

Background: Mitochondrial dysfunction may influence myocardial remodeling in diabetes.
Results: Impaired respiratory complex activity and energy depletion without mitochondrial uncoupling was identified in a new conplastic rat model of diabetes.
Conclusion: mtDNA mutations are a risk factor for developing myocardial dysfunction in diabetes.
Significance: Identifying mtDNA background is important for recognizing patients at risk for heart diseases.
Keywords: ATP, Biomarkers, Cardiomyopathy, Diabetes, Heart Failure, Mitochondrial DNA, Conplastic Animal
Abstract
Myocardial remodeling and dysfunction are serious complications of type 2 diabetes mellitus (T2DM). Factors controlling their development are not well established. To specifically address the role of the mitochondrial genome, we developed novel conplastic rat strains, i.e. strains with the same nuclear genome but a different mitochondrial genome. The new animals were named T2DNmtFHH and T2DNmtWistar, where the acronym T2DN denotes their common nuclear genome (type 2 diabetic nephropathy (T2DN) rats) and mtFHH or mtWistar the origin of their mitochondria, Fawn Hooded Hypertensive (FHH) or Wistar rats, respectively. The T2DNmtFHH and T2DNmtWistar showed a similar progression of diabetes as determined by HbA1c, cholesterol, and triglycerides with normal blood pressure, thus enabling investigation of the specific role of the mitochondrial genome in cardiac function without the confounding effects of obesity or hypertension found in other models of diabetes. Echocardiographic analysis of 12-week-old animals showed no abnormalities, but at 12 months of age the T2DNmtFHH showed left ventricular remodeling that was verified by histology. Decreased complex I and complex IV but not complex II activity within the electron transport chain was found only in T2DNmtFHH, which was not explained by differences in protein content. Decreased cardiac ATP levels in T2DNmtFHH were in agreement with a lower ATP synthetic capacity by isolated mitochondria. Together, our data provide experimental evidence that mtDNA sequence variations have an additional role in energetic heart deficiency. The mitochondrial DNA background may explain the increased susceptibility of certain T2DM patients to develop myocardial dysfunction.
Introduction
Type 2 diabetes mellitus (T2DM)2 is a complex and heterogeneous disorder with a prevalence nearing epidemic proportions worldwide. Myocardial dysfunction and heart failure are serious complications developing in many cases independently of coronary artery disease and hypertension. It is considered that molecular alterations at the myocyte level may be a key factor in the development of myocardial dysfunction. The pathogenesis of ventricular dysfunction has been examined previously in genetic animal models of T2DM (1–3). Because these models present with a variety of metabolic disorders, it has been impossible to isolate the influence of intrinsic mitochondrial impairments in disease mechanisms.
A new rat model of type 2 diabetes mellitus was developed by crossing diabetic male Goto-Kakizaki (GK) rats with female Fawn Hooded Hypertensive (FHH) rats (4). This new T2DM rat model was named T2DN, which is a non-obese rat model that develops diabetes spontaneously as GK rats but with proteinuria and abnormal renal histological findings (4). Genetic analysis indicated that the two major differences between GK and T2DN are passenger loci (3%) and mtDNA (4). Specific sequence analysis of mtDNA showed that the T2DN rats carry mitochondria from FHH, whereas the GK carries mitochondria from Wistar Kyoto (WKY/NCrl) rats (5). To better understand the relationship between different mtDNA genomes and phenotypical manifestations in the setting of diabetes, we transferred the mitochondrial genome from GK to T2DN rats. This crossing produced a progeny with comparable nuclear genome (T2DN) as judged by genome scan but with a total change in mtDNA from FHH to Wistar. In this work, we refer to these new conplastic models as T2DNmtFHH and T2DNmtWistar, which denote their common nuclear genome but different mitochondrial background.
Previous work has shown that mild cardiac dysfunction in GK/UK (University of Wales College of Medicine, Cardiff, UK) can be induced by hypoxia supporting the notion that functional alteration at the mitochondrial level is a proximal cause of myocyte dysfunction (6). In patients with diabetes, the prevalent utilization of fatty acids to glucose is thought to promote energy deficiency resulting from mitochondrial lipotoxicity, decreased biogenesis, and uncoupling (1, 7). Whether a defective ability of the heart mitochondria to supply critical energetic demand plays a role in cardiac remodeling is still an unresolved matter. The availability of T2DNmtFHH and T2DNmtWistar rats offers a unique opportunity to address the influence of mitochondrial genome in the cardiac energy state and the susceptibility to ventricular dysfunction in the setting of T2DM. Here, we report that T2DNmtFHH is energetically deprived and demonstrates adverse cardiac remodeling when compared with T2DNmtWistar hearts. The cardiac remodeling develops with an increase in myocyte diameter but in the absence of myocardial fibrosis. These data provide experimental evidence for the concept that mtDNA influences energetic deficiency leading to an increased susceptibility to cardiac dysfunction.
EXPERIMENTAL PROCEDURES
Animals
All animal use protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the Medical College of Wisconsin and conform to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication no. 85-23, revised 1996). Rats were maintained with 12-h light/12-h dark periods with free access to water and standard chow (Purina). Young 12-week-old males were used for the biochemical studies. Both 12-week- and 12-month-old animals were used for the echocardiographic studies.
Blood Analysis
All procedures were performed in the mornings at the same time to minimize diurnal variation. Blood was obtained at the moment of tissue collection by heart puncture after fasting overnight. Measurement of plasma glucose, serum cholesterol, and triglyceride concentrations were performed by Marshfield Laboratories (Marshfhield, WI) as described previously (8). Plasma glucose levels were measured by standard procedures as described (4). Glycosylated hemoglobin (HbA1c) was measured using in2itTM (Bio-Rad) according to the manufacturer's protocol. T2DN rats were subjected to an intraperitoneal glucose tolerance test. Before the test, all animals were subjected to two to three training sessions. After the determination of fasting (12 h) glucose levels, the animals received an injection of 1.0 g/kg of glucose intraperitoneally. Approximately 10-μl blood samples were collected from an incision in the tail at 30, 60, 90, and 120 min after the glucose load. Glucose levels were measured using reagent strips read with a Bayer® glucometer (Elkhart, IN).
Assessment of Cardiac Function by Echocardiography
Twelve-week- and 12-month-old male T2DN rats were evaluated by noninvasive two-dimensional echocardiography. Echocardiograms were recorded under isoflurane anesthesia using a General Electric Vivid 7 system (Waukesha, WI) equipped with an 11 MHz M12L linear transducer. In brief, standard parasternal short axis images were obtained at the mid-left ventricular level (papillary muscles served as markers) by two-dimensional echocardiography with image depth at 2–2.5 cm, 234–256 frames/s acquisition, harmonic imaging, and with electrocardiographic gating. The images were transferred to a workstation for image analysis (EchoPAC workstation with Q analysis software, General Electric, Waukesha, WI). Anatomical M-mode from the two-dimensional images was performed to measure LV dimensions in diastole and systole as well as anterior and posterior wall thickness in diastole. Fractional shortening (FS) was calculated as follows: %FS = (end diastolic LV dimension − end systolic LV dimension)/(end diastolic LV dimension) × 100%. Left ventricular mass (LVM) was calculated using the following formula: LVM = 0.8(1.04{(posterior wall thickness + LV internal diameter + anterior thickness)3 − LV internal diameter thickness3}) + 0.6 (9). Ejection fraction was derived using the Teicholz formula. Global radial and circumferential strain were averaged over six equidistant left ventricular regions using speckle tracking strain imaging as described previously (10–12). Investigators remained blind as to group allocation throughout the echocardiographic measurements.
Histology
Cardiac histology was assessed in perfused T2DN rats at 12 months of age. Whole hearts were excised, weighed, and fixed in 10% formalin solution. Post-fixation, samples were embedded in paraffin, cut into 4-μm-thick sections and stained with hematoxylin & eosin and Masson's trichrome. The sections were examined by light microscopy at ×10 and ×20 magnification. The diameters of the myocytes were analyzed at the longitudinal line of the myocyte nucleus. Approximately 100 cells were measured and analyzed in each section using MetaMorph®.
Transmission Electron Microscopy
Freshly isolated hearts before fixation were perfused by 1% glutaraldehyde and 4% paraformaldehyde in 0.1 m sodium phosphate buffer, pH 7.4. A portion of apex was rapidly cut into 1-mm3 pieces and placed in cold 2.5% glutaraldehyde fixative in 0.1 mol/liter cacodylate buffer, pH 7.4, and stored at 4 °C. Post-fixation, tissues were embedded in EmBED812 epoxy resin, and sections were cut at a thickness of 60 nm and mounted on 200 mesh copper grids, stained in uranyl acetate and lead citrate, observed, and digital images recorded using ×8000 and ×29,000 magnifications at the MCW Electron Microscopy Core Facility.
Biochemical Measurements
Adenine Nucleotide Levels
The tissue adenine nucleotide pool was quantified by high performance liquid chromatography (HPLC) with UV detection (Agilent 1100, diode array) in freshly isolated hearts. Briefly, a section of beating heart tissue from 12-week-old T2DN rats (<50 mg) was transferred to a 2-ml microcentrifuge tube containing ice-cold perchloric acid (5%) (13). The tissue was immediately homogenized, and the acidic homogenate was kept on ice for 30 min. It was then neutralized with 1 m K2HPO4 and kept on ice for an additional 30 min to promote protein precipitation. Finally, the mixture was centrifuged at 12,000 × g for 10 min. Supernatants were used for HPLC assay, and the pellet was used for protein assay (13). Samples were resolved on a Kromasil C-18 column using a gradient of tetrabutylammonium bisulfate in phosphate buffer, pH 6.0, and methanol as described before (14). Pellets were dissolved in 0.5 m NaOH and protein content was determined using the Bio-Rad protein assay protocol. Concentrations of ATP, ADP, and AMP were calculated using authentic standards.
PCR Analysis of mtDNA
The copy number of mtDNA and nuclear DNA was determined by quantitative real time PCR, and the ratio of genomic nuclear DNA (nDNA) to mtDNA was calculated. Amplification of specific mtDNA sequences was optimized, and the best results were obtained with the following primers: forward, 5′-CAATTCTCCTAGCACAAGTG, and reverse, 5′-CCCAACCGAAATTTTTTAGTTC, which amplified mitochondrial 16 S rRNA. The Sh2b3 genomic sequence was amplified utilizing the following primers: forward, 5′-GAGCTTGAGTCTGTGAGCAGTG and reverse, 5′-AATGAAGCTACAGGGAAGGACA, which served as an internal control. Amplification curves generated using QuantiTect SYBR Green kit (Qiagen) were analyzed using qBase software (15).
Mitochondrial Isolation
Hearts from 12-week- and 12-month-old rats were processed immediately after excision for mitochondrial isolation at 4 °C, which was performed using standard differential centrifugation techniques as described with slight modifications (16). Briefly, heart tissue was collected and homogenized in modified Chappell Perry medium: 10 mm HEPES, 100 mm KCl, 1 mm EGTA, 5 mm MgSO4, 1 mm ATP, and 0.2% BSA, pH 7.4. Homogenates were centrifuged at 700 × g for 15 min at 4 °C (Sorvall High speed, SA600 rotor). The supernatant was transferred to a cold clean tube (Nalgene) and subjected to a high speed centrifugation (10,000 × g, 15 min, 4 °C). The final supernatant was discarded, and the mitochondrial pellet was washed twice. Finally, the pellets were resuspended in storage buffer: 10 mm HEPES, 100 mm KCl, and 1 mm EGTA, pH 7.4. Mitochondrial preparation was used immediately for functional assays or stored at −80 °C for immunoblot and enzyme assay.
Mitochondrial Proteins Immunoblot
The expression levels of total respiratory complexes in the mitochondria of the T2DN rats were analyzed by Western blotting using the total OXPHOS rodent Western blot antibody mixture (Mitosciences MS604). Briefly, mitochondrial proteins were solubilized into RIPA buffer (50 mm Tris-HCl, pH 7.4, 0.1 mm EDTA, 0.1 mm EGTA, 0.1% SDS, 0.1% deoxycholic acid, 1% IGEPAL CA630, and protein inhibitor mixture). Samples were loaded onto 10–20% Ready gels (Bio-Rad) using a loading buffer without heat treatment. Resolved proteins were transferred to a nitrocellulose membrane and probed with specific antibody mixture. Detection of the reactive protein-antibody complex was performed according to the manufacturer's instructions using the ECL Western blot detection kit and the autoradiographic signals analyzed by ALPHA imager.
Measurement of Mitochondrial Enzyme Activities
The electron transport complex activities in mitochondrial heart preparation were measured spectrophotometrically (17, 18).
NADH:Coenzyme Q Reductase (Complex I + III)
Activity was measured by incubating mitochondrial protein (25 μg) in the presence of 50 mm KH2PO4, 5 mm MgCl2, 5 g/liter BSA, 0.2 mm KCN, and 0.12 mm ferricytochrome c, pH 7.5. NADH (0.15 mm) was added, and the oxidation of NADH by complex I was monitored at 340 nm for 10 min before and after the addition of rotenone (2 mg/liter), and the rotenone-sensitive activity was calculated from the linear portion of the slope using an extinction coefficient of 6.2 mm−1 cm−1.
Succinate:Cytochrome c Reductase (Complex II + III)
Mitochondrial protein (25 μg) was added to the buffer consisting of 50 mm KH2PO4, 5 mm MgCl2, 5 g/liter BSA, 0.2 mm KCN, 30 mm succinate, 2 mg/liter rotenone, and 0.12 mm ferricytochrome c, pH 7.5. Enzyme-catalyzed reduction of cytochrome c was monitored at 550 nm for 10 min. Concentration was calculated from the initial linear portion of the slope using an extinction coefficient of 21 mm−1 cm−1.
Cytochrome c Oxidase (Complex IV)
Activity was measured following the oxidation of ferrocytochrome c. Ascorbic acid in 10 mm phosphate buffer, pH 7.0, was used to reduce cytochrome c. Excess ascorbate was removed by dialyzing against phosphate buffer for 18–24 h at 4 °C with three changes of buffer. Mitochondrial protein (25 μg) was incubated in 50 mm KH2PO4, pH 7.5, 0.03 mm ferrocytochrome c, and 2 mg/liter rotenone. Complex IV activity was measured at 550 nm for 10 min, and the rate was calculated from the initial linear portion of the slope using an extinction coefficient of 21 mm−1 cm−1.
Mitochondrial Respiration
Mitochondrial oxygen consumption was monitored at 37 °C in a thermostatically controlled chamber with magnetic stirring equipped with a Clarke oxygen electrode (Rank Brother's Ltd., Digital Model 10) (16). Freshly isolated mitochondria (1 mg/ml) were resuspended in respiration buffer: 10 mm Tris-HCl, 5 mm MgCl2, 2 mm KH2PO4, 20 μm EGTA, and 250 mm sucrose, pH 7.4 (16). Glutamate (10 mm), malate (5 mm), succinate (5 mm), and palmitoylcarnitine (1 mm) were used to initiate maximal respiration, and state-3 respiration was obtained in the presence of ADP (100 μm) and state-4 respiration after ADP exhaustion. The respiratory rates were normalized to protein content.
Rate of ATP Production in Isolated Mitochondria
Freshly isolated mitochondria (1 μg/μl) in storage buffer: 10 mm HEPES, 100 mm KCl, and 1 mm EGTA, pH 7.4, was incubated in ATP assay mix (Sigma, Luciferase Assay Kit) containing luciferase, luciferin, MgSO4, DTT, EDTA, bovine serum albumin (BSA), and Tricine buffer salts as per manufacturer's protocol. ATP production was stimulated by addition of glutamate (10 mm), malate (5 mm), and ADP (10 μm) at room temperature. ATP-stimulated luciferase activity was followed by the emitted light from luciferin adenylate oxidation, and measurements were performed in a luminometer (Beckman Coulter, DTx 880, Multimode detector) every 20 s for 10 min. A calibration curve was obtained using authentic ATP in the range of 5–25 μm following the same procedure.
Statistics
Results are reported as means ± S.D. Student's paired t test was used to compare continuous variables, and differences between means were considered statistically significant at p values <0.05.
RESULTS
Metabolic Parameters in T2DN Rats
Fasting blood glucose and HbA1c levels indicated the influence of mitochondrial genomes (mtFHH versus mtWistar) on the severity of diabetes. We found similar increased plasma glucose levels along with HbA1c in both strains indicating that these animals endure comparable extended periods of hyperglycemia at 12 weeks of age (supplemental Table S1). Also, intraperitoneal glucose tolerance test at age 52 weeks (12 months) revealed equivalent responses between them (supplemental Fig. S1). The levels of triglycerides were found to be elevated but not significantly different. Body weight was maintained in the normal ranges at all ages (supplemental Table 2).
Assessment of Heart Function
Echocardiographic analysis was carried out on 12-week- and 12-month-old T2DN male rats. Analysis of mean arterial blood pressure through a catheter implanted into the left femoral artery was normal, and heart rate showed no significant differences between the two age groups (supplemental Table S2). At 12 weeks, there was no evidence of cardiac remodeling. However, at 12 months, T2DNmtFHH showed an increase in left ventricular (LV) diastolic and systolic dimension signifying cardiac dilation and remodeling (Fig. 1 and supplemental Table S2). The LV alterations did not coincide with the atrial dimensions suggesting that the adverse LV remodeling has not caused enough left atrium pressure increase to result in a marked left atrium enlargement (Fig. 1). Despite the dilation in the left ventricle, there was preservation of overall left ventricular systolic function as measured by fractional shortening, ejection fraction (supplemental Table S2), global radius, and global longitudinal strain 36 ± 6 versus 35 ± 4% (p = 0.86) and −14 ± 2 versus −12 ± 1% (p = 0.43) in T2DNmtWistar versus T2DNmtFHH, respectively. Left ventricular mass was not significantly different between the two groups, despite the increased LV size in T2DNmtFHH rats.
FIGURE 1.

Cardiac remodeling evidenced as increased left ventricular internal diameter in diastole and systole in 12-month-old T2DNmtFHH by two-dimensional echocardiography analysis. Cardiac function and structural features were assessed in 12-week- and 12-month-old T2DN rats fed a regular diet. Echocardiograms were recorded under anesthesia and temperature controlled conditions. Increased left ventricular internal diameter (LVID) dimensions at 12 months were observed in T2DNmtFHH rats at systole (A) and diastole (B). Left atrial dimension (LAD) was unchanged in T2DNmtFHH hearts (C). The ratio of ventricular weight to body weight in 12-week- and 12-month-old T2DN rats remained unaltered at both age groups (D). Values are mean ± S.E., n = 4; *, p < 0.05.
Myocardial Fibrosis
Histological analysis of heart tissue obtained from 12-month-old rats was performed to establish whether LV remodeling in T2DNmtFHH detected in the echocardiographic analysis could be explained by abnormal myocardial fibrosis. There was no significant evidence of increased collagen deposition in the heart samples (Fig. 2). A significant increase in myocyte diameter, however, was found in the T2DNmtFHH rats in comparison with T2DNmtWistar. This result coincides with the increase in LV dimensions observed in echocardiographic analysis and is consistent with previous findings that increased cardiomyocyte length is associated with eccentric left ventricular remodeling and dilation (19).
FIGURE 2.

Histological analysis of cardiac phenotype in conplastic T2DN rats indicates cardiac remodeling without significant fibrosis. A, hematoxylin & eosin-stained sections showed no significant fibrosis in T2DN hearts. B, Masson trichrome-stained sections ×10 magnification (panels a and b) and ×20 magnification (panels c and d) confirmed minor changes in collagen accumulation. C, heart weight (HWt) to body weight (BWt) ratio was comparable between T2DNmtFHH and T2DNmtWistar rats at 12 months of age. D, quantitative analysis at ×20 magnification of Masson trichrome-stained sections using MetaMorph® revealed increased myocyte diameter in 12-month-old T2DNmtFHH compared with T2DNmtWistar at the same age and same degree of diabetes. Values represent mean ± S.D., *, p < 0.05.
Mitochondrial Biogenesis
Slides from two transmission electron microscopy nonserial sections at ×8000 and ×29,000 magnification were analyzed for mitochondrial number, distribution, and morphology (Fig. 3). Although there were no major morphological changes in either of the sections, we found that mitochondria from T2DNmtFHH were disarranged in comparison with T2DNmtWistar (Fig. 3A). At a larger magnification (Fig. 3A, panels c and d), mitochondria from T2DNmtFHH appeared swollen along with an enlargement of the myofibrils that may reflect the changes in myocyte dimension. There was also a small increase in glycogen deposits in the T2DNmtFHH rats that are not found in the T2DNmtWistar rats (Fig. 3A). The distribution and content of mitochondria in both the sarcolemmal and interfibrillar areas were equivalent as was verified by counting. Total mitochondrial number, an index of mitochondrial biogenesis, was also determined by counting the number of mitochondria present in a given area at comparison with ×8000 magnification. As shown in supplemental Fig. S2, no significant differences were found. We further verified the differences in the mitochondrial number by measuring the ratio of mtDNA relative to genomic nuclear DNA (mtDNA/nDNA), which was 3.7 ± 0.28 and 3.4 ± 0.21 in T2DNmtWistar and T2DNmtFHH, respectively (Fig. 3B). These findings showed that mitochondrial biogenesis in the heart of these two models occurs at similar rates.
FIGURE 3.

Difference between mitochondrial genome in diabetic T2DN rats is associated with variation in cardiac morphology but not mitochondrial DNA content. A, representative transmission electron micrographs from left ventricles of 12-month-old T2DNmtFHH and T2DNmtWistar rats. Tissue sections were analyzed at ×8000 magnification (panels a and b) showing normal myocardial fine structure with myofibrils presenting regular and continuous sarcomere in T2DNmtWistar rats that are disrupted in T2DNmtFHH rats presenting randomly distributed mitochondria between poorly organized myofibrils (thick black arrow); panels c and d, ×29,000 magnification showing rows of mildly electron-dense T2DNmtFHH mitochondria with accumulation of glycogen granules (G, thin black arrows). B, nonsignificant (N.S.) differences in left ventricular mitochondrial DNA copy number between T2DNmtFHH and T2DNmtWistar as verified by quantitative real time PCR. Values are mean ± S.E., n = 5. *, p < 0.05.
Myocardial Energy State in T2DNmtFHH and T2DN mtWistar
The steady state level of total adenine nucleotides (TAN) was examined to evaluate the energetic proficiency of the hearts. TAN was detected by HPLC in freshly isolated left ventricles where phosphorylated nucleotides were quenched by acid treatment during isolation (13). It was determined that TAN in T2DNmtFHH was decreased by ∼35% compared with T2DNmtWistar (Fig. 4A). This decrease was mainly due to a significant decrease in the ATP. The ADP and AMP levels were comparable in both strains. The same TAN values were found if the beating hearts were snap-frozen before perchloric acid treatment. It was found, however, that TAN values in nondiabetic 12-week-old rat hearts were significantly higher (50.8 ± 8.3 nmol of ATP/mg of protein, 35.4 ± 18.1 nmol of ADP/mg of protein, and 5.4 ± 0.5 nmol of AMP/mg of protein) than in T2DN hearts and that T2DNmtFHH is significantly energy impaired (data not shown).
FIGURE 4.
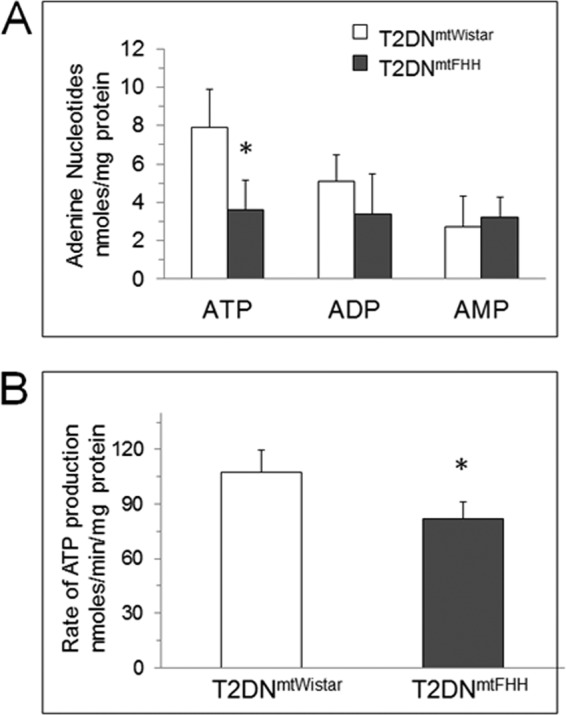
T2DNmtFHH has lower steady state cardiac ATP levels and ATP synthetic capacity than T2DNmtWistar. Steady state concentrations of adenine nucleotides in 12-week-old hearts were determined in freshly obtained tissue immediately stabilized in ice-cold 5% perchloric acid. A, samples were analyzed by reverse phase HPLC and UV integrated values compared with standards are expressed in nanomoles (ATP, ADP, AMP, respectively)/mg of protein. Values represent mean ± S.D., n = 6 hearts; *, p < 0.05. B, ATP synthesis rate determined in freshly isolated mitochondria in isolation buffer: 10 mm HEPES, 100 mm KCl, 1 mm EGTA, 5 mm MgSO4, 1 mm ATP, and 0.2% BSA, pH 7.4, and diluted in storage buffer: 10 mm HEPES, 100 mm KCl, and 1 mm EGTA, pH 7.4. ATP synthesis was stimulated by addition of ADP (10 μm) to reaction mixture containing glutamate (10 mm), malate (5 mm) at room temperature. ATP-dependent bioluminescence was measured using luciferase-luciferin assay (Sigma) in manufacturer-provided buffer containing MgSO4, DTT, EDTA, bovine serum albumin (BSA), and Tricine buffer, pH 7.8. Results are expressed as mean ± S.D., n = 4. *, p < 0.05.
Isolated Mitochondrial ATP Production and Oxygen Consumption
Mitochondrial uncoupling may explain the relative energetic deficiency in the T2DNmtFHH hearts. Thus, we examined both ATP synthesis and oxygen consumption rates in freshly isolated heart mitochondria. Using glutamate + malate as respiratory substrates, it was found that T2DNmtFHH mitochondria consumed oxygen at a lower rate than T2DNmtWistar. The ADP-stimulated mitochondrial oxygen consumption (State 3 respiration) was decreased by 21%, and no differences were found in ADP-exhausted respiration (State 4 respiration) (Table 1). Interestingly, this decrease in respiration rate was evident only with glutamate and malate and not with succinate or palmitoylcarnitine. This suggests that a decrease in oxygen consumption was more pronounced through complex I-dependent respiration as glutamate + malate provide electrons to complex I and succinate provides electrons to complex II. Also, palmitoylcarnitine via β-oxidation and the TCA cycle provide electrons through both complex I and II. Therefore, even if complex I is compromised, complex II-mediated respiration occurs at normal rates. The maximal ATP synthetic capacity of the T2DNmtFHH mitochondria measured with glutamate + malate as substrate was also decreased with respect to T2DNmtWistar mitochondria (Fig. 4B). The decreased oxygen consumption followed by a decreased maximal rate of ATP synthesis using glutamate + malate indicates that mitochondrial uncoupling is unlikely the mechanism of defective energy production in the T2DNmtFHH hearts.
TABLE 1.
Reduced cardiac T2DNmtFHH mitochondrial State 3 respiration rates with glutamate + malate but not succinate and palmitoylcarnitine + malate attest to the deficient mitochondrial complex I activity
Respiration rates were determined in freshly isolated mitochondria from T2DN rat hearts using isolation buffer: 10 mm HEPES, 100 mm KCl, 1 mm EGTA, 5 mm MgSO4, 1 mm ATP, and 0.2% BSA, pH 7.4. Final purified mitochondrial pellets were resuspended in respiration buffer: 10 mm Tris-HCl, 5 mm MgCl2, 2 mm KH2PO4, 20 mm EGTA, and 250 mm sucrose, pH 7.4. Respiration assays were performed at 37 °C with mitochondrial incubations supplemented with the following: (i) 10 mm glutamate and 5 mm malate; (ii) 5 mm succinate; or (iii) 1 mm palmitoylcarnitine plus 5 mm malate and State 3 respiration initiated by the addition of 10 μm ADP. State 4 respiration was calculated after ADP exhaustion from the medium. Values are in nanomoles of oxygen per min per mg of mitochondrial protein. Calculated values represent means ± S.D. from four samples in each group. Respiratory control ratio (RCR) represents the ratio between respiration rate in State 3 and State 4.
| Respiration | Glutamate + malate |
Succinate |
Palmitoylcarnitine + malate |
|||
|---|---|---|---|---|---|---|
| T2DNmtWistar | T2DNmtFHH | T2DNmtWistar | T2DNmtFHH | T2DNmtWistar | T2DNmtFHH | |
| nmol of oxygen/min/mg protein | ||||||
| State-3 | 66.4 ± 4.5 | 54.6 ± 8.6a | 73.0 ± 5.7 | 68.4 ± 6.0 | 60.8 ± 9.2 | 56.5 ± 9.0 |
| State-4 | 16.5 ± 4.8 | 18.3 ± 3.6 | 17.8 ± 6.5 | 22.8 ± 5.5 | 18.2 ± 3.7 | 23.7 ± 4.0 |
| RCR | 4.2 ± 0.9 | 3.1 ± 0.9 | 4.5 ± 1.7 | 3.2 ± 1.1 | 3.5 ± 1.2 | 2.4 ± 0.4 |
a p < 0.05.
Mitochondrial Enzyme Activities in Mitochondria from T2DN Rats
Knowing that mtDNA of FHH carries mutations affecting mitochondrially encoded NADH dehydrogenase 2 (mt-ND2), NADH dehydrogenase 4 (mt-ND4), cytochrome b (mt-CyB), and ATP synthase 6 (mt-ATP6) genes, which are not found in mtDNA from Wistar (5), we hypothesized that changes in ATP production were linked to defective respiratory complex activity. It was established that mitochondrial respiratory complex expression levels were similar between both T2DNmtWistar and T2DNmtFHH mitochondria (supplemental Fig. S3). Furthermore, on examination of the activity of respiratory complexes, it was found that NADH consumption in mitochondrial preparations supplemented with cytochrome c as an acceptor proceeded at lower rates in the T2DNmtFHH than the T2DNmtWistar rats (Fig. 5). This activity was inhibited by rotenone, which indicated the specific contribution of complex I. The succinate-dependent reduction of cytochrome c (complex II + III) was the same between these mitochondria. Cytochrome c oxidase activity (complex IV) was measured by determining the cytochrome c oxidation, which was also decreased in the T2DNmtFHH mitochondria when compared with the T2DNmtWistar. Thus, T2DNmtFHH mitochondria show a significant loss of respiratory complex activity compared with T2DNmtWistar. In summary, a defective energy supply in the T2DNmtFHH is related to a change in respiratory complex activity. This finding is the most significant observation that can sustain myocardial remodeling in T2DNmtFHH hearts, which otherwise diabetes alone cannot fully explain.
FIGURE 5.
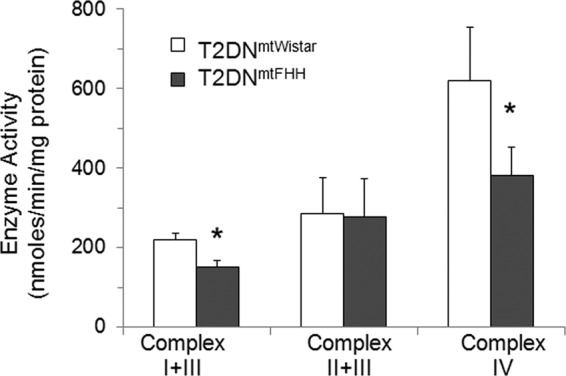
Reduced electron transport chain complex I and complex IV activity in T2DNmtFHH cardiac mitochondria compared with T2DNmtWistar. Mitochondria were isolated from 12-week-old T2DN left ventricles and homogenized in isolation buffer: 10 mm HEPES, 100 mm KCl, 1 mm EGTA, 5 mm MgSO4, 1 mm ATP, and 0.2% BSA, pH 7.4. The purified mitochondrial pellet was dissolved in storage buffer: 10 mm HEPES, 100 mm KCl, and 1 mm EGTA, pH 7.4. Respiratory complex I + III activities were measured spectrophotometrically at 340 nm in assay buffer: 50 mm KH2PO4, 5 mm MgCl2, 5 g/liter BSA, pH 7.5, supplemented with 0.2 mm KCN, 0.12 mm ferricytochrome c, and 0.15 mm NADH. Complex II + III activities were measured in the same assay buffer supplemented with 0.2 mm KCN, 30 mm succinate, 2 mg/liter rotenone, and 0.12 mm ferricytochrome c. The reduction of ferricytochrome c was monitored at 550 nm. Complex IV activity was measured in assay buffer supplemented with 0.03 mm ferrocytochrome c. The reaction was followed at 550 nm, and the amount of ferrocytochrome c oxidized was calculated using an extinction coefficient of 21 mm−1 cm−1. Values represent mean ± S.D., n = 5 mitochondrial preparations in each group. *, p < 0.05.
DISCUSSION
The diabetic heart endures several metabolic challenges during the progression from early to established disease. Generally hyperglycemia and dyslipidemia have been connected to mechanisms of ventricular dysfunction in the absence of coronary artery disease or hypertension (20). Directing these adaptive responses to variations in energy sources is mitochondria itself. Previous work by others supports the notion that decreased glucose utilization in favor of fatty acid by the mitochondria explains cellular damage and loss of function of the heart (6, 21, 22). Mitochondrial dysfunction appears to be an accompanying phenomenon via increased expression of uncoupling proteins and oxidative stress (23, 24). Other mechanisms of mitochondrial dysfunction are related to altered biogenesis (1, 25, 26). Several of these mechanisms have been examined in animal models of diabetes following detailed metabolic and functional assessment. A question that remains not properly addressed, however, is what factor(s) determine(s) patient susceptibility to develop cardiac dysfunction considering that not all diabetic patients will develop cardiac dysfunction and some patients develop dysfunction even with their diabetes being well controlled (27, 28).
Molecular clinical findings indicate that some forms of dilated cardiomyopathy are linked to mitochondrial mtDNA defects (29, 30). The changes at the mtDNA level vary from deletions to point mutations, which generally correspond with the severity of disease. Deletions in mtDNA cause severe forms of the disease with early lethality, and point mutations can cause a whole range of disorders depending on their impact in specific protein and respiratory complexes (30). The significance of mtDNA point mutations in disease is often overlooked as a mechanism because it is difficult to study. In this work, we reasoned that the T2DN rat model of type 2 diabetes mellitus with an identical nuclear genome and a similar level of diabetes but different susceptibility to cardiac remodeling as shown by the echocardiographic analysis could be explained by their different mitochondrial background.
Twelve weeks (when both hyperglycemia and dyslipidemia are borderline high) seems to be an appropriate age to examine the mitochondrial impact on cardiac energy supply. At this age, metabolic changes are just occurring; thus, it is expected to be less influential to the overall energetic proficiency compared with 12-month-old hearts that have undergone changes to accommodate their long periods of hyperglycemia and dyslipidemia. We found that T2DNmtFHH hearts have a decreased tissue ATP level that appears connected to mitochondrial inability to maintain ATP production. Previous work with diabetic mitochondria has indicated the occurrence of mitochondrial uncoupling as a mechanism for low ATP synthetic capacity, mitochondrial damage, and cardiomyocyte death (31, 32). Increased oxygen consumption in T2DNmtFHH mitochondria compared with T2DNmtWistar could not be found indicating that uncoupling is not a likely mechanism. Lower ATP synthetic capacity with diminished activity of the mitochondrial respiratory proteins was also evident. The specific activity of both complex I and complex IV was found to be decreased in T2DNmtFHH with respect to T2DNmtWistar. These changes, however, were not related to a different protein expression levels. Also, this was not associated with alterations in mitochondrial biogenesis, mitochondrial number, or sarcolemmal and interfibrillar distribution.
Sequencing data showed variants in the mtFHH DNA in two genes (mt-ND2 and mt-ND4) that code for proteins forming part of complex I that is constituted by 25 total subunits of which 7 are encoded by mtDNA. Here, we showed that these mutations lead to impaired NADH electron transport ability of the complex resulting in low complex I activity and also decreased ATP synthesis. However, mutations in CytB did not show any effect on the activity of complex III. Although there were no sequence variants for complex IV, its activity was significantly decreased in T2DNmtFHH mitochondria. Furthermore, the decrease in complex IV activity was more marked than for complex I activity. The exact reasons for this difference are not presently known, but the interdependence in activity of the respiratory complexes has been reported previously (33). This phenomenon establishes that complex I influences the behavior of other respiratory complexes typically complex III and/or complex IV. This observation lends support to the notion that formation of supercomplexes of mitochondrial respiratory complexes (respirasomes) is logical for optimization of their electron transfer and energy production (34, 35). A similar mechanism may explain our data on loss of complex IV activity in T2DNmtFHH mitochondria. Further studies on the organization of mitochondrial respirasomes in the T2DNmtFHH hearts will address this important possibility.
We have demonstrated that T2DNmtFHH rats harboring mtDNA point mutations resulting in amino acid substitutions in mt-ND2 (A265T, T304M, and *318H), mt-ND4 (I23T and T356M), mt-cyB (D214N), and mt-ATP6 (E35K) show a defective mitochondrial ATP production. The mutations affecting complex I are suggested to exert a dominant negative effect over the activity of other respiratory complexes like complex IV activity in T2DNmtFHH, which cannot be explained by mutations or mitochondrial uncoupling. The overall effect is to worsen energetic deficiency in the already compromised diabetic hearts.
Although hyperglycemia is an important factor in cardiac remodeling, it is clear that it is not the only driving factor because T2DNmtWistar has a better outcome than T2DNmtFHH hearts. Recent studies checking for pathogenic mutations have identified nonsynonomous mutations in mitochondrial genes encoding mt-ND4 and mt-CyB, cytochrome c oxidase I (mt-CO1), and mt-ATP6 in patients with confirmed and suspected cardiomyopathy (36). Like in T2DNmtFHH, the presence of these mutations in humans may indicate increased susceptibility to undergo cardiac remodeling in a setting of T2DM. Thus, it is possible that mtDNA mutations are the predisposing factor for the development of diabetic cardiomyopathy and further characterization may be helpful in distinguishing patients at increased risk.
Acknowledgments
We are grateful to Dr. K. Broniowska and C. W Wells (MCW Electron Microscopy Core Facility) for their assistance with ATP measurements and transmission electron microscopy, respectively.
This work was supported, in whole or in part, by National Institutes of Health Grant HL-67244 (to J. V. V.). This work was also supported by Postdoctoral Fellow Grant 11POST5480016 (to S. S.) from the American Heart Association.
This article was selected as a Paper of the Week.

This article contains supplemental Figs. S1–S3 and Tables S1 and S2.
- T2DM
- type 2 diabetes mellitus
- GK
- Goto Kakizaki
- T2DN
- type 2 diabetic nephropathy
- FHH
- Fawn Hooded Hypertensive
- mtDNA
- mitochondrial DNA
- CytB
- cytochrome b
- LV
- left ventricular
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- TAN
- total adenine nucleotide.
REFERENCES
- 1. Boudina S., Sena S., Theobald H., Sheng X., Wright J. J., Hu X. X., Aziz S., Johnson J. I., Bugger H., Zaha V. G., Abel E. D. (2007) Mitochondrial energetics in the heart in obesity-related diabetes. Direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 56, 2457–2466 [DOI] [PubMed] [Google Scholar]
- 2. Huss J. M., Kelly D. P. (2005) Mitochondrial energy metabolism in heart failure. A question of balance. J. Clin. Invest. 115, 547–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Asbun J., Villarreal F. J. (2006) The pathogenesis of myocardial fibrosis in the setting of diabetic cardiomyopathy. J. Am. Coll. Cardiol. 47, 693–700 [DOI] [PubMed] [Google Scholar]
- 4. Nobrega M. A., Fleming S., Roman R. J., Shiozawa M., Schlick N., Lazar J., Jacob H. J. (2004) Initial characterization of a rat model of diabetic nephropathy. Diabetes 53, 735–742 [DOI] [PubMed] [Google Scholar]
- 5. Schlick N. E., Jensen-Seaman M. I., Orlebeke K., Kwitek A. E., Jacob H. J., Lazar J. (2006) Sequence analysis of the complete mitochondrial DNA in 10 commonly used inbred rat strains. Am. J. Physiol. Cell Physiol. 291, C1183–C1192 [DOI] [PubMed] [Google Scholar]
- 6. El-Omar M. M., Yang Z. K., Phillips A.O., Shah A.M. (2004) Cardiac dysfunction in the Goto-Kakizaki rat. A model of type II diabetes mellitus. Basic Res. Cardiol. 99, 133–141 [DOI] [PubMed] [Google Scholar]
- 7. Mazumder P. K., O'Neill B. T., Roberts M. W., Buchanan J., Yun U. J., Cooksey R. C., Boudina S., Abel E. D. (2004) Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes 53, 2366–2374 [DOI] [PubMed] [Google Scholar]
- 8. Kwitek A. E., Jacob H. J., Baker J. E., Dwinell M. R., Forster H. V., Greene A. S., Kunert M. P., Lombard J. H., Mattson D. L., Pritchard K. A., Jr., Roman R. J., Tonellato P. J., Cowley A. W., Jr. (2006) BN phenome. Detailed characterization of the cardiovascular, renal, and pulmonary systems of the sequenced rat. Physiol. Genomics 25, 303–313 [DOI] [PubMed] [Google Scholar]
- 9. Devereux R. B., Alonso D. R., Lutas E. M., Gottlieb G. J., Campo E., Sachs I., Reichek N. (1986) Echocardiographic assessment of left ventricular hypertrophy. Comparison with necropsy findings. Am. J. Cardiol. 57, 450–458 [DOI] [PubMed] [Google Scholar]
- 10. Migrino R. Q., Zhu X., Pajewski N., Brahmbhatt T., Hoffmann R., Zhao M. (2007) Assessment of segmental myocardial viability using regional two-dimensional strain echocardiography. J. Am. Soc. Echocardiogr. 20, 342–351 [DOI] [PubMed] [Google Scholar]
- 11. Migrino R. Q., Zhu X., Morker M., Brahmbhatt T., Bright M., Zhao M. (2008) Myocardial dysfunction in the peri-infarct and remote regions following anterior infarction in rats quantified by two-dimensional radial strain echocardiography. An observational cohort study. Cardiovasc. Ultrasound 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pieper G. M., Shah A., Harmann L., Cooley B. C., Ionova I. A., Migrino R. Q. (2010) Speckle-tracking two-dimensional strain echocardiography. A new noninvasive imaging tool to evaluate acute rejection in cardiac transplantation. J. Heart Lung Transplant. 29, 1039–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zur Nedden S., Eason R., Doney A. S., Frenguelli B. G. (2009) An ion-pair reversed-phase HPLC method for determination of fresh tissue adenine nucleotides avoiding freeze-thaw degradation of ATP. Anal. Biochem. 388, 108–114 [DOI] [PubMed] [Google Scholar]
- 14. Ganzera M., Vrabl P., Wörle E., Burgstaller W., Stuppner H. (2006) Determination of adenine and pyridine nucleotides in glucose-limited chemostat cultures of Penicillium simplicissimum by one-step ethanol extraction and ion-pairing liquid chromatography. Anal. Biochem. 359, 132–140 [DOI] [PubMed] [Google Scholar]
- 15. Hellemans J., Mortier G., De Paepe A., Speleman F., Vandesompele J. (2007) qBase relative quantification framework and software for management and automated analysis of real time quantitative PCR data. Genome Biol. 8, R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Frezza C., Cipolat S., Scorrano L. (2007) Organelle isolation. Functional mitochondria from mouse liver, muscle, and cultured fibroblasts. Nat. Protoc. 2, 287–295 [DOI] [PubMed] [Google Scholar]
- 17. Wibom R., Hagenfeldt L., von Döbeln U. (2002) Measurement of ATP production and respiratory chain enzyme activities in mitochondria isolated from small muscle biopsy samples. Anal. Biochem. 311, 139–151 [DOI] [PubMed] [Google Scholar]
- 18. Barrientos A., Fontanes F., Diaz F. (2009) Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr. Protoc. Hum. Genet. Chapter 19, Unit 19.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ryan T. D., Rothstein E. C., Aban I., Tallaj J. A., Husain A., Lucchesi P. A., Dell'Italia L. J. (2007) Left ventricular eccentric remodeling and matrix loss are mediated by bradykinin and precede cardiomyocyte elongation in rats with volume overload. J. Am. Coll. Cardiol. 49, 811–821 [DOI] [PubMed] [Google Scholar]
- 20. Bell D. S. (2003) Diabetic cardiomyopathy. Diabetes Care 26, 2949–2951 [DOI] [PubMed] [Google Scholar]
- 21. Shoghi K. I., Finck B. N., Schechtman K. B., Sharp T., Herrero P., Gropler R. J., Welch M. J. (2009) In vivo metabolic phenotyping of myocardial substrate metabolism in rodents. Differential efficacy of metformin and rosiglitazone monotherapy. Circ. Cardiovasc. Imaging 2, 373–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tan Y., Ichikawa T., Li J., Si Q., Yang H., Chen X., Goldblatt C. S., Meyer C. J., Li X., Cai L., Cui T. (2011) Diabetic down-regulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes 60, 625–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boudina S., Sena S., O'Neill B. T., Tathireddy P., Young M. E., Abel E. D. (2005) Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 112, 2686–2695 [DOI] [PubMed] [Google Scholar]
- 24. How O. J., Aasum E., Severson D. L., Chan W. Y., Essop M. F., Larsen T. S. (2006) Increased myocardial oxygen consumption reduces cardiac efficiency in diabetic mice. Diabetes 55, 466–473 [DOI] [PubMed] [Google Scholar]
- 25. Ventura-Clapier R., Garnier A., Veksler V., Joubert F. (2011) Bioenergetics of the failing heart. Biochim. Biophys. Acta 1813, 1360–1372 [DOI] [PubMed] [Google Scholar]
- 26. Holloway G. P., Gurd B. J., Snook L. A., Lally J., Bonen A. (2010) Compensatory increases in nuclear PGC1α protein are primarily associated with subsarcolemmal mitochondrial adaptations in ZDF rats. Diabetes 59, 819–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Diamant M., Lamb H. J., Groeneveld Y., Endert E. L., Smit J. W., Bax J. J., Romijn J. A., de Roos A., Radder J. K. (2003) Diastolic dysfunction is associated with altered myocardial metabolism in asymptomatic normotensive patients with well controlled type 2 diabetes mellitus. J. Am. Coll. Cardiol. 42, 328–335 [DOI] [PubMed] [Google Scholar]
- 28. Kosmala W., Colonna P., Przewlocka-Kosmala M., Mazurek W. (2004) Right ventricular dysfunction in asymptomatic diabetic patients. Diabetes Care 27, 2736–2738 [DOI] [PubMed] [Google Scholar]
- 29. Arbustini E., Diegoli M., Fasani R., Grasso M., Morbini P., Banchieri N., Bellini O., Dal Bello B., Pilotto A., Magrini G., Campana C., Fortina P., Gavazzi A., Narula J., Viganò M. (1998) Mitochondrial DNA mutations and mitochondrial abnormalities in dilated cardiomyopathy. Am. J. Pathol. 153, 1501–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Silvestri G., Santorelli F. M., Shanske S., Whitley C. B., Schimmenti L. A., Smith S. A., DiMauro S. (1994) A new mtDNA mutation in the tRNA (Leu(UUR)) gene associated with maternally inherited cardiomyopathy. Hum. Mutat. 3, 37–43 [DOI] [PubMed] [Google Scholar]
- 31. Barouch L. A., Gao D., Chen L., Miller K. L., Xu W., Phan A. C., Kittleson M. M., Minhas K. M., Berkowitz D. E., Wei C., Hare J. M. (2006) Cardiac myocyte apoptosis is associated with increased DNA damage and decreased survival in murine models of obesity. Circ. Res. 98, 119–124 [DOI] [PubMed] [Google Scholar]
- 32. Cai L., Li W., Wang G., Guo L., Jiang Y., Kang Y. J. (2002) Hyperglycemia-induced apoptosis in mouse myocardium mitochondrial cytochrome c. Mediated caspase-3 activation pathway. Diabetes 51, 1938–1948 [DOI] [PubMed] [Google Scholar]
- 33. Schäfer E., Seelert H., Reifschneider N. H., Krause F., Dencher N. A., Vonck J. (2006) Architecture of active mammalian respiratory chain supercomplexes. J. Biol. Chem. 281, 15370–15375 [DOI] [PubMed] [Google Scholar]
- 34. Suthammarak W., Yang Y. Y., Morgan P. G., Sedensky M. M. (2009) Complex I function is defective in complex IV-deficient Caenorhabditis elegans. J. Biol. Chem. 284, 6425–6435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schägger H., Pfeiffer K. (2001) The ratio of oxidative phosphorylation complexes I–V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J. Biol. Chem. 276, 37861–37867 [DOI] [PubMed] [Google Scholar]
- 36. Zaragoza M. V., Brandon M. C., Diegoli M., Arbustini E., Wallace D. C. (2011) Mitochondrial cardiomyopathies: how to identify candidate pathogenic mutations by mitochondrial DNA sequencing, MITOMASTER and phylogeny. Eur. J. Hum. Genet. 19, 200–207 [DOI] [PMC free article] [PubMed] [Google Scholar]