2X2 receptors in the brain is unclear. Although P2X2 receptors are highly expressed in many parts of the central nervous system (10, 11), with the assays used so far most regions of the brain where these receptors are expressed function normally in P2X2 knock-out mice (12).
The molecular basis for high potency zinc potentiation of rP2X2 is zinc binding to His-120 and His-213, which lie on opposite sides of each subunit interface (13, 14). However, the molecular mechanism for high potency zinc inhibition of hP2X2 is unclear. All of the extracellular histidines of hP2X2 have been mutated (7) and two sites were identified (His-204 and His-209) at which replacement with an alanine resulted in a modest decrease in the extent of zinc inhibition. However, in neither of these mutants nor in the H204A/H209A double mutant was the potency of zinc dramatically shifted from wild-type hP2X2.
One striking feature of the structure of zP2X4.1 is the presence of three cavities referred to as vestibules that run down the 3-fold symmetric central axis from the top of the molecule to just above the pore region in the membrane (2). In the ligand-free closed state that was crystallized, the upper and middle vestibules are separated by a constriction that would prevent ion flow, and a relatively narrow region that might impede ion flow also separates the middle vestibule from the lowest cavity, which is referred to as the extracellular vestibule. The reason for this designation is that just above the membrane there are three fenestrations between subunits that appear large enough to allow the unimpeded flow of ions directly into the extracellular vestibule even when the channel gate (which lies midway through the membrane) is closed. Two recent papers have supported the idea that the pathway for ion movements through open P2X channels is via the fenestrations (15, 16). However, there must be a pathway that allows ions to enter the middle vestibule, because Gd3+, an inhibitor of zP2X4.1 channel function was shown to bind to Glu-98 from all three subunits at the top of the middle vestibule (2). It is possible that the movements associated with channel opening might also allow access to the upper vestibule. Thus these vestibules are potential locations for the high potency inhibitory zinc-binding site of hP2X2.
Our goal for this study was to use a homology model of hP2X2 based on the structure of zP2X4.1 to suggest candidate residues for involvement in inhibitory zinc binding, with the hope of identifying mutations that dramatically enhance or attenuate zinc inhibition of hP2X2. Identification of such mutants might provide a framework for understanding the molecular basis of zinc inhibition, and also offer the possibility of using them as tools to probe the role of P2X receptors in vivo.
EXPERIMENTAL PROCEDURES
Homology Modeling of Human P2X2 Receptor
The human P2X2 homology model was generated using the SWISS-MODEL server “Automated Protein Modeling Server” (EXPASY (17, 18). The amino acid sequence for hP2X2b (GenBankTM AAF74202.1) was used as the input and the template was the single chain structure of zebrafish P2X4.1 (PDB 3H9V (2)). The program COOT (Crystallographic Object-Oriented Toolkit (19)) was then used to superimpose the hP2X2b monomer single chain onto the zP2X4.1 trimer template (PDB 3I5D) to generate the hP2X2b trimer structure. Molecular graphics images of the structure were drawn using the UCSF Chimera software from the Resources for Biocomputing, Visualization and Informatics at the University of California, San Francisco, CA.
Site-directed Mutagenesis
The source of the human P2X2b and rat P2X2a cDNAs has been previously described (7). Mutations were made using the QuikChange mutagenesis kit (Agilent Technologies, Santa Clara, CA) and referred to by the original single letter amino acid codon followed by the residue number and the substituted single letter codon. All mutations were confirmed by DNA sequencing from the University of Michigan DNA sequencing core.
Expression
RNAs encoding wild-type and mutant P2X2 receptors were synthesized using the mMessage Machine T7 kit (Invitrogen) and expressed in stage V-VI Xenopus laevis oocytes. Oocytes were surgically extracted and harvested following methods approved by the University of Michigan Committee on the Use and Care of Vertebrate Animals. Each oocyte was injected with 50 nl of RNA (50–100 ng/μl unless it is stated that a lower concentration was used).
Cross-linking and Western Blotting
Prior to protein isolation, oocytes injected with RNA (50 ng/μl) encoding either wild-type or mutant hP2X2 receptors were exposed at room temperature to ND96 buffer alone (96 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 5 mm sodium pyruvate, and 5 mm HEPES, pH 7.6) or to ND96 buffer supplemented with either the long arm Cys-reactive cross-linker BM(PEG)3 (1,11-bismaleimidotriethyleneglycol (1 mm), Thermo Fisher Scientific), the short arm Cys-reactive cross-linker BMOE (bismaleimidoethane (1 mm), Thermo Fisher Scientific), or H2O2 (0.1% w/v) for 10 min. Oocytes were washed five times in ND96 buffer and homogenized in buffer H (100 mm NaCl, 20 mm Tris·Cl, pH 7.4, 1% (w/v) Triton X-100, 10 μl/ml of protease inhibitor mixture (P8340, Sigma) in a volume of 10 μl/oocyte. The samples were centrifuged at 16,000 × g at 4 °C for 2 min and the supernatant was transferred into clean microcentrifuge tubes. 15 μl of the supernatant was mixed with 4× SDS-PAGE loading sample buffer (50 mm Tris·Cl, pH 6.8, 50 mm DTT, 2% (w/v) SDS, 10% (v/v) glycerol) and stored on ice for SDS-PAGE analysis of total protein. Protein samples were heated to 95 °C for 3 min, and loaded on pre-cast NuPAGE 4–12% (w/v) BisTris gels (Invitrogen). Gels were transferred to nitrocellulose membranes and probed using a polyclonal antibody (1:200) against an epitope in the extracellular domain of human P2X2 (Santa Cruz Biotechnology, Santa Cruz, CA) and then visualized by chemiluminescence with the SuperSignal West Pico Chemiluminescent Substrate kit (Thermo Fisher Scientific) that was detected on film.
In Fig. 2, for each construct, the treated and untreated samples illustrated were always taken from adjacent lanes on the same gel and shown at identical exposures, so there is no separation between them. However, white bars separate the images from different constructs to indicate that they were selected from films of several different gels exposed for different periods of time, so that a similar amount of P2X2 immunoreactivity could be illustrated in each lane. This was necessary because the amount of protein expressed varied widely among mutants (likely because of variation in the quality of the RNA injected). The position of molecular mass markers (in kDa) are shown on the left, and because commercial precast gels were always run for the same length of time, their positions on all gels were superimposable.
FIGURE 2.

Biochemical test of accessibility of H204C or H209C to H330C of the adjacent subunit. A, lack of effect of the cysteine reactive cross-linker BM(PEG)3 on the indicated single cysteine mutant constructs. The positions of the molecular mass markers (in kDa) shown on the left apply to panels A–C. The double headed arrows between panels A and B and panels B and C indicate the expected position of monomers (light gray arrows), dimers (dark gray arrows), and trimers (black arrows). B, effect of BM(PEG)3 on double cysteine mutants. C, effect of the oxidizing agent H2O2 and the cysteine reactive cross-linker BMOE on double cysteine mutants.
Electrophysiological Recordings
Two-electrode voltage clamp studies were performed on oocytes expressing wild-type or mutant receptors 1–5 days after injection. For all recordings, the holding potential was −50 mV. Recording electrodes consisted of thin-walled borosilicate glass pipettes pulled on a P-97 Flaming Brown puller (Sutter Instrument Company, Novato, CA) that had resistances of 0.5–1 MΩ. Currents were recorded using a Turbo-Tec3 or Turbo-Tec10 amplifier (npi electronic GmBH, Tamm, Germany). Data acquisition was done using a Digidata 1322A interface controlled by Clampex 9 or 10 (Molecular Devices, Sunnyvale, CA).
Solutions
The external recording solution consisted of (in mm): 90 NaCl, 1 KCl, 1.3 MgCl2, and 10 HEPES, pH 7.5. Disodium ATP (Sigma) was prepared as a 100 mm stock in external recording solution and stored at −20 °C. The ATP solutions were made by diluting the stock in external recording solution and adjusting the pH to 7.5 to obtain the desired concentrations. Recording electrodes were filled with an internal solution of 3 m KCl. Zinc chloride was prepared as a 10 mm stock in external recording solution that was acidified with 0.01 m HCl to prevent precipitation.
At the flow rates typically used, the time constant for solution exchange in the recording chamber was ∼3 s. In most experiments, a computer controlled, gravity fed 8-valve array (ALA Scientific Instruments, Farmingdale, NY) regulated solution delivery. For electrophysiological studies of the effect of modification of accessible cysteines, the oocytes were studied while impaled in the recording chamber, but to conserve materials, the flow through the recording chamber was stopped, and a concentration of reagent sufficient to cause maximum modification within a 2-min incubation was added from a P-200 pipetter in a volume sufficient to cause a nearly complete solution change within a few seconds. At the end of the incubation period, a wash of at least 30 s occurred before recording was resumed. The modifiers tested were BMOE, BM(PEG)3, and MTSET ((2-(trimethylammonium)ethyl)methanethiosulfonate chloride, Toronto Research Chemicals, North York, ON, Canada). The modifier stocks were made in dimethyl sulfoxide and kept on ice, and then freshly diluted at least 1:100 into room temperature recording solution immediately before testing.
Data Analysis
Preliminary data analysis was performed using Clampfit 10 (Molecular Devices, Sunnyvale, CA), with additional analysis done with Microsoft Excel. Concentration-response relationships were fit using the nonlinear curve fitting program of Sigmaplot 9.0 or 10.0 (Systat Software, San Jose, CA). For ATP, the 3 parameter Hill equation was used. For zinc inhibition the 3 parameter Hill equation was used when the inhibition was complete at high zinc, and the 4 parameter Hill equation was used when the maximal effective concentration of zinc did not completely eliminate the ATP activated current.
For analysis of the magnitude of zinc inhibition, the data from each cell were normalized to the maximum value of the ATP response, which was assigned a value of 100%. When parameters of the fit are given in text or figure legends, they were obtained by finding the best IC50 for each cell, and then reporting the average IC50 ± S.E. and the sample size. For display in figures, the normalized points at each zinc concentration from all cells were averaged and plotted with error bars indicating the mean ± S.E. The fits shown on the graphs are to these averaged data, and therefore the IC50 of these fits sometimes differed slightly from the mean value of the IC50 reported in text.
Correcting Zinc IC50 for Changes in ATP Potency
The IC50 for zinc differs depending on whether one tests hP2X2 at EC10, EC50, or EC90 for ATP (7). To test for differences in zinc potency between mutants, we normally tested each construct for zinc inhibition at its own EC10 for ATP. Accomplishing this required pre-screening, because the ATP concentration-response relationship of oocytes expressing P2X2 varies considerably, so that when the average EC50 concentration is used, individual oocytes respond from 20 to 75% of the maximal current (20). For the pre-screen, a series of oocytes were tested with a concentration of ATP that was the average EC10 for a population of cells expressing that construct and with saturating ATP, and only the oocytes that had ATP responses between 5 and 15% of maximum were used for studying zinc inhibition.
Chemical modification of free cysteines by MTSET or cross-linkers sometimes changed the potency of ATP. Managing the number of solutions needed to test oocytes at both their pre- and post-MTSET EC10 values for ATP would have been very difficult to accomplish with the available equipment, and would have been impossible for mutants like P206C, whose sensitivity to ATP changed rapidly following MTSET modification. Instead, we simplified the experiment by using the same ATP concentration (the pre-screen EC10) before and after modification. We then used data in Fig. 3 of Tittle and Hume showing the relationship between zinc potency and the dose of ATP used (7) to develop a correction for changes in ATP potency. By fitting a power function to these data, we were able to define a scaling factor based on the percentage of the maximum current (ECx) that the ATP concentration used in each experiment produced after modifier treatment. The scaling factor was calculated by the equation,
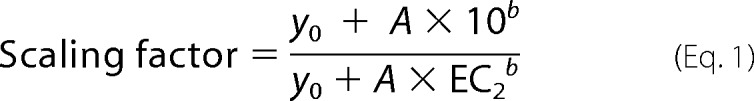 |
where EC2 is the ECx for ATP after modifier treatment (and so would be 80 if cells were studied at their EC80) and y0, A, and b are empirical constants determined from fitting the data from wild-type hP2X2 (y0 = 8.29, A = 1.19 × 10−4, and b = 2.98). Multiplying the scaling factor by the measured IC50 for zinc after modifier treatment gave the expected IC50 at the post-modification EC10 for ATP. This procedure perfectly corrected for the change in zinc potency of wild-type hP2X2 when the ATP concentration was raised.
FIGURE 3.

Effect of elimination of the three clustered histidines. A, two electrode voltage clamp recordings from Xenopus oocytes expressing hP2X2 or the hP2X2 H204A/H209A/H330A mutant. In all experiments, the holding potential was −50 mV. The arrow indicates the current at the end of the period of zinc application. B, concentration-response relationship for a series of oocytes studied as in A. In these experiments, a range of concentrations of zinc were tested, whereas the ATP concentration was held constant at 2 μm. The data were fit to the 3 parameter Hill equation (wild-type, IC50 = 11 μm; H204A/H209A/H330A, IC50 = 979 μm).
RESULTS
Homology Model for hP2X2 Suggests That His-204, His-209, and His-330 Lie Close Together Across the Subunit Interface
In rP2X2, the zinc-binding site that causes potentiation includes two histidines on opposite sides of the subunit interface. When the sequence of hP2X2 was used to create a homology model based on the structure of zP2X4.1, His-330 from one subunit was predicted to lie close to His-204 and His-209 across the interface between adjacent subunits (Fig. 1A), raising the possibility that this histidine cluster might contribute to the zinc-binding site responsible for the high potency zinc inhibition seen in hP2X2 receptors. If these residues are part of the inhibitory zinc-binding site, it might also explain why zinc inhibition of rP2X2 has such low potency, because in rP2X2 the residue equivalent to His-209 is a lysine (Lys-197), which would be expected to disrupt zinc binding.
FIGURE 1.

Location of candidate residues for participation in the inhibitory zinc binding of hP2X2. A, homology model of hP2X2 based on the structure of zP2X4.1 in the closed state. The three identical subunits of this trimeric protein are shown in pink, green, and light blue. The positions shown in orange (His-132 and Arg-225) are equivalent to the two histidines of rP2X2 that are required for the potentiating effect of zinc. The positions of the three histidines to be tested for participation in the inhibitory zinc-binding site are shown in red. The white box shows the region illustrated in B. B, higher resolution view of the region around the three candidate histidines. His-204 and His-209 are close to each other on the light blue subunit, whereas the nearest His-330 is on the adjacent green subunit. In this closed state model, Pro-206 (yellow) of the light blue subunit partially shields His-330 from access to His-204 and His-209. The black arrow indicates the top of the fenestration that provides access to the extracellular vestibule. Several residues on the inside of this vestibule arising from the pink subunit are visible. The white arrow indicates a smaller fenestration into the middle vestibule. The visible residue arising from the pink subunit at the back of the middle vestibule is Ser-76. Another middle vestibule residue visible through this opening is Glu-75 (dark blue) from the same subunit as His-204 and His-206.
A challenge to the idea that this histidine cluster contributes to zinc binding is that in the homology model, Pro-206 is positioned in a way that would likely interfere with interactions between His-204 or His-209 on one side of the subunit interface and His-330 on the other (Fig. 1B). However, the homology model was based on the unliganded, closed channel so the interference might be absent in other conformations. To test if hP2X2 can take on a conformation in which His-330 closely approaches His-204 or His-209, we expressed the double cysteine mutants H204C/H330C and H209C/H330C in oocytes and then used physiological and biochemical methods to explore whether these positions were close enough to form ectopic disulfide bonds.
In some cases an ectopic disulfide bond across a subunit interface can form spontaneously and constrain the conformation of a channel such that the electrophysiological properties are altered. For instance, the H120C/H213C double mutant in rP2X2 dramatically right shifts the ATP concentration-response curve by spontaneously forming disulfide bonds (14). However, the hP2X2 double mutants H204C/H330C and H209C/H330C responded normally to ATP. Both of these mutants still showed potent zinc inhibition. Indeed, when tested in parallel with wild-type in the same batch of oocytes, both had an IC50 that was slightly left shifted (wild-type, IC50 = 12.9 ± 0.9 μm, n = 17; H204C/H330C, IC50 = 1.8 ± 0.2 μm; H209C/H330C, IC50 = 2.0 ± 0.3 μm, n = 6 for each). Thus, either ectopic disulfide bonds did not form, or bonds at these positions have little effect on function. To distinguish between these possibilities, total proteins from oocytes expressing either wild-type hP2X2, single cysteine mutants, or double cysteine mutants were extracted and run on Western blots (Fig. 2).
In the absence of an exogenous cross-linking agent, the vast majority of hP2X2 protein in wild-type and all single and double mutants was at the monomer size, indicating that spontaneous cross-linking was rare. Occasionally there was a small amount of material the size expected for a dimer, but trimer-sized material was never observed. Because dimer was observed in some preparations of wild-type hP2X2, whatever caused it was not a consequence of the added cysteines. Very different results were observed when cysteine-reactive cross-linkers were used. BM(PEG)3 had no effect on the size of the P2X2 proteins extracted from any of the single cysteine mutants (Fig. 2A), nor on the material extracted from the H204C/H209C double mutant (as expected because in this mutant both cysteines are on the same side of the subunit interface, so cross-linking these residues would not create bonds that join two subunits together). In contrast, BM(PEG)3 shifted most of the hP2X2 protein from H204C/H330C and H209C/H330C to the size of trimers (Fig. 2B), demonstrating that the distance between these residues across the subunit interface must be less than the extended length of BM(PEG)3 (17.8 Å). To further probe the distance between these residues, we used H2O2, an oxidizing agent that can promote disulfide bond formation between cysteines that are close to each other but do not react spontaneously, and BMOE (8 Å), a cross-linker with a shorter span than BM(PEG)3 (Fig. 2C). H2O2-treated material was monomer sized, whereas BMOE shifted most of the hP2X2 protein from H204C/H330C and H209C/H330C to trimer size. These results indicate that hP2X2 can take on a conformation in which the distance across the subunit interface between these cysteine pairs is less than the span of BMOE (8 Å), but that these cysteines are farther apart than the maximum distance that allows a disulfide bridge to form between cysteines (2–3 Å).
Mutation of the Histidine Cluster Can Dramatically Alter Zinc Inhibition of hP2X2
A challenge to the idea that histidine cluster His-204/His-209/His-330 contributes to zinc inhibition is that it was previously reported that the H204A and H209A mutants have an IC50 for zinc only slightly higher than wild-type hP2X2 (7). We replicated these results and extended them to H330A. When H330A was studied near its EC10 for ATP, zinc caused inhibition with an IC50 similar to wild-type hP2X2 (Table 1). Similarly, at the EC10 for ATP, the double mutant H209A/H330A had a zinc IC50 similar to wild-type (5.5 ± 0.3 μm, n = 5) as previously been reported for the H204A/H209A double mutant (7). Although wild-type hP2X2 and the other mutants reported in this study showed only zinc inhibition at all ATP levels tested, when H330A was tested with ATP at the EC40 or above, addition of zinc produced transient inhibition that was followed by growth. At low zinc concentrations the balance at steady state favored inhibition, whereas at higher zinc there was modest potentiation. For example, as compared with the amplitude before zinc, the currents at the most effective inhibitory concentration (20 μm zinc) become stable at 73 ± 8% and the currents at the most effective potentiating concentration (500 μm) become stable at 223 ± 75% (n = 5 experiments, each with 4–5 cells at each concentration). Potential explanations for this unexpected observation are considered under “Discussion.”
TABLE 1.
Potency of zinc inhibition for equivalent mutations in human and rat P2X2
All constructs were tested at the EC10 concentration for ATP.
| Human P2X2 | IC50 for zinc | Number of cells | Rat equivalent | IC50 for zinc | Number of cells |
|---|---|---|---|---|---|
| μm | μm | ||||
| WT | 13 ± 1 | 17 | H120A/K197H/H213A (humanized rP2X2) | 19 ± 2 | 15 |
| H209K | 77 ± 8 | 10 | H120A/H213A | 121 ± 9 | 8 |
| H204A | 19 ± 1 | 5 | H120A/H192A/K197H/H213A | 27 ± 2 | 4 |
| H209A | 25 ± 1 | 5 | H120A/K197A/H213A | 116 ± 12 | 7 |
| H330A | 13 ± 5 | 4 | H120A/K197H/H213A/H319A | 16 ± 3 | 4 |
In contrast to the relatively normal zinc potency of the single and double alanine mutants at the histidine cluster, the H204A/H209A/H330A triple mutant gave a very different result. Although this mutant had a nearly normal EC50 for ATP (20 ± 2 μm, n = 24), when studied at its EC10 for ATP the H204A/H209A/H330A triple mutant had very low zinc potency, with its IC50 shifted about 100-fold to the right as compared with wild-type (Fig. 3). Thus mutations in the region of the histidine cluster can dramatically alter zinc inhibition of hP2X2.
Reciprocal Modification of Potency of Zinc Inhibition in Rat P2X2 and Human P2X2
If the difference in the potency of zinc inhibition between hP2X2 and rP2X2 is due to the fact that rP2X2 receptors lack a histidine in a key position (the homolog of hP2X2 His-209 is rP2X2 Lys-197) then placing a histidine at this position of rP2X2 should mimic the high potency zinc inhibition seen in hP2X2 receptors. To test this possibility, it was necessary to first eliminate high potency zinc potentiation in rP2X2, by mutating the two histidines essential for potentiation (14). As expected based on previous work, in the rP2X2 H120A/H213A double mutant, zinc potentiation was absent and zinc inhibition was of low potency with an IC50 of more than 100 μm. When K197H was introduced into this background, the rH120A/K197H/H213A mutant, which will be referred to as humanized rP2X2, responded to zinc with much higher potency, which was quite similar to wild-type hP2X2 (Fig. 4A, Table 1). The opposite mutation in hP2X2 (H209K) reciprocally converted zinc potency from high to low (Fig. 4B, Table 1).
FIGURE 4.

Reciprocal effects of mutations of rP2X2 and hP2X2 on zinc inhibition. In both panels the arrow indicates the direction of the change in zinc potency when the endogenous residue was changed to the residue found in the same position in the other species. A, zinc concentration-response relationship for two variants of rP2X2 that lacks critical residues at the potentiating zinc-binding site (H120A/H213A). The residue at position 197 was either the normal lysine, or was mutated to a histidine, as is found at the equivalent site in hP2X2. B, zinc concentration-response relationship for wild-type hP2X2 (same data as Fig. 3, so only the fit is shown) and for a mutant in which His-209 was changed to lysine, as found at the equivalent site (position 197) of rat P2X2.
To further explore similarities between the mechanism of zinc inhibition of rP2X2 and hP2X2 we made alanine mutations of the rat P2X2 residues equivalent to H204A, H209A, and H330A and compared them to humanized rP2X2 (Fig. 5 and Table 1). rH120A/K197H/H213A/H319A was inhibited by zinc with an IC50 nearly identical to humanized rP2X2, just as hH330A was inhibited by zinc with an IC50 nearly identical to wild-type hP2X2. rH120A/H192A/K197H/H213A had a zinc IC50 slightly higher than humanized rP2X2, as was also the case for hH204A compared with wild-type hP2X2. For both species, the largest effect on zinc inhibition resulted from mutation of the middle histidines. The IC50 of hH209A was shifted about 2-fold to the right, whereas the IC50 of rH120A/H197A/H213A was shifted over 8-fold to the right, and was as zinc insensitive as rH120A/H213A.
FIGURE 5.

Potency of zinc inhibition in a series of rP2X2 mutants. All mutants were made deficient in zinc potentiation by the presence of the H120A and H213A mutations. Effect of substituting an alanine at His-192, Lys-197, or His-319 (equivalent residues to hP2X2 His-204, His-209, or His-330). The bold part of each label indicates the change from humanized rP2X2. The thick lines without points are reprints of the fits to data from H120A/H213A and H120A/K197H/H213A that were presented in Fig. 4.
Tests of Potential Mechanisms by Which Mutations in the Histidine Cluster Alter Zinc Inhibition
The observation that zinc inhibition is barely changed by single alanine mutations to the histidine cluster (H204A, H209A, H330A) and is still present but dramatically right shifted in the H204A/H209A/H330A mutant led us to consider whether this region might be essential for zinc inhibition, and yet not be the zinc-binding site. A possible explanation could be that the residues of this cluster control access of zinc to a site elsewhere.
The residues of the histidine cluster sit at a position at which they potentially could control entry into the middle vestibule. We therefore tested whether the inhibitory binding site for zinc might be in the upper or middle vestibule by mutating candidate zinc-binding residues.
In the upper vestibule of hP2X2 there are no histidines or cysteines, but there are several negative charges that might potentially participate in binding zinc. When hP2X2 residues Glu-96, Glu-97, and Glu-103 were simultaneously mutated to alanines, the IC50 of zinc inhibition was very slightly right shifted as compared with that seen in wild-type hP2X2 (Fig. 6A). When oocytes from the same day were compared, the IC50 was 16.2 ± 1.0 μm for hP2X2 and 20.6 ± 1.5 μm for E96A/E97A/E103A (n = 5 each). Similarly, when the rP2X2 homologs of these residues (Glu-84, Glu-85, and Glu-91) plus Asp-82 (the homolog of human Asp-94) were all mutated to alanine in the humanized rP2X2 background, there was no change in zinc inhibition (Fig. 6B). Therefore, it seems unlikely that any of the negatively charged residues in the upper vestibule are part of the inhibitory zinc-binding site.
FIGURE 6.

Effect of removal of negative charges from the upper vestibule on zinc inhibition. A, comparison of the zinc concentration-response relationship for wild-type hP2X2 and a variant in which three negative charges in the upper vestibule were mutated to alanine. B, comparison of the zinc concentration-response relationship for humanized rP2X2 (H120A/K197H/H213A) and humanized rP2X2 in which all four negative charges in the upper vestibule were mutated to alanine. The average IC50 for the mutant lacking negative charges in the upper vestibule was 11 ± 3 μm, n = 5.
In the middle vestibule of hP2X2, there are no cysteines, and the only histidine is His-330. However, there are two negative charges (Glu-75 and Asp-326) that might potentially participate in zinc binding. The absence of a negative charge at another middle vestibule position is noteworthy. The crystal structure of zP2X4.1 identified three negatively charged residues within the middle vestibule that could bind Gd3+ (Glu-98 from all three subunits). However, the hP2X2 residue at the homologous position is Gly-104, which is not a zinc binding candidate. As Asp-326 is predicted to form a salt bridge with Arg-324 we also mutated this residue. The R324A mutant responded to ATP similar to wild-type (EC50 of 14.9 ± 1.2 μm, n = 5), and D326A had a somewhat left-shifted ATP concentration-response relationship (EC50 of 2.6 ± 0.6 μm, n = 25). However, when studied at their respective EC10 for ATP, both mutants caused only a very slight decrease in the potency of zinc inhibition (Fig. 7A). The E75A mutant had slightly enhanced ATP potency (3.7 ± 0.6 μm, n = 4), but zinc inhibition in this mutant was substantially less potent than wild-type (Fig. 7B).
FIGURE 7.

Effect of removal of negative charges from the middle vestibule on zinc inhibition. All mutants were made in hP2X2. Data from five cells were averaged for each mutant. A, zinc concentration-response relationship for R324A (IC50 = 20.1 ± 1.7 μm) and D326A (IC50 = 17.9 ± 0.9 μm). B, zinc concentration-response relationship for Glu-75 mutants (IC50 values were E75A = 68.2 ± 7.0 μm; E75H = 1.6 ± 0.1 μm).
If either Asp-326 or Glu-75 participates in zinc binding or is near the binding site then replacing it with a histidine might enhance zinc inhibition. This was not the case for D326H, which like D326A had an IC50 slightly right shifted (21.0 ± 0.8 μm, n = 5) compared with wild-type. However, the zinc potency of the E75H mutant was 10-fold greater than for wild-type hP2X2 (Fig. 7B).
Effects of Mutations of Pro-206 on Zinc Inhibition
In the homology model of the hP2X2 receptor, Pro-206 protrudes from the middle vestibule into the space between the three residues of the histidine cluster (Fig. 1B). If this proline impedes interactions between these histidines, then its replacement with other residues might increase the potency of zinc inhibition. As an initial test of this hypothesis, the glycine mutant of Pro-206 was made to minimize potential steric hindrance. The zinc potency of the P206G mutant (6.1 ± 0.8 μm, n = 7) was similar to wild-type hP2X2 (Fig. 8A). However, P206G was unlike wild-type hP2X2 (and most other mutants characterized in this study) in one feature. When tested at EC10 for ATP, wild-type hP2X2 showed no residual ATP-dependent current at saturating zinc, but in P206G at saturating zinc the residual ATP-dependent current was about 25% of the response to ATP alone (Fig. 8A). The failure of saturating zinc to completely inhibit EC10 ATP in P206G is similar to results seen in wild-type hP2X2 when high ATP was used. For instance, at the EC90 for ATP, maximal zinc did not eliminate ATP responses, but rather only reduced them to 29% of the response without zinc (7). The explanation proposed for this was that the inhibition by zinc was allosteric. That is, zinc binding lowered the potency of ATP, rather than blocking the channel. Viewed in this way, the lowest concentration of ATP that can begin to overcome maximal zinc is an indicator of the efficacy of the allosteric interaction. As EC10 ATP could partially overcome maximal zinc in P206G but not in wild-type, these results show that it is possible for the potency of zinc to be much higher than wild-type, and yet the efficacy of allosteric zinc coupling to ATP binding to be lower than wild-type.
FIGURE 8.

Effect of mutations at hP2X2 Pro-206 on zinc potency. A, zinc inhibition of P206G. Because the final value at high zinc was not 0, the four-parameter version of the Hill equation was used to fit these data. The thick line without points in panels A–C is the response of wild-type hP2X2 initially shown in Fig. 4. B, zinc inhibition of P206H (IC50 = 0.09 ± 0.02 μm, n = 13). C, zinc inhibition of P206C.
The P206H mutant increased the potency of zinc inhibition about 100-fold compared with wild-type hP2X2 and was completely inhibited by saturating zinc (Fig. 8B). Possible explanations for this effect are that a histidine at position 206 enhances the potency of the endogenous zinc-binding site, or that it creates a novel zinc-binding site in this region. If either idea is correct then cysteine, which can also ligate zinc, might have a similar effect on zinc potency and covalent modification of this cysteine might modify zinc potency. As a covalent modifier, we used MTSET, which was originally tested on cysteine-modified nicotinic acetylcholine receptors (21) and has subsequently been used extensively to probe the accessibility of specific residues of many ion channels, including P2X receptors (15, 16). One key property of MTSET is that it reacts specifically and rapidly with the thiols of free cysteines to form mixed disulfides and so blocks any function that requires a free thiol (such as metal binding). A second is that because MTSET is positively charged and relatively bulky, it can often alter permeation through narrow regions of channels due to steric hindrance or charge repulsion of permeant cations. Finally, the mixed disulfide bond is usually stable in the absence of an exogenous reducing agent, producing quasi-irreversible modification (21).
The ATP potency of P206C (5.7 ± 0.5 μm, n = 47) was similar to wild-type hP2X2. When cells were tested at their EC10 for ATP, the zinc potency of the P206C mutant was extremely high (IC50 = 11.2 ± 1.6 nm, n = 17, Fig. 8C). Thus P206C has zinc potency 1,000 times higher than wild-type hP2X2 and about 10 times higher than P206H. However, like the P206G mutant, the ATP-dependent currents did not decline to 0 at saturating zinc (average steady-state current was 24 ± 3% of the peak).
An unexpected feature of the P206C mutation was that the responses to saturating ATP were usually small (<3 μA). In contrast, wild-type P2X2 injected with comparable amounts of RNA typically give saturating ATP responses over 30 μA. This was not because the mutant protein had failed to be highly expressed on the cell surface, as when MTSET was applied with the goal of covalently modifying this cysteine, the ATP-evoked currents were massively increased (Fig. 9). For a population of cells studied near their EC10 for ATP prior to MTSET application, the average potentiation was 116 ± 14-fold (n = 76). It previously had been demonstrated that MTSET had no significant effect on wild-type hP2X2 (7). As a further control that the effect of MTSET on P206C was due to modification of the cysteine at this position, we also applied MTSET to P206H. After MTSET treatment there was no significant change in the amplitude of the peak current elicited by saturating ATP, the EC50 for ATP or the IC50 for zinc in oocytes expressing P206H, so the effect on P206C was specific.
FIGURE 9.

Effect of MTSET on responses of hP2X2 P206C to ATP and zinc. A, massive, but transient potentiation of ATP responses by MTSET. Top, after an initial 10-s application of ATP (gray box) MTSET was applied for 2 min (black box), and after a 30-s washout, 10-s ATP pulses were given once per minute. After 15 ATP pulses, MTSET was reapplied, and then a final ATP pulse was given. Bottom, the first ATP pulse after MTSET was delayed until 15 min after the washout of MTSET. B, histogram of the time constant of loss of potentiation after MTSET for a series of cells tested as in the top panel of A. C, more rapid loss of potentiation when a long ATP pulse was used. D, histogram of the fold-potentiation after MTSET when tested with ATP concentrations near the EC10 prior to MTSET treatment (top) or with ATP concentrations that caused a nearly maximal response prior to MTSET treatment (bottom). E, traces illustrating the shift in the ATP concentration-response relationship after MTSET treatment. At both concentrations 200 μm produced a maximal response. MTSET treatment shifted the response to 1 μm from 9% of maximal to 81% of maximal. The traces inset within the box show the responses to 200 μm normalized to the same peak amplitude, to accentuate the relative change in the response to 1 μm ATP. F, histogram of the EC50 for ATP before and immediately after MTSET from experiments done as in panel E. G, recordings illustrating the change in zinc inhibition immediately after MTSET treatment. For each panel 4–5 traces obtained under the indicated conditions were normalized to the peak current and then averaged. The gray traces in the panels with zinc represent the extrapolated response amplitudes with ATP only. The top and bottom of the vertical dashed lines indicate the amplitude of the responses with and without zinc that were used to calculate the extent of zinc inhibition. In all panels, ATP was 1 μm. The time calibration applies to all panels. H, zinc concentration-response relationship measured after MTSET calculated from data collected and measured as in G, The thick black line is the fit from Fig. 8C. The thin black line is the fit to the model described in text. The concentration of ATP used in this experiment was the EC10 for ATP prior to MTSET, so it was near the EC80 after MTSET.
Although the effect of MTSET usually reverses very slowly in the absence of an exogenous reducing agent, this was not the case at P206C. When cells were tested with 10-s ATP pulses once per minute, the ATP-dependent currents declined back toward the pre-MTSET amplitude along a relatively rapid, exponential time course (Fig. 9A). The time constant of this return to baseline varied substantially (Fig. 9B) but the average time constant (2.6 ± 0.6 min, n = 17) indicated that by 15 min after MTSET treatment most cells tested with this protocol would be close to the original baseline. The decline of current during a long, continuous application of ATP (Fig. 9C) was much faster (time constant = 32.7 s ± 0.7 s, n = 5) than when ATP was applied as a series of 10-s pulses, suggesting that the decline in potentiation might be dependent on activation of the channel by ATP. To test this possibility, in a series of cells the first ATP test pulse after MTSET was delayed until 15 min of washout had occurred (Fig. 9A, bottom). For a series of five cells in which the average potentiation above the pre-MTSET response was 49.8-fold, 15 10-s pulses of ATP at 1 per min brought the average potentiation down to only 2.9-fold above the pre-MTSET response. In contrast, waiting 15 min without applying ATP left the average potentiation at 53.8-fold (n = 4) suggesting that no recovery occurred in the absence of ATP. Once the currents had returned to near baseline, they could be re-potentiated by a second dose of MTSET (Fig. 9A, top). For the set of five cells that had declined to 2.9-fold potentiation at 15 min post-MTSET, the second dose of MTSET took the currents back up to 22.7-fold potentiation. Thus, either the bond is rapidly cleaved due to the conformation taken on when ATP is present, or MTSET causes its potentiating effect by interacting with P206C in a noncovalent manner and can only be released when the channel enters the activated state.
The fold-potentiation by MTSET depended on the concentration of ATP at which the measurement was made (Fig. 9D) and was much greater at lower ATP, but substantial potentiation was still present when a concentration that produced a maximal ATP response prior to MTSET treatment was used (11.9 ± 1.3-fold, n = 65).
When low and high concentrations of ATP were applied (Fig. 9E) it became clear that ATP was much more potent immediately after MTSET. In the example shown, 1 μm, a concentration that produced about an EC10 response prior to MTSET instead produced an EC80 response. From a series of two pulse experiments we estimated the ATP potency. Although there was considerable cell to cell variability in the EC50 for ATP, on average MTSET treatment caused a 10-fold increase in the potency of ATP (Fig. 9F) from 5.7 ± 0.4 (n = 58) to 0.57 ± 0.04 μm (n = 56).
A similar two-pulse paradigm allowed us to estimate the potency of zinc inhibition. For these experiments we used an ATP concentration near the pre-MTSET EC10 (0.5 or 1 μm). Prior to MTSET treatment this concentration of ATP gave nearly stable responses, and 200 nm zinc was sufficient to maximally decrease the response, which as noted above plateaus at about 25% of the initial amplitude (Fig. 9G, left panels). After MTSET treatment, the response to even a brief application of ATP ran down as receptors returned to the unmodified state, so measuring the extent of zinc inhibition required us to compare the current amplitude at the end of the zinc application to the amplitude that would have occurred had ATP alone been applied (Fig. 9G, right panels). Oocytes treated with ATP plus 200 nm zinc showed a small, but significant decline. However, the extent of the decline did not change in a concentration-dependent manner as the zinc concentration increased from 200 nm to 20 μm (Fig. 9H). To exceed 50% inhibition shortly after MTSET treatment required over 500 μm zinc.
The complex shape of the zinc concentration-response relationship was readily explained with a model that took into account the time course of decay of potentiation in the presence of ATP (Fig. 9C) and the observation that at maximal ATP, channels in the MTSET-modified state carry about 10 times as much current as unmodified channels when studied at equivalent points on the concentration-response relationship. At the time we applied zinc (30 s of washout plus 20 s of ATP alone), the decay time constant indicated that the population consisted of ∼20% modified receptors and 80% receptors that had already returned to the unmodified state. Even though the majority of receptors were in the unmodified state, the amplitude of the current at this time was dominated by the modified receptors, because of their 10 times larger maximal current. The unmodified receptors were maximally inhibited (see Fig. 8C) by the lowest concentration of zinc we tested after MTSET (200 nm) so the concentration-response curve did not begin to fall until the unmodified receptors began to be affected. Fitting this model gave zinc potency for the MTSET-modified receptors of 577 ± 78 μm, which is a 50,000-fold decrease in potency from the pre-MTSET condition.
Because of the shift in ATP potency after MTSET treatment, these measurements were made near the EC80 for ATP. It was not practical to examine the zinc concentration-response relationship before and after MTSET at the post-MTSET EC10, because untreated P206C expressing oocytes did not reliably give measurable responses to ATP at the post-MTSET EC10. When we corrected for the effect of using a relatively high ATP concentration of ATP as described under “Experimental Procedures,” the predicted zinc IC50 at the post-MTSET EC10 for ATP was 76 μm, which is still a decrease in zinc potency of over 6,000-fold.
Lack of Effect of Cross-linking on Zinc Inhibition
The growth of current in the P206C mutant after MTSET is a property shared with some other cysteine-modified residues in the immediate vicinity. In hP2X2, MTSET modification of H209C caused a substantial increase in current, although modification of H204C with this reagent produced inhibition of current (7). Similarly, in rP2X2, MTSET modification of H319C, the residue equivalent to human H330C, caused a modest (less than 2-fold) increase in current (15). We therefore tested the effect of MTSET on hP2X2 H330C. When tested before and after MTSET at the pre-MTSET EC10 for ATP, the amplitude of currents from H330C expressing oocytes were potentiated 12.4 ± 1.2-fold (n = 7). In part this was due to an increase in ATP potency from an EC50 of 7.0 ± 0.6 μm (n = 4) to 1.5 ± 0.8 μm (n = 5), but the current at maximal ATP was also increased 2.2 ± 0.4-fold. The IC50 of zinc inhibition was slightly right shifted after MTSET treatment (from 3.4 ± 0.3 to 6.5 ± 0.9 μm). However, when the change in the ATP concentration-response relationship was corrected, the predicted zinc potency at the post-MTSET EC10 for ATP was nearly 5-fold higher (0.69 μm). Thus modifying this cysteine to give it a substantially larger volume did not impair zinc inhibition.
Finally, we tested whether BMOE and BM(PEG)3, two reagents that were demonstrated in Fig. 2 to form cross-links across the subunit interfaces, altered receptor function (Table 2). BMOE had no effect on any of the measured properties of the H209C/H330C mutant, even though the companion biochemical studies indicated that these double mutant receptors were extensively cross-linked by this treatment. In contrast, for the H204C/H209C and H204C/H330C mutants BMOE reduced the currents in response to saturating ATP to ∼50% of controls, providing physiological evidence that BMOE binding had occurred. However, again there was no significant change in either the ATP or zinc concentration-response relationship. One possible explanation for the lack of change is that the BMOE-modified H204C/H209C and H204C/H330C receptors were completely nonfunctional. Another is that cross-linking at this location did not alter ATP or zinc potency. The results were less ambiguous for BM(PEG)3. This compound had little effect on wild-type hP2X2, as expected because there are no free cysteines on the extracellular side. As with BMOE, there was no change in zinc inhibition in either double mutant after exposure to BM(PEG)3. However, in this case we know that BM(PEG)3-bound receptors were functional, as after treatment the amplitude of the ATP-evoked currents from both H204C/H209C and H204C/H330C were dramatically potentiated, similar to the effect of MTSET on H209C and H330C. Therefore we conclude that zinc inhibition does not require these cysteines to be unmodified, nor to move any further than BM(PEG)3 would allow and infer that this is also the case for the endogenous histidines.
TABLE 2.
Effect of the cross-linkers BMOE and BM(PEG)3 on zinc inhibition of double C mutants in the histidine cluster
The same oocytes were studied before and after treatment.
| Construct | Current at maximal ATP |
ATP EC50 |
Zinc IC50 |
Number of cells | ||
|---|---|---|---|---|---|---|
| Post-BMOE/Pre-BMOE | Before BMOE | After BMOE | Before BMOE | After BMOE | ||
| μm | μm | |||||
| H204C/H209C | 44 ± 6% | 9.9 ± 1.6 | 11.3 ± 0.9 | 11.2 ± 1.8 | 6.5 ± 0. 6 | 5 |
| H204C/H330C | 47 ± 4% | 7.6 ± 0.6 | 9.2 ± 0.3 | 6.0 ± 0.4 | 4.8 ± 0. 7 | 3 |
| H209C/H330C | 97 ± 8% | 5.5 ± 0.6 | 7.9 ± 0.7 | 8.3 ± 0.8 | 8.2 ± 1.2 | 4 |
| Construct | Current at 10 μm ATP |
Zinc IC50 |
Number of cells | |
|---|---|---|---|---|
| Post-BM(PEG)3/Pre-BM(PEG)3 | Before BM(PEG)3 | After BM(PEG)3 | ||
| μm | ||||
| hP2X2 | 90 ± 1% | 19.5 ± 3.1 | 19.8 ± 1.3 | 3 |
| H204C/H330C | 363 ± 59% | 3.3 ± 0.4 | 1.7 ± 0.4 | 3 |
| H209C/H330C | 338 ± 31% | 3.8 ± 0.5 | 6.0 ± 0.7 | 3 |
DISCUSSION
The biological role of zinc inhibition of hP2X2 is unknown, in part because no mutations that significantly alter this modulation had previously been described (7). In seeking a region of the receptor that might be essential for zinc inhibition, our attention was focused on the histidine cluster (His-204/His-209/His-330) because the homology model to zP2X4.1 suggested that these potential zinc-binding residues were close to each other across a subunit interface, and because there was an interesting amino acid difference in this region between the human receptor, which has high potency zinc inhibition and the rat receptor, which has much lower potency zinc inhibition. By focusing on this region of hP2X2, we have been able to produce receptors that respond relatively normally to ATP, and yet spread the sensitivity to inhibition by zinc over a 100,000-fold range. At one extreme, P206C has an IC50 of about 10 nm and at the other extreme H204A/H209A/H330A has an IC50 of about 1 mm. Other mutant receptors spread zinc potency fairly evenly across this range (P206H near 100 nm, E75H near 1 μm, wild-type near 10 μm, and H209K and E75A near 100 μm). Two issues of interest are what these results indicate about the nature of zinc binding to hP2X2, and how these mutant receptors might be used to further our understanding of the in vivo role of P2X2 receptors.
One surprising feature that we have not yet explored experimentally is that unlike wild-type hP2X2 and the other mutants studied, in H330A modest potentiation by zinc was uncovered at high ATP and zinc. A possible explanation for this result is that some of the residues that bind zinc to cause potentiation in rP2X2 are retained in hP2X2 (7) and that under appropriate conditions H330A allows a residual bit of activity at this site to be detected. Potentially relevant to this issue is that His-330 (and its rat equivalent His-319) is known to be required for pH potentiation, and so this site clearly has an influence on the gating machinery (7, 13). Alternatively, it is possible that in the presence of zinc, the H330A channels are able to slowly enter the same enhanced conductance state that results from binding MTSET to H209C, H330C, or P206C. Whatever the cause, the 2-fold potentiation shown by this mutant indicates that it is very inefficient at potentiation as compared with zinc potentiation of rat P2X2 and MTSET potentiation of H209C, H330C, or P206C, which can be over 100-fold when low concentrations of ATP are used.
Although mutations in the vicinity of the histidine cluster had a dramatic effect on zinc potency, it remains unclear how they did this. One possible interpretation of the results with the H204A/H209A/H330A mutant is that these histidines constitute most of the high potency binding site, and the very low potency of zinc inhibition in this mutant represents residual binding to other residues near them. A related possibility is that these residues are the entire high potency site, and that the residual inhibition is caused by low potency binding elsewhere. Consistent with these ideas, when reciprocal changes were made at the position in the histidine cluster that varies between humans and rats (hHis-209 or rLys-197), the zinc potency was reciprocally shifted. Similarly, when Pro-206, which in the closed state model sits between these histidines, was changed to known zinc binding residues (His or Cys) the potency of zinc inhibition was greatly increased. Finally, MTSET modification of P206C mutant receptors reversibly decreased zinc potency by more than a factor of 50,000. However, two results previously reported as well as several additional results reported here are difficult to explain if this cluster is the zinc-binding site. First, Tittle and Hume (7) found that no single His to Ala mutation had more than a modest effect on zinc inhibition, nor did the double mutant H204A/H209A. Second, they reported that when either His-204 or His-209 are mutated to cysteine, the mutants retain potent zinc inhibition that is only subtly altered after MTSET is bound. In contrast, in the high potency potentiating zinc-binding site of rP2X2, mutating single essential histidines nearly obliterates the modulation and binding MTSET to these sites greatly attenuates it (14). We verified the previous results and extended them by showing that MTSET did not significantly interfere with the ability of H330C to respond to zinc and might even enhance it. Furthermore, cross-linking with BMOE or BM(PEG)3 failed to interfere with zinc inhibition. Finally, although BMOE cross-linking showed that cysteines at these locations can come within 8 Å of each other, to bind zinc they would have to come within 2–3 Å (22). The S-S distance in a disulfide bond is also between 2 and 3 Å, so the failure of H2O2 to accomplish cross-linking suggests that H330C does not get close enough to either H204C or H209C to directly engage in zinc binding.
If the histidine cluster is not the zinc-binding site, how might modifications to this region increase or decrease the potency of zinc inhibition and where might the binding site be? The homology model suggests that His-204 and His-209 sit just outside a potential entry into the middle vestibule, and His-330 sits just inside it, so these residues are perfectly positioned to control access of zinc to a binding site in the middle or upper vestibule. We therefore tested all of the most common zinc-binding residues in these vestibules. The inhibitory zinc-binding site is unlikely to be in the upper vestibule, as simultaneous mutation of all candidate zinc-binding residues had virtually no effect in either hP2X2 or in humanized rP2X2. As far as the middle vestibule, it is noteworthy that in zP2X4.1 (2) this region has a gadolinium-binding site that is believed to be the cause of Gd3+ inhibition of this channel. The homologous position of hP2X2 is a glycine, and so zinc cannot bind there. Of the highest likelihood candidates for binding zinc within the middle vestibule, mutation of Glu-326 and His-330 had only minor effects on zinc potency but the substantial effects of mutation of Glu-75 make it an appealing candidate for participation in zinc binding. If so, additional residues would be needed, and the most likely untested candidates are serines, which although extremely rare in nanomolar affinity structural zinc-binding sites (<0.1%) contribute to zinc binding in about 4% of the lower affinity catalytic zinc sites (23). Candidates are Ser-76, Ser-77, and Ser-106, all of which are close to Glu-75 in our homology model. In summary, a plausible explanation for the loss of zinc potency in H204A/H209A/H330A and MTSET-modified P206C is that in these mutants zinc cannot enter the middle vestibule, even though the binding site is intact.
Considerable evidence suggests that in the mammalian central nervous system P2X2 receptors are expressed both on neurons and glia (10, 24), but the CNS phenotype of mouse P2X2 knock-outs is quite modest (12). The mutants we have characterized have several potential uses in understanding the role of P2X receptors, and how this role is modulated by zinc. It is estimated that the typical concentration of free extracellular zinc in the brain is ∼20 nm (25), which would have no detectable effect on wild-type hP2X2 or rP2X2. However, the zinc concentration in the synaptic cleft beneath terminals that express the ZNT-3 vesicular zinc transporter has been estimated to go above 10 μm (26), so any wild-type P2X2 receptors on neurons or glia close to such synapses would be expected to be modulated whenever ATP and zinc are simultaneously present. Three mutants seem most promising. First, hP2X2 H204A/H209A/H330A responds normally to ATP, but is effectively zinc insensitive over the range that can occur in the brain. Thus expressing this mutant as a replacement for wild-type mouse P2X2 in knock-in replacement animals or in acutely transfected slices from P2X2 knock-out mice seems a potentially useful way to explore what occurs without zinc modulation. Conversely, P206H has greatly heightened zinc sensitivity without compromising ATP responsiveness, such that it would be expected to be inhibited (although not maximally) by basal zinc levels, and thus could be used to chronically lower P2X2 “tone.” Last, P206C is sufficiently zinc sensitive that it should be essentially nonfunctional at basal zinc levels. However, when modified by MTSET it can be reversibly converted to a state that is nearly zinc insensitive, but has enhanced ATP sensitivity. Thus, if a way could be found to express these P206C receptors in a system where access by exogenously applied substances is possible (such as in thin brain slices or in cell culture), it would be possible to observe the same set of interactions with and without functional P2X2 receptors within just a few minutes by manipulating the zinc and MTSET levels.
Acknowledgments
We thank Connie Truong for preparing oocytes and other skilled technical assistance. We thank Drs. Haoxing Xu, Matthew Chapman, and Gabrielle Rudenko for helpful comments on the studies reported here.
This work was supported, in whole or in part, by National Institutes of Health Grant NS039196 (to R. I. H.) and an Okkelberg Fellowship from the University of Michigan (to S. P.).
- rP2X2
- rat P2X2 receptor
- hP2X2
- human P2X2 receptor
- BM(PEG)3
- 1,11-bismaleimidotriethyleneglycol
- BMOE
- bismaleimidoethane
- MTSET
- (2-(trimethylammonium)ethyl)methanethiosulfonate chloride
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. North R. A. (2002) Molecular physiology of P2X receptors. Physiol. Rev. 82, 1013–1067 [DOI] [PubMed] [Google Scholar]
- 2. Kawate T., Michel J. C., Birdsong W. T., Gouaux E. (2009) Crystal structure of the ATP-gated P2X4 ion channel in the closed state. Nature 460, 592–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Coddou C., Stojilkovic S. S., Huidobro-Toro J. P. (2011) Allosteric modulation of ATP-gated P2X receptor channels. Rev. Neurosci. 22, 335–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nakazawa K., Ohno Y. (1996) Dopamine and 5-hydroxytryptamine selectively potentiate neuronal type ATP receptor channels. Eur. J. Pharmacol. 296, 119–122 [DOI] [PubMed] [Google Scholar]
- 5. Wildman S. S., King B. F., Burnstock G. (1998) Zn2+ modulation of ATP-responses at recombinant P2X2 receptors and its dependence on extracellular pH. Br. J. Pharmacol. 123, 1214–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clyne J. D., LaPointe L. D., Hume R. I. (2002) The role of histidine residues in modulation of the rat P2X2 purinoceptor by zinc and pH. J. Physiol. 539, 347–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tittle R. K., Hume R. I. (2008) Opposite effects of zinc on human and rat P2X2 receptors. J. Neurosci. 28, 11131–11140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Palmiter R. D., Huang L. (2004) Efflux and compartmentalization of zinc by members of the SLC30 family of solute carriers. Pflugers Arch. 447, 744–751 [DOI] [PubMed] [Google Scholar]
- 9. Mathie A., Sutton G. L., Clarke C. E., Veale E. L. (2006) Zinc and copper. Pharmacological probes and endogenous modulators of neuronal excitability. Pharmacol. Ther. 111, 567–583 [DOI] [PubMed] [Google Scholar]
- 10. Burnstock G., Knight G. E. (2004) Cellular distribution and functions of P2 receptor subtypes in different systems. Int. Rev. Cytol. 240, 31–304 [DOI] [PubMed] [Google Scholar]
- 11. Köles L., Leichsenring A., Rubini P., Illes P. (2011) P2 receptor signaling in neurons and glial cells of the central nervous system. Adv. Pharmacol. 61, 441–493 [DOI] [PubMed] [Google Scholar]
- 12. Cockayne D. A., Dunn P. M., Zhong Y., Rong W., Hamilton S. G., Knight G. E., Ruan H. Z., Ma B., Yip P., Nunn P., McMahon S. B., Burnstock G., Ford A. P. (2005) P2X2 knock-out mice and P2X2/P2X3 double knock-out mice reveal a role for the P2X2 receptor subunit in mediating multiple sensory effects of ATP. J. Physiol. 567, 621–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clyne J. D., Wang L. F., Hume R. I. (2002) Mutational analysis of the conserved cysteines of the rat P2X2 purinoceptor. J. Neurosci. 22, 3873–3880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nagaya N., Tittle R. K., Saar N., Dellal S. S., Hume R. I. (2005) An intersubunit zinc binding site in rat P2X2 receptors. J. Biol. Chem. 280, 25982–25993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kawate T., Robertson J. L., Li M., Silberberg S. D., Swartz K. J. (2011) Ion access pathway to the transmembrane pore in P2X receptor channels. J. Gen. Physiol. 137, 579–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Samways D. S., Khakh B. S., Dutertre S., Egan T. M. (2011) Preferential use of unobstructed lateral portals as the access route to the pore of human ATP-gated ion channels (P2X receptors). Proc. Natl. Acad. Sci. U.S.A. 108, 13800–13805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace. A web-based environment for protein structure homology modeling. Bioinformatics. 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 18. Bordoli L., Kiefer F., Arnold K., Benkert P., Battey J., Schwede T. (2009) Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 4, 1–13 [DOI] [PubMed] [Google Scholar]
- 19. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Clyne J. D., Brown T. C., Hume R. I. (2003) Expression level dependent changes in the properties of P2X2 receptors. Neuropharmacology. 44, 403–412 [DOI] [PubMed] [Google Scholar]
- 21. Akabas M. H., Stauffer D. A., Xu M., Karlin A. (1992) Acetylcholine receptor channel structure probed in cysteine-substitution mutants. Science 258, 307–310 [DOI] [PubMed] [Google Scholar]
- 22. Alberts I. L., Nadassy K., Wodak S. J. (1998) Analysis of zinc-binding sites in protein crystal structures. Protein Sci. 7, 1700–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Andreini C., Bertini I., Cavallaro G. (2011) Minimal functional sites allow a classification of zinc sites in proteins. PloS One 6, e26325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Illes P., Alexandre Ribeiro J. (2004) Molecular physiology of P2 receptors in the central nervous system. Eur. J. Pharmacol. 483, 5–17 [DOI] [PubMed] [Google Scholar]
- 25. Frederickson C. J., Giblin L. J., Krezel A., McAdoo D. J., Muelle R. N., Zeng Y., Balaji R. V., Masalha R., Thompson R. B., Fierke C. A., Sarvey J. M., de Valdenebro M., Prough D. S., Zornow M. H. (2006) Concentrations of extracellular free zinc (pZn)e in the central nervous system during simple anesthetization, ischemia, and reperfusion. Exp. Neurol. 198, 285–293 [DOI] [PubMed] [Google Scholar]
- 26. Sindreu C., Storm D. R. (2011) Modulation of neuronal signal transduction and memory formation by synaptic zinc. Front. Behav. Neurosci. 5, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]