

Background: Restitution is a method for maintaining and repairing the intestinal epithelial barrier.
Results: CXCL12 stimulated E-cadherin relocalization and improved barrier function in the reparative epithelium.
Conclusion: E-cadherin plays an important role in basal and CXCL12-induced restitution.
Significance: Understanding how cell-cell adhesion is regulated during sheet migration is crucial to enhancing treatment of gut barrier defects.
Keywords: Actin, Chemokines, Cxcr4, Intestinal Epithelium, Rho, Adherens Junction Regulation, Epithelial Sheet Migration
Abstract
Chemokines and other immune mediators enhance epithelial barrier repair. The intestinal barrier is established by highly regulated cell-cell contacts between epithelial cells. The goal of these studies was to define the role for the chemokine CXCL12 in regulating E-cadherin during collective sheet migration during epithelial restitution. Mechanisms regulating E-cadherin were investigated using Caco2BBE and IEC-6 model epithelia. Genetic knockdown confirmed a critical role for E-cadherin in in vitro restitution and in vivo wound repair. During restitution, both CXCL12 and TGF-β1 tightened the monolayer by decreasing the paracellular space between migrating epithelial cells. However, CXCL12 differed from TGF-β1 by stimulating the significant increase in E-cadherin membrane localization during restitution. Chemokine-stimulated relocalization of E-cadherin was paralleled by an increase in barrier integrity of polarized epithelium during restitution. CXCL12 activation of its cognate receptor CXCR4 stimulated E-cadherin localization and monolayer tightening through Rho-associated protein kinase activation and F-actin reorganization. These data demonstrate a key role for E-cadherin in intestinal epithelial restitution.
Introduction
The human intestine is lined by a layer of highly polarized, continuously renewing, and actively migrating epithelial cells. Intestinal epithelial cells comprise a physical and ion-selective barrier separating the lumen from the submucosa. This barrier is established through a series of protein interactions between neighboring cells. The sites of interactions are localized at the tight junctions and the adherens junction (1). The adherens junction results primarily from homotypic interactions between the E-cadherin transmembrane proteins of adjacent cells. E-cadherin is, in turn, linked to the actin cytoskeleton through a series of adaptor proteins (2, 3). Importantly, the adherens junction plays a key regulatory role in barrier integrity through its temporal and spatial coordination of the tight junction (2–5). Genetic analyses indicate the accumulation of varying mutations within the CDH1 gene encoding E-cadherin in humans with ulcerative colitis or celiac disease (6–9), a defect that is correlated with increased intestinal permeability in those patients. Further, Crohn's disease patients and unaffected family members have increased intestinal permeability that reflects defects in the epithelial barrier, including altered junctional protein expression (10–12).
The epithelium receives a barrage of luminal and mucosal insults, both physical and chemical, that result in damage. Loss of barrier function because of injury is inherently followed by resealing of the epithelium. The initial step in injury repair occurs through a rapid migration response of the epithelial sheet. This process, termed restitution, occurs independently of proliferation and minimizes entry of potentially dangerous stimuli (13, 14). Results from cell culture and mouse models indicate that the constitutive migration response, which we term basal restitution, can be inducibly regulated and enhanced by several effectors of the mucosal immune response. Thus, TGF-β1, EGF, members of the trefoil factor family, and chemokines stimulate restitution (14–18). Recent work from our laboratory has revealed that although inducible effectors of restitution increase epithelial cell migration, the mechanisms whereby they elicit this response are distinct (19–21). Sheet migration characteristic of epithelial restitution is typical of tissue development and angiogenesis and is distinct from migration of fibroblasts and leukocytes in that epithelial cells maintain cell-to-cell contacts (22–24). Thus, in restitution, collective sheet migration involves maintenance of the intraepithelial junctions to minimize the space between cells in coordination with active migration. The cellular and molecular mechanisms orchestrating junctional stability in the dynamic epithelial monolayer during basal and inducible restitution remain complex and poorly understood.
We have demonstrated previously that the chemokine CXCL12 increases the velocity and magnitude of barrier formation as well as epithelial cell migration (18, 19). With this report we have discovered that restitution is an E-cadherin-dependent process. Further, cytokine stimuli differed in their ability to seal the barrier and improve function, with CXCL12 superior to TGF-β1 in directing the localization of E-cadherin to the lateral membranes between migrating cells. Chemokine-directed E-cadherin relocalization was via Rho-associated protein kinase (ROCK2)-dependent F-actin reorganization. These studies resulted in a model in which immune effectors function as redundant systems to seal epithelial barrier injuries. The potential for pharmacological intervention to increase E-cadherin localization would be expected to increase tightening and improve mucosal barrier integrity.
EXPERIMENTAL PROCEDURES
Materials
Recombinant human CXCL12, EGF, TGF-β1, and purified mouse monoclonal IgG2B were purchased from R&D Systems (Minneapolis, MN). Src family kinase inhibitor PP2, ROCK inhibitor Y27632, the Gαi inhibitor pertussis toxin (PTx) and actin polymerization inhibitor cytochalasin D (CytoD) were purchased from EMD Calbiochem (San Diego, CA). Murine monoclonal E-cadherin and N-cadherin antibodies were purchased from BD Biosciences. AMD3100, secondary goat anti-mouse polyclonal Alexa Fluor 488, and Alexa Fluor 594-conjugated wheat germ agglutinin were from Sigma. Alexa Fluor 568 phalloidin was obtained from Invitrogen.
Wound Assay
Non-transformed normal rat small intestinal IEC-6 cells (CRL-1592) and human Caco2BBE intestinal carcinoma cells were cultured as detailed previously (18, 21). Confluent IEC-6 cell monolayers grown in 60-mm dishes were serum-starved for 24–48 h. Cells were pretreated for 30 min with 2 mm EGTA at 37 °C. Cells were wounded with a sterile razor blade and incubated for 20 h with titrated concentrations of EGTA at 37 °C in 10% CO2 for calcium depletion assays. In separate experiments, IEC-6 cells were pretreated for 30 min with titrated concentrations of the Src inhibitor PP2 or CytoD before wounding and stimulation with cytokines. Images were obtained (Nikon Eclipse TE2000-U) at ×100 magnification, and the number of migrated cells was enumerated by counting nucleated cells that crossed the wound edge. The area of the intercellular space between cells was quantified with MetaMorph software (Molecular Devices, Sunnyvale, CA).
Polarized Caco2BBE cells were grown to confluence and maintained as monolayers in 12-well transwell inserts (pore size 0.4 μm, Corning, Danvers, MA). After 19–21 days, monolayers were serum-starved for 24 h and wounded with a 0.1- to 10-μl plastic pipette tip (USA Scientific, Ocala, FL) connected to a benchtop vacuum aspirator as defined previously (19). Injured polarized epithelia were stimulated daily with serum-free medium containing CXCL12 (20 ng/ml) or TGF-β1 (5 ng/ml). Photomicrographs of the circular wounds were taken daily using an inverted Nikon Eclipse TE2000-U microscope, and the area of each wound was defined using MetaMorph. In parallel, transepithelial resistance (Ωcm2) was assessed using a 12-well, 12-mm Endohm voltmeter (World Precision Instruments, Sarasota, FL).
Immunofluorescence
To define the paracellular space, IEC-6 cells were grown to 100% confluence on glass eight-well chamber slides (LabTek, Scotts Valley, CA) and serum-starved for 24–48 h. Cells were wounded with a sterile razor blade purchased from OLFA (Rosemont, IL), rinsed with PBS, and stimulated with CXCL12, TGF-β1, or 50 ng/ml EGF in serum-free medium. Twenty hours after wounding, cells were fixed with 1% (w/v) paraformaldehyde and stained for 10 min with 5 μg/ml of Alexa Fluor 594 wheat germ agglutinin. In separate experiments, cells were pretreated with AMD3100 (5 μg/ml) or Y27632 (10 μm) for 30 min prior to wounding and stimulation.
To define localization of E-cadherin or F-actin, confluent IEC-6 cells were serum-starved and wounded with a sterile razor, rinsed with PBS, and treated with CXCL12 and TGF-β1. In separate experiments, cells were pretreated with PTx (100 ng/ml), CytoD (30 nm), or Y27632 for 30 min prior to wounding. Wounded IEC-6 cells were fixed with 1% (w/v) paraformaldehyde at 4, 8, and 16 h after wounding. Cells were blocked with 5% (w/v) BSA in PBS with 0.3% (v/v) Triton X-100 for 1 h. Cells were incubated with primary antibodies overnight at 4 °C, washed, and incubated with the appropriate secondary antibody for 2 h at room temperature.
Images were obtained using a fluorescence microscope (Nikon Eclipse) at ×600 magnification. The space between migrating cells was quantified using MetaMorph image analysis software. Relative intensity of E-cadherin immunofluorescence was determined by a line scan from the nucleus of the leading migrating cell to the nucleus of the following cell. The cell-cell membrane was then aligned in the center of a 50-pixel total length line. To obtain the relative intensity at the cell wound edge, the 50-pixel line scan was repeated and aligned from the nucleus of the leading migrating cell into the adjacent denuded space.
Immunoblot Analysis
Serum-starved IEC-6 cells were treated with CXCL12 or TGF-β1. To inhibit the Src family of kinases, cells were pretreated for 30 min with 1 μm PP2 at 37 °C before being injured. Cells were separately pretreated with PTx, CytoD, or Y27632 to inhibit Gαi, actin polymerization, or ROCK signaling, respectively. Wounded IEC-6 cell monolayers were microdissected using an inverted microscope (Nikon Eclipse TS100) at ×100 into stationary and migratory subcell populations with a cell scraper. Cells located beyond the wound edge, as premarked on the tissue culture dish, were defined as migratory populations. Stationary populations were defined as cells located at least 10 mm away from the wound. Cells were lysed with modified radioimmune precipitation assay lysis buffer and protein levels determined as defined previously (21). Equal protein levels were size-separated using reducing SDS-PAGE and electrophoretically transferred to PVDF membranes. E-cadherin was detected using specific mouse monoclonal antibody. Proteins were visualized with appropriate HRP-conjugated secondary antibodies and SuperSignal West Pico chemiluminescence substrate (Pierce).
E-cadherin Knockdown
E-cadherin shRNA was generated using the Lentilox3.7 system (Addgene, Cambridge, MA) as described recently (21). E-cadherin protein levels were assessed using immunoblot analysis and immunofluorescence microscopy. For wounding assays, confluent E-cadherin-depleted Caco2BBE cells were serum-starved, injured with a sterile razor, and treated with serum-free medium or CXCL12 or TGF-β1.
Experimental Animals
Mutant mice in which exons 6–10 of the E-cadherin gene (CDH1) were flanked by loxP recognition elements (CDH1flox/flox or CDH1flox/+) were genotyped as described previously (25). CDH1flox/flox mice were crossed with heterozygous mice expressing the Cre transgene under the transcriptional control of the mouse villin promoter (26). Hematoxylin and eosin staining was performed on 4-μm sections from paraffin-embedded tissue as described previously (27, 28).
In Vivo Confocal Microscopy
Experimental mice were anesthetized with an intraperitoneal injection of ketamine (75 mg/kg) and xylazine (25 mg/kg). A midline laparotomy exposed a 5-cm loop of intestinal jejunum that was cannulated at the proximal and distal ends with silicone tubing of 0.8 mm internal diameter. Hanks' balanced salt solution (Invitrogen) was warmed to 37 °C and perfused through the jejunal loop at 0.5 ml/min for 10 min using a peristaltic pump (model EP-1, Bio-Rad). Acriflavine at 0.05% (w/v) in Hanks' balanced salt solution was perfused through the intestine at 37 °C in a recirculating manner. Input was via the proximal tube and output via the distal tube at a rate of 1 ml/min for 30 min. A 1-cm jejunal loop was exteriorized and opened longitudinally on the antimesenteric border. The luminal surface of the loop was placed, lumen side down, on a glass-bottom 35-mm culture dish (MatTek Corp., Ashland, MA) and imaged using a two-photon laser confocal microscope (Zeiss LSM 510 META NLO) with a visible and multiphoton laser and outfitted with an XL3 incubator chamber for a controlled environment. Individual cells were exposed to 6-μs bursts of the Chameleon Ultra II Ti sapphire laser at 800 nm to reproducibly create 12-μm wounds. Each exteriorized intestine was injured three to five times over a 3-h period. Sheet migration after wounding was followed by time series imaging at 1-min intervals using 488-nm excitation.
Statistical Analyses
Differences between varying stimuli were analyzed using an unpaired Student's t test (SigmaStat, Jandel Scientific Software, San Rafael, CA). Multiple comparisons between groups were analyzed using a two-way analysis of variance with a Bonferroni post hoc test to identify pair-wise differences (GraphPad Prism 4, La Jolla, CA). Statistical significance was set at p ≤ 0.05.
RESULTS
E-cadherin Is Required for Intestinal Epithelial Wound Healing
To ascertain the role for E-cadherin in intestinal epithelial restitution, we first examined mice with the targeted deletion of E-cadherin in the cells of the murine intestinal epithelium. We noted an increase in crypt branching within small intestines of mice with the heterozygous deletion of E-cadherin (Fig. 1, A and B) compared with control mice lacking Cre expression (C and D). In vivo two-photon confocal microscopy was therefore used next to monitor restitution of laser-wounded epithelium in real time (29, 30). The expulsion of wounded enterocytes from heterozygous E-cadherin knockout mice was significantly faster than that of control mice (Fig. 1, E and F). These data suggest that enterocyte migration was increased or that cellular adhesiveness was diminished in E-cadherin knockout epithelium.
FIGURE 1.

Heterozygous deletion of intestinal epithelial E-cadherin results in disrupted epithelial homeostasis and wound repair. A and B, increased crypt branching in mice lacking E-cadherin (CDH1flox/+ villin-Cre) (A) compared with control (CDH1flox/flox) mice (B). Small intestine sections were stained with hematoxylin and eosin stain. Enumeration of the number of crypt branches revealed that control mice with intact E-cadherin expression had 1.01 ± 0.41 branches per 100 crypts CDH1flox/flox. In contrast, Cre-expressing CDH1flox/+ mice had a significant (p = 0.036) increase in malformed crypts, with 3.11 ± 0.61 branches per 100 crypts. Images are representative of three to four individual mice. The rightmost panels in A and B are a higher magnification (×400) of the dotted box region in the left-hand panels (×100). C, increased shedding of damaged epithelium in conditional E-cadherin-deficient mice. Laser-injured enterocytes were increasingly shed into the bowel lumen of CDH1flox/+ villin-Cre mice compared with control (CDH1flox/flox) animals. The small intestine epithelium (E) stained with acriflavin was wounded using a two-photon photobleach (the wound is denoted with a white arrow). Images of damaged epithelium were taken every 1 min for 40 min as wounded cells detached from the mucosal lamina propria (M) into the lumen (L). Images were taken at ×400. Images are representative of three to four mice. D, quantification of damaged cell shedding from the epithelium after wounding. Values are mean ± S.E. of three to four experiments. **, p ≥ 0.01.
To determine the potential role for E-cadherin in collective sheet migration, a standard calcium chelation assay was employed next. Chelation of extracellular calcium induces retraction of calcium-dependent E-cadherin from the cell membrane and, hence, disrupts epithelial cell-cell adhesion (31). IEC-6 cells were pretreated with 2 mm EGTA to reduce extracellular calcium levels to ∼0.54 μm and were injured with a sterile razor. Restitution of injured monolayers was monitored in cells incubated with titrated concentrations of EGTA and compared with cells without pretreatment (supplemental Fig. 1, A–C). Consistent with a role for E-cadherin in epithelial restitution, the number of migrating cells was dose-dependently decreased as EGTA levels increased (supplemental Figs. 1, A and B). Similarly, the space between cells was also increased with EGTA treatment (supplemental Fig. 1C). Indeed, the increased expulsion of E-cadherin knockout enterocytes in vivo (Fig. 1E) was mirrored in vitro by the appearance of phase bright cells detaching from the wound edge of IEC-6 monolayers (supplemental Fig. 1).
Taking advantage of human-specific lentiviral shRNA probes, we next moved to Caco2BBE cells to more closely define the role for E-cadherin in epithelial migration and barrier functions. Immunoblot analysis indicated nearly 90% knockdown in E-cadherin protein in two separate E-cadherin-depleted human Caco2BBE colonic epithelial cell lines (Figs. 2, A and B). Immunofluorescence microscopy confirmed the successful depletion of E-cadherin compared with wild-type, puromycin empty vector, or scramble hairpin-transduced control cells (Fig. 2B). Adapting the scrape wound assay to human colonic Caco2BBE cells, we demonstrated that depletion of E-cadherin significantly impaired basal migration compared with control cells (Fig. 2, C and D). Thus, E-cadherin has an essential role in migration of intestinal epithelia both in vivo and in vitro.
FIGURE 2.

Decreased restitution in E-cadherin knockdown Caco2BBE cells. A, densitometry confirmed the 89 and 87% depletion knockdown of E-cadherin protein from Caco2BBE cells inoculated with lentiviral particles encoding specific shRNA (Ecad sh1 and Ecad sh2) compared with the wild type and cells inoculated with empty vector (Puro) or scramble vector (Scram). E-cadherin protein levels were normalized to the loading control GAPDH. B, immunofluorescence confirmed knockdown of E-cadherin within E-cadherin-specific shRNA-treated cells (E-cadherin sh1 and sh2). Cells inoculated with specific shRNA to E-cadherin (green) demonstrated marked depletion compared with wild-type, empty vector (Puromycin) and scramble vector-treated (Scramble) cells. F-actin staining was visualized using Alexa Fluor phalloidin 568 (red). Cell nuclei were visualized with DAPI (blue). Images are representative of three separate experiments. Scale bar = 50 μm. C, two separate Caco2BBE cells inoculated with lentiviral particles encoding E-cadherin-specific shRNA exhibited a decreased level of basal restitution compared with wild-type cells 20 h after wounding. Control inoculation of scramble shRNA (Scramble) and empty (Puromycin) did not inhibit basal restitution. Direction of migration is from left to right, where the white line marks the wounded edge. Scale bar = 200 μm. Images are representative of four separate experiments. The level of basal migration was quantified as the area of migration across the wounded edge 20 h after wounding for wild-type, scramble (scram), empty (Puro), and two separate Caco2BBE cells inoculated with lentiviral particles encoding E-cadherin-specific shRNA (sh1 and sh2). Values are mean ± S.E. of four experiments. **, p ≤ 0.01.
CXCL12 Enhanced Restitution Requires E-cadherin
The roles for CXCL12 (18–21, 28), TGF-β1 (14), and EGF (17) in inducible restitution are well established. We next tested whether extracellular migration stimulants could rescue E-cadherin knockdown abrogation of wound healing. Caco2BBE E-cadherin-depleted cells were stimulated with either CXCL12 or TGF-β1. Interestingly, although TGF-β1 significantly increased sheet migration within E-cadherin knockdown cells (Figs. 3, B and D), CXCL12 was unable to increase restitution compared with control cells (A and C). CXCL12 and TGF-β1 both significantly increased epithelial cell migration in wild-type and scramble-transduced controls (Fig. 3). These findings suggest CXCL12 and TGF-β1 utilize distinct mechanisms to drive restitution.
FIGURE 3.
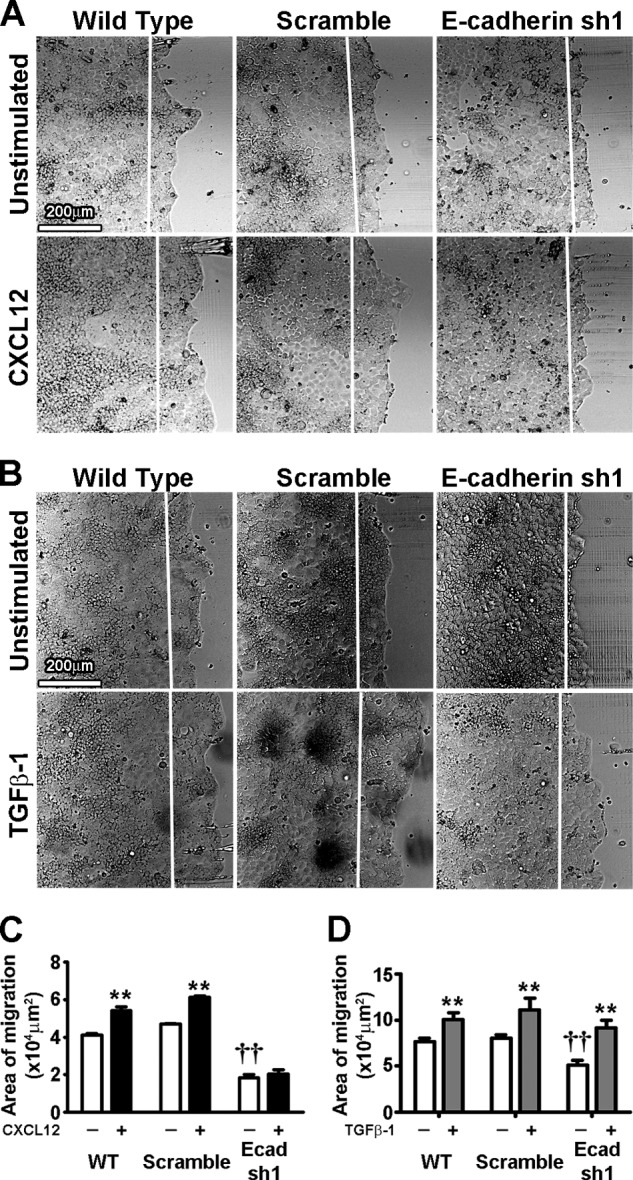
E-cadherin knockdown ablates CXCL12- but not TGF-β1-stimulated restitution in Caco2BBE cells. A, E-cadherin knockdown (E-cadherin sh1) ablates CXCL12 (20 ng/ml)-induced restitution, whereas WT and scramble shRNA-treated (Scramble) cells showed increased restitution with CXCL12. B, TGF-β1-stimulated (5 ng/ml) restitution was not ablated by E-cadherin knockdown (E-cadherin sh1) compared with wild-type and scramble-treated shRNA (Scramble) cells. Direction of migration was from left to right. The white line marks the initial wound edge. Scale bar = 200 μm. Images are representative of four separate experiments. C, restitution was quantified by the area of migration from left to right across the white line denoting the wounded edge 20 h after wounding. Values are mean ± S.E. of five experiments. **, p ≤ 0.01 compared with unstimulated cells within same cell lines; ††, p ≤ 0.01 compared with unstimulated WT cells.
CXCL12 Activation of CXCR4-stimulated Paracellular Space Closure during Restitution
Previous studies in our laboratory have shown that CXCL12 and TGF-β1 stimulate wound healing via distinct signaling pathways. Trefoil factor family 3 also evokes collective cell migration, whereas EGF-treated cells migrate individually like fibroblasts (32). Thus, to test the hypothesis that CXCL12 and TGF-β1 stimulation induce distinct migration phenotypes, the number and area of the space between cells were quantified. Fluorescence microscopy revealed that treatment with CXCL12 significantly decreased the number and size of the paracellular spaces between actively migrating epithelial sheets (Fig. 4). Interestingly, CXCL12-treated cells exhibited a more robust decrease in the number of uncovered spaces between cells within the monolayer than that of TGF-β1 (Fig. 4B). EGF-treated cells migrated with the largest number and size of the uncovered region between cells within the migration sheet (Fig. 4, A–C). CXCR4 specific antagonist AMD3100 prevented CXCL12-induced changes in the number of uncovered regions between cells without affecting control or TGF-β1-stimulated effects (Fig. 4D).
FIGURE 4.

CXCL12 and TGF-β1 increase paracellular sealing within migrating sheets. A, CXCL12 (20 ng/ml) and TGF-β1 (5 ng/ml) stimulation after wounding confluent IEC-6 monolayers decreased paracellular space compared with unstimulated cells and EGF (50 ng/ml) 20 h after wounding. Wheat germ agglutinin (WGA) was used to stain the cell membrane. Direction of migration was from left to right. Specificity was determined by absence of lectin staining (No WGA). Nuclei of No WGA imaged cells were visualized using DAPI staining (inset). Scale bar = 50 μm. The arrows denote paracellular space within the migrating sheet. Images are representative of three separate experiments. Paracellular sealing was assessed by determining the number (B) or area (C) of the paracellular space between cells within migrating sheet. Values are mean ± S.E. of three experiments. *, p ≤ 0.05; **, p ≤ 0.01. D, CXCR4-specific inhibitor AMD3100 (5 μg/ml) abolished CXCL12-induced paracellular sealing within migrating sheets. Paracellular sealing of untreated or TGF-β1 stimulated cells were not affected by AMD3100. Values are mean ± S.E. of three experiments. **, p ≤ 0.01 compared with unstimulated cells not treated with AMD3100. ##, p ≤ 0.01 compared with unstimulated cells treated with AMD3100.
Increased E-cadherin Protein Levels within the Migrating Epithelium
E-cadherin protein levels were assessed in unwounded monolayers stimulated with CXCL12 and TGF-β1 to determine whether decreased paracellular space is dependent upon E-cadherin-based adherens junction formation during restitution. CXCL12 stimulation did not alter levels of total E-cadherin protein compared with unstimulated cells (Figs. 5, A and B). In contrast, TGF-β1 stimulation reduced E-cadherin protein within IEC-6 cells in agreement with its known functions (33, 34). Protein levels were further examined within stationary and migratory cell populations in CXCL12- and TGF-β1-treated IEC-6 cells. CXCL12 stimulation evoked an increase in E-cadherin protein within the migratory cell population (Fig. 5, C and D). As noted in unwounded monolayers CXCL12 did not significantly alter the levels of total E-cadherin protein in stationary cells. In marked contrast with the observed decrease in the intercellular space between cells, TGF-β1 significantly decreased E-cadherin protein levels in both stationary and migratory cells (Fig. 5, E and F). Further, unlike its effects on E-cadherin or the paracellular space, TGF-β1was unable to increase expression of N-cadherin (supplemental Fig. 2).
FIGURE 5.

Increased E-cadherin protein levels in CXCL12-stimulated migrating cells. A, of TGF-β1 (5 ng/ml) stimulation decreased E-cadherin protein levels within confluent IEC-6 monolayers compared with unstimulated monolayers after 20 h. CXCL12-stimulated (20 ng/ml) monolayers were not significantly different compared with unstimulated monolayers. B, quantification of the immunoblot analysis of E-cadherin from the unwounded IEC-6 monolayer treated with CXCL12 and TGF-β1 normalized to unstimulated E-cadherin protein level (Unstim). Values are mean ± S.E. from four experiments. **, p ≤ 0.01. C, CXCL12 stimulation increased E-cadherin protein levels within the migrating subcell population 20 h after wounding compared with unstimulated cells, whereas no significant difference was observed within the stationary subcell population. D, densitometry of E-cadherin from total protein lysates of the stationary and migratory subpopulation of wounded IEC-6 treated with CXCL12. Values are mean ± S.E. of four experiments. **, p ≤ 0.01. E, TGF-β1 stimulation decreased E-cadherin protein levels within both the stationary and migratory subcell population 20 h after wounding compared with unstimulated cells. F, quantification of the immunoblot analysis of E-cadherin from total protein lysates of the stationary and migratory subpopulation of wounded IEC-6 treated with CXCL12. Values are mean ± S.E. of four experiments. *, p ≤ 0.05; **, p ≤ 0.01.
Increased E-cadherin Localization to the Cell-Cell Membrane during Restitution
Localization of E-cadherin was next analyzed to characterize whether the increase in CXCL12-stimulated E-cadherin protein levels correlate to cell-cell membrane localization and decreased space between cells. Immunofluorescence microscopy demonstrated biphasic E-cadherin localization within unstimulated wounded epithelium (Fig. 6). CXCL12-stimulated IEC-6 cells consistently exhibited elevated levels of membrane-localized E-cadherin within cells at the leading edge of the migrating epithelial sheet (Fig. 6A). The absolute peak intensity of E-cadherin in CXCL12-stimulated cells was significantly higher compared with unstimulated cells at 4 and 16 h after wounding (Fig. 6B). In contrast to CXCL12, there was a significant decrease in E-cadherin localization to the cell-cell membrane within migrating cells treated with TGF-β1 compared with control cells (supplemental Fig. 3).
FIGURE 6.

CXCL12 stimulates E-cadherin localization to the cell-cell membrane during restitution. A, representative immunofluorescence images of E-cadherin detected 4, 8, and 16 h after wounding confluent IEC-6 monolayers with or without CXCL12 (20 ng/ml) stimulation. Compared with unstimulated cells, CXCL12 increased E-cadherin localization to the cell-cell membrane between leading migrating and following cells 4 and 16 h after wounding. Direction of migration was from left to right. Data are representative of three separate experiments. Scale bar = 50 μm. B, relative intensity of E-cadherin immunofluorescence across a 50-pixel line scan was determined as defined under “Experimental Procedures.” Relative peak intensity at the cell-cell membrane was derived from the line scan. Values are mean ± S.E. of three experiments. *, p ≤ 0.05; **, p ≤ 0.01.
CXCL12 and TGF-β1 Differentially Regulate Transepithelial Resistance during Wound Healing
Given the roles for adherens junctions in coordination and maintenance of the tight junctions (2–4), we next asked if CXCL12 and TGF-β1 evoked parallel changes in wound closure and barrier integrity in model polarized epithelia. As shown in Fig. 7, A and C, CXCL12 and TGF-β1 induced comparable purse-string wound closure in Caco2BBE monolayers. However, CXCL12 treatment markedly accelerated barrier restoration, as shown by the significant increase in transepithelial resistance relative to control and TGF-β1-stimulated cells (Fig. 7B). These latter data are consistent with the discovery that CXCL12 stimulates increased expression and membrane localization of E-cadherin during restitution. Together, these results suggest that although both signaling factors induce an increase in cell migration, CXCL12-induced restitution additionally enhances paracellular tightening, which is dependent upon expression and localization of E-cadherin.
FIGURE 7.
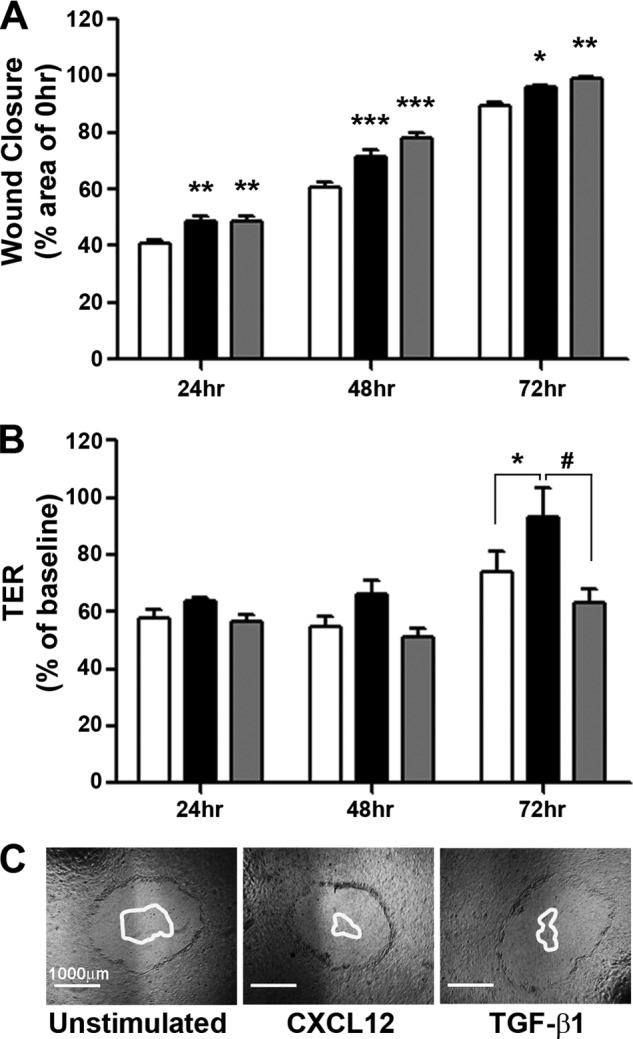
CXCL12 stimulation increases wound closure and transepithelial resistance (TER) of polarized Caco2BBE monolayers. A, CXCL12 (20 ng/ml) (black) and TGF-β1 (5 ng/ml) (gray) stimulated significant wound closure at 24, 48, and 72 h post-wounding compared with unstimulated (white) monolayers. Data are percent closure relative to the 0 h wound area. Values are mean ± S.E. of five experiments. *, p ≤ 0.05; **; p ≤ 0.01; ***, p ≤ 0.001. B, wounded monolayers stimulated with CXCL12 (black) had significantly higher TER than unstimulated (white) or TGF-β1 (gray) treated monolayers 72 h after wounding. Data are percentage of TER prior to wounding (baseline). Values are mean ± S.E. of five experiments. * p ≤ 0.05; #, p ≤ 0.05. C, representative images of 0-hour wounds with a superimposed outline of the remaining wound at 72 h. Scale bar = 1000 μm.
Inhibition of Src Kinases Abrogated E-cadherin Localization and Restitution
Src family kinases are well characterized in the regulation of adherens junction stabilization and maintenance of E-cadherin and β-catenin (35, 36). CXCL12 has also been shown to activate Src in various cell lines (37–40). In agreement with those reports, basal and inducible restitution of IEC-6 cells was dose-dependently inhibited by the Src kinase inhibitor PP2 (supplemental Figs. 4, A and B). Next, pretreatment with 1 μm PP2 blocked CXCL12-stimulated membrane localization of E-cadherin (Fig. 8, A–C). Congruent with those data, we observed that PP2 treatment was associated with increased localization of E-cadherin at the cell membrane in contact with the denuded wound surface (Fig. 8D). Immunoblot analysis indicated that PP2 treatment of confluent IEC-6 cells did not significantly alter total E-cadherin protein levels (supplemental Fig. 5, A and B). Our data suggest CXCL12 induced redistribution of E-cadherin to the membranes of actively migrating cells during restitution is dependent upon Src activation.
FIGURE 8.
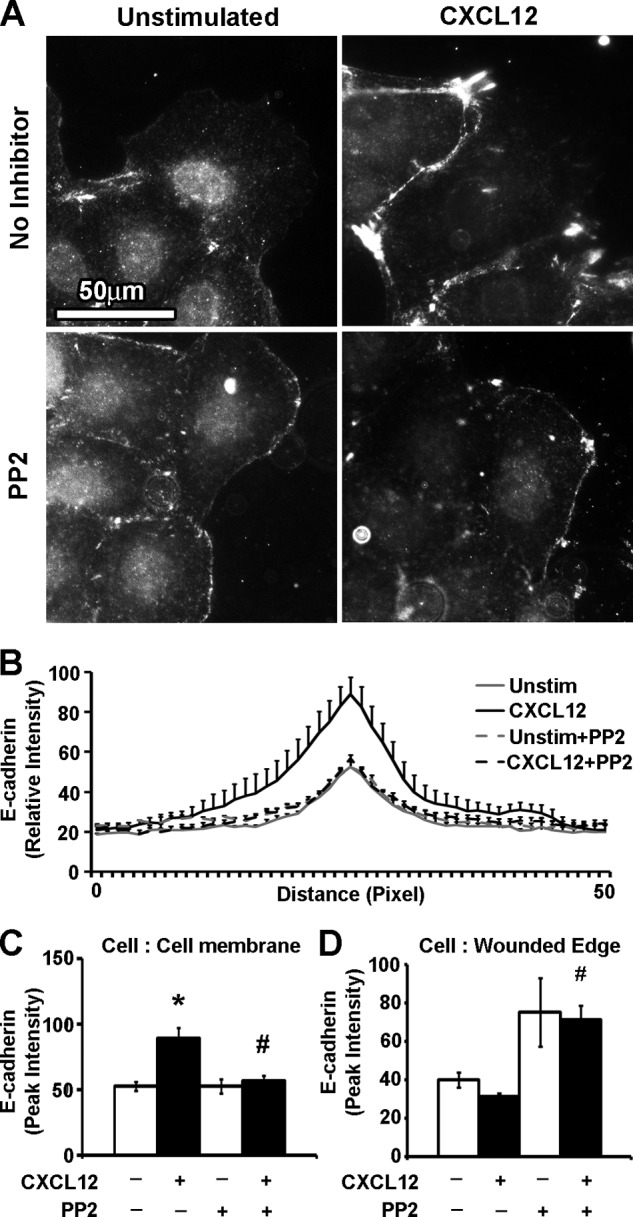
Src family kinase inhibitors decreased sheet migration and E-cadherin membrane localization. A, representative immunofluorescence images indicate that PP2 (1 μm) treatment abrogated CXCL12 (20 ng/ml) stimulated cell-cell membrane localization of E-cadherin. Scale bar = 50 μm. B, quantification of relative E-cadherin immunofluorescence intensity along a 50-pixel line scan revealed that inhibition with the Src inhibitor PP2 reversed CXCL12-induced localization of E-cadherin to the cell-cell membrane (C) and resulted in increased accumulation of E-cadherin at the edge of the cell in communication with the denuded wound edge (D). Values are mean ± S.E. of three experiments. *, statistically significant difference (p ≤ 0.05) between CXCL12-stimulated and control untreated cells. #, p ≤ 0.05 to cells treated with CXCL12.
CXCL12 Stimulated E-cadherin Localization Is Dependent upon ROCK and F-actin
Src not only regulates E-cadherin but has been shown to coordinate F-actin reorganization and plasma membrane protrusions in tandem with RhoA and ROCK (41–43). Moreover, within the adherens junction, E-cadherin interaction with the cortical F-actin cytoskeleton plays a significant role in initiation and stabilization of the complex (44–47). As highlighted in supplemental Fig. 6, immunofluorescence microscopy revealed the colocalization of E-cadherin and F-actin to the membranes of adjacent cells within the migrating epithelial sheet.
Disruption of F-actin accumulation with CytoD blocked IEC-6 wound healing in a dose-dependent manner (supplemental Fig. 4, C and D). Using the lowest concentration shown to block migration, we found that 30 nm CytoD inhibited CXCL12-induced accumulation of F-actin and E-cadherin to the cell membrane of migrating cells (Fig. 9). CytoD did not significantly change protein expression levels of E-cadherin (supplemental Fig. 5, C and D) or alter its localization between stationary cells 10–20 cells distant from the wound edge (supplemental Fig. 5G). The increased reorganization of both CXCL12-induced E-cadherin and F-actin was sensitive to PTx (supplemental Fig. 6), consistent with our previous findings for CXCR4 signaling (18). Immunoblot analysis showed that PTx alone had little effect on total E-cadherin protein levels in unwounded monolayers (supplemental Fig. 5, C and D).
FIGURE 9.

CXCL12-stimulated coaccumulation of E-cadherin and F-actin is disrupted with Cytochalasin-D treatment. A, representative images revealed that CytoD (30 nm) inhibition of F-actin (red) accumulation resulted in the concomitant loss of CXCL12 (20 ng/ml) induced E-cadherin (green) localization at the cell-cell membranes between adjacent cells. Nuclei were visualized with DAPI (blue). Direction of migration was from left to right. Images are representative of three separate experiments. Scale bar = 50 μm. B, quantification of relative E-cadherin immunofluorescence intensity was determined across a 50-pixel line scan as described under “Experimental Procedures.” C, relative peak immunofluorescence intensity of E-cadherin at the cell-cell membrane was derived from the line scan. Values are mean ± S.E. of three experiments. *, p ≤ 0.05. D, CytoD treatment decreased the relative intensity of phalloidin across a 50-pixel line scan as described under “Experimental Procedures.” E, relative peak immunofluorescence intensity of F-actin-bound phalloidin at the cell-cell membrane was derived from the line scan. Data are mean ± S.E. of three experiments. *, p ≤ 0.05; #, p ≤ 0.05 to cells not treated with CytoD.
Our previous work has shown that CXCL12-CXCR4 activation led to a ROCK-dependent increase in F-actin accumulation within migrating intestinal epithelial sheets (19). ROCK inhibition with Y27632 was performed next to determine the downstream effector regulating CXCL12 stimulated F-actin and E-cadherin reorganization. Inhibition of ROCK did not significantly modify overall E-cadherin protein levels (supplemental Fig. 5, E and F). However, Y27632 prevented CXCL12-stimulated accumulation of E-cadherin and F-actin to the cell membranes of the leader migrating cells (Fig. 10, A–E). Furthermore, Y27632 not only abrogated CXCL12-stimulated paracellular sealing during sheet migration but also increased the number and area between cells compared with both CXCL12-stimulated and unstimulated controls (Fig. 10, E–G). Together, these data suggest that Src and ROCK-mediated F-actin accumulation regulates E-cadherin localization to the membranes of epithelial cells during CXCL12-stimulated sheet migration.
FIGURE 10.

CXCL12-stimulated E-cadherin localization and paracellular tightening is dependent upon ROCK activation. A, representative immunofluorescence images indicate that inhibition of ROCK using Y27632 (10 μm) resulted in the loss of CXCL12-induced (20 ng/ml) E-cadherin localization and F-actin accumulation. Cell nuclei were visualized with DAPI (blue). Direction of migration was from left to right. Images are representative of three separate experiments. Scale bar = 50 μm. Quantification of the relative intensity of E-cadherin immunofluorescence demonstrated the significant decrease in membrane localized E-cadherin across a 50-pixel line scan (B) and peak intensity (C) as defined under “Experimental Procedures.” C, relative peak intensity values are mean ± S.E. of three experiments. *, p ≤ 0.05. Quantification of the relative intensity of phalloidin fluorescence demonstrated the significant decrease in cortical F-actin accumulation across a 50-pixel line scan (D) and peak intensity (D) as defined under “Experimental Procedures.” B–E, values are mean ± S.E. of three experiments. *, p ≤ 0.05; significant difference (p ≤ 0.05) to cells not treated with Y27632. F, inhibition of ROCK via Y27632 treatment abrogated CXCL12-stimulated paracellular tightening. Wheat germ agglutinin (WGA) was used to stain the cell membrane. Direction of migration was from left to right. Scale bar = 50 μm. The arrows denote paracellular spaces between cells within the migrating sheet. Images are representative of three separate experiments. Paracellular sealing was assessed by determining the number (G) or area (H) of the intercellular space between cells within the migrating sheet. Values are mean ± S.E. of three experiments. *, p ≤ 0.05; #, p ≤ 0.05 to cells not treated with Y27632.
DISCUSSION
E-cadherin is known to play a key role in epithelial maintenance and enterocyte shedding within the murine intestine (30). Moreover, mutations in Cadherin-1 (Cdh-1) have been linked with barrier weakness in patients with inflammatory bowel disease (6–9). In normal rat gastric mucosal cells, E-cadherin has been shown to be essential for sheet migration during wound healing (48). However, the role for E-cadherin and the mechanisms regulating its functions in reparative migration responses in the small or large intestine remain unexplored. Our data establish a model wherein CXCL12-increased cell-cell adhesion corresponds with decreased wound size and reduced paracellular permeability, minimizing the potential for noxious environmental stimuli to cross the wound.
Examination of barrier integrity and restitution in experimental mice was restricted to analyzing those with the heterozygous deletion of epithelial E-cadherin as homozygous epithelial knockout results in a neonatal fatal phenotype. Initial morphological characterization of the intestine revealed increased crypt branching, a finding similar to that previously noted in N-cadherin mutant animals (49), p120catenin-deficient mice (50), and APC(Min) mice (51). Subsequent work using an inducible Cre mouse indicates that targeted deletion of E-cadherin from epithelial cells results in marked inflammatory responses, disruption in organized enterocyte migration and increased bacterial translocation into mucosal tissues (52). In agreement with that report, we noted that in vivo two-photon-induced micro-lesion of the intestinal epithelium invoked an increased rate of cell detachment from the intestinal epithelium of E-cadherin knockout mice compared with controls. These latter data are consistent with the notion that cells were less faithfully attached to each other and support previous data from N-cadherin mutant animals in which crypt-villus migration rates were increased (49). Additionally, we found that genetic depletion of E-cadherin using RNAi or pharmacological blockade using EGTA similarly reduced baseline restitution, suggesting a significant role for E-cadherin in epithelial sheet migration. Cumulatively, data from these in vivo and in vitro experiments reveal a critical role for E-cadherin not only in maintenance of the epithelial barrier but, significantly, in the repair and likely restoration of barrier functions.
Studies of E-cadherin in migrating cells have focused largely on its roles in development as well as its disassembly during neoplasia (23, 53). The currently accepted model of cell migration postulates that depolarization of intestinal epithelial cells surrounding the wounded area results in the adoption of a migratory phenotype akin to mesenchymal cells. This process has been categorized as a partial epithelial-to-mesenchymal transition (EMT) (54, 55). EMT is correlated with the increased expression and responsiveness to TGF-β1 and is tightly linked with transcriptional repression of E-cadherin, resulting in diminished cell-cell adhesiveness and a promigration phenotype (56). Given that EMT is promoted through the down-regulation of E-cadherin and the concordant increase in N-cadherin (34), we postulated that TGF-β1-induced migration reflected induction of an EMT phenotype during restitution. In agreement with that notion, TGF-β1 decreased total protein level and was ineffective in altering membrane localization of E-cadherin during restitution. However, N-cadherin levels were unchanged following epithelial wounding or stimulation with either CXCL12 or TGF-β1. The absence of alterations in N-cadherin protein levels suggests that intestinal epithelial cells surrounding the wound may depolarize and migrate in an intermediate EMT phenotype absent of ”cadherin switching” (57). Thus, the cellular mechanisms whereby TGF-β1 decreases the space between cells during restitution are independent of homotypic adherens junction proteins characteristic of EMT.
Studies of intestinal restitution focus predominantly on the mechanisms regulating constitutive cellular movement. Although an array of extracellular mediators accelerates restitution, it remains to be seen how redundancy in extracellular effectors orchestrate beneficial responses. Ongoing studies in our laboratory reveal that chemokines are distinct from established TGF-β1 in the mobilization of intracellular calcium (20, 21), suggesting that redundancy in extracellular mediators is correlated with activation of parallel signaling pathways. Wounded IEC-6 monolayers treated with CXCL12 and TGF-β1 both increased paracellular sealing within migrating sheets. Consistent with our previous work showing comparable CXCL12 effects and regulatory mechanisms in multiple model epithelia, we found that CXCL12 regulated paracellular tightening and barrier functions in both IEC-6 and Caco2BBE cells (18–21, 28). Further, we demonstrated that CXCL12 stabilized E-cadherin localization at cell-cell junctions while simultaneously increasing overall levels of protein within actively migrating epithelia. CXCL12 stabilization of membrane-localized E-cadherin during migration further suggests that down-regulation of cell-cell adhesion is not the major determinant in acquiring a migratory phenotype for intestinal epithelial cells.
CXCL12-CXCR4 activation concomitantly increased F-actin accumulation and E-cadherin localization to the cell membranes of adjacent migrating cells. These data are consistent with our previous work and suggest that G-protein coupled receptor-mediated F-actin signaling may play a key role in E-cadherin organization during restitution (19). Stable E-cadherin anchorage to the F-actin cytoskeleton is well characterized (58–60). It is understood that the interaction between E-cadherin at the cell membrane and peripheral cortical F-actin stabilizes the adherens junction and, hence, cell-cell adhesion (44–47). Several reports, however, argue that interactions between the F-actin cytoskeleton and adherens junction proteins are more dynamic than previously modeled. For example, α-catenin may not be the sole linker between E-cadherin and the F-actin cytoskeleton (61). Further, different diffusion rates between adherens junction proteins and cortical F-actin argue against static E-cadherin to F-actin anchorage (59). We have shown using CytoD and Y27632 inhibitors that CXCL12-induced localization of E-cadherin depends upon F-actin polymerization. Efforts to disrupt E-cadherin-based cell-cell adhesion with CytoD have shown that within transient and initial cell-cell contacts, E-cadherin localization to cell membranes are CytoD-sensitive (62). However, within established confluent monolayers, CytoD inhibition of F-actin polymerization had a minimal effect on E-cadherin localization to sites of cell-cell adhesion (63). Consistent with that work, our data show that although CytoD treatment had a minimal effect on E-cadherin localization within stationary cells, inhibition of F-actin polymerization disrupted E-cadherin localization within migrating cells. Cumulatively, our data support a model wherein cell-cell adhesion during collective sheet migration is dynamic.
Our studies, along with others, illustrate a complex signaling circuit involving cadherin-based cell-cell adhesion and RhoA/ROCK activation (64, 65). In our studies, we used a dose of the ROCK inhibitor we have established previously to prevent CXCL12-induced migration without significantly impacting basal migration (19). With this level of ROCK inhibition, we determined that although cell migration was unimpaired, the number and size of the gaps between cells were increased significantly. The increased incidence of gaps within the collectively migrating sheet is comparable with that observed with EGTA treatment and supports a link between RhoA/ROCK and E-cadherin-dependent cell adhesion during restitution. Further support for this model stems from a recent conditional knockout study showing a loss of adherens junction formation in the absence of RhoA (66). Similar sheet migration defects, such as loss of cell-cell adhesion, were seen during zebrafish gastrulation or in actin-depolymerizing factor cofilin knockout mice (67). Further deletion and knockdown studies of zyxin, a key regulator of actin assembly, decreases the rate of E-cadherin-mediated adhesion and closing of small circular wounds (68). Taken together, these genetic studies dovetail with our pharmacological inhibitor data and support a model in which RhoA and ROCK coordinate E-cadherin localization during sheet migration.
CXCL12- and TGF-β1-stimulated restitution reflects intracellular calcium-dependent and calcium-independent signaling pathways (20, 21) that intersect to increase F-actin formation and drive restitution. However, although both stimulants reduced the macroscopic number and size of the paracellular space between migrating sheets, only CXCL12 effectively increased barrier function, as shown by increased transepithelial resistance. Together, these data indicate a further sealing between neighboring intestinal epithelial cells at a molecular level that is regulated largely by the G-protein-coupled chemokine receptor. Cumulatively, our data demonstrate that E-cadherin plays a key role in basal and inducible restitution. We postulate that restitution likely reflects epithelial “pulling” via strengthened adherens junctions between cells. The ability of CXCL12 to stabilize E-cadherin localization resulted in a functional tightening of the barrier and may be unique relative to other growth factor restitution motogens in the coincident tightening of barrier integrity with active migration to cover the denuded surface.
Therapeutic strategies to improve epithelial repair in human intestinal disease are largely focused on decreasing the motility of damage-inducing immune cells into the inflamed mucosa (24). With this work, we demonstrate a critical role for E-cadherin in restitution and indicate that in contrast to growth factors, adherens junction remodeling is preferentially regulated by chemokines. Our study is the first to show that an increase in total protein level and localization of E-cadherin corresponds to an increase in collective cell migration and barrier integrity in restitution.
Acknowledgments
We thank Dr. Stephen Duncan (Medical College of Wisconsin) for the kind gift of shRNA constructs and Dr. Michele Battle (Medical College of Wisconsin) for the E-cadherin-floxed experimental animals.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 DK062066 (to M. B. D.).

This article contains supplemental Figs. 1–6.
- ROCK
- Rho-associated protein kinase
- PTx
- pertussis toxin
- CytoD
- Cytochalasin-D
- EMT
- epithelial-to-mesenchymal transition.
REFERENCES
- 1. Turner J. R. (2009) Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 9, 799–809 [DOI] [PubMed] [Google Scholar]
- 2. Itoh M., Nagafuchi A., Moroi S., Tsukita S. (1997) Involvement of ZO-1 in cadherin-based cell adhesion through its direct binding to α catenin and actin filaments. J. Cell Biol. 138, 181–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yamada A., Irie K., Fukuhara A., Ooshio T., Takai Y. (2004) Requirement of the actin cytoskeleton for the association of nectins with other cell adhesion molecules at adherens and tight junctions in MDCK cells. Genes Cells 9, 843–855 [DOI] [PubMed] [Google Scholar]
- 4. Tunggal J. A., Helfrich I., Schmitz A., Schwarz H., Günzel D., Fromm M., Kemler R., Krieg T., Niessen C. M. (2005) E-cadherin is essential for in vivo epidermal barrier function by regulating tight junctions. EMBO J. 24, 1146–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Umeda K., Ikenouchi J., Katahira-Tayama S., Furuse K., Sasaki H., Nakayama M., Matsui T., Tsukita S., Furuse M., Tsukita S. (2006) ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell 126, 741–754 [DOI] [PubMed] [Google Scholar]
- 6. Jankowski J. A., Bedford F. K., Boulton R. A., Cruickshank N., Hall C., Elder J., Allan R., Forbes A., Kim Y. S., Wright N. A., Sanders D. S. (1998) Alterations in classical cadherins associated with progression in ulcerative and Crohn's colitis. Lab. Invest. 78, 1155–1167 [PubMed] [Google Scholar]
- 7. Barshack I., Goldberg I., Chowers Y., Weiss B., Horowitz A., Kopolovic J. (2001) Immunohistochemical analysis of candidate gene product expression in the duodenal epithelium of children with coeliac sprue. J. Clin. Pathol. 54, 684–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gassler N., Rohr C., Schneider A., Kartenbeck J., Bach A., Obermüller N., Otto H. F., Autschbach F. (2001) Inflammatory bowel disease is associated with changes of enterocytic junctions. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G216–28 [DOI] [PubMed] [Google Scholar]
- 9. Azarschab P., Porschen R., Gregor M., Blin N., Holzmann K. (2002) Epigenetic control of the E-cadherin gene (CDH1) by CpG methylation in colectomy samples of patients with ulcerative colitis. Genes Chromosomes Cancer 35, 121–126 [DOI] [PubMed] [Google Scholar]
- 10. Hollander D., Vadheim C. M., Brettholz E., Petersen G. M., Delahunty T., Rotter J. I. (1986) Increased intestinal permeability in patients with Crohn's disease and their relatives. A possible etiologic factor. Ann. Intern. Med. 105, 883–885 [DOI] [PubMed] [Google Scholar]
- 11. Katz K. D., Hollander D., Vadheim C. M., McElree C., Delahunty T., Dadufalza V. D., Krugliak P., Rotter J. I. (1989) Intestinal permeability in patients with Crohn's disease and their healthy relatives. Gastroenterology 97, 927–931 [DOI] [PubMed] [Google Scholar]
- 12. Kucharzik T., Walsh S. V., Chen J., Parkos C. A., Nusrat A. (2001) Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am. J. Pathol. 159, 2001–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lacy E. R., Ito S. (1984) Rapid epithelial restitution of the rat gastric mucosa after ethanol injury. Lab. Invest. 51, 573–583 [PubMed] [Google Scholar]
- 14. Dignass A. U., Podolsky D. K. (1993) Cytokine modulation of intestinal epithelial cell restitution. Central role of transforming growth factor β. Gastroenterology 105, 1323–1332 [DOI] [PubMed] [Google Scholar]
- 15. Blay J., Brown K. D. (1985) Epidermal growth factor promotes the chemotactic migration of cultured rat intestinal epithelial cells. J. Cell Physiol. 124, 107–112 [DOI] [PubMed] [Google Scholar]
- 16. Ciacci C., Lind S. E., Podolsky D. K. (1993) Transforming growth factor β regulation of migration in wounded rat intestinal epithelial monolayers. Gastroenterology 105, 93–101 [DOI] [PubMed] [Google Scholar]
- 17. Polk D. B. (1998) Epidermal growth factor receptor-stimulated intestinal epithelial cell migration requires phospholipase C activity. Gastroenterology 114, 493–502 [DOI] [PubMed] [Google Scholar]
- 18. Smith J. M., Johanesen P. A., Wendt M. K., Binion D. G., Dwinell M. B. (2005) CXCL12 activation of CXCR4 regulates mucosal host defense through stimulation of epithelial cell migration and promotion of intestinal barrier integrity. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G316–26 [DOI] [PubMed] [Google Scholar]
- 19. Moyer R. A., Wendt M. K., Johanesen P. A., Turner J. R., Dwinell M. B. (2007) Rho activation regulates CXCL12 chemokine stimulated actin rearrangement and restitution in model intestinal epithelia. Lab. Invest. 87, 807–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vongsa R. A., Zimmerman N. P., Dwinell M. B. (2009) CCR6 regulation of the actin cytoskeleton orchestrates human β defensin-2- and CCL20-mediated restitution of colonic epithelial cells. J. Biol. Chem. 284, 10034–10045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Agle K. A., Vongsa R. A., Dwinell M. B. (2010) Calcium mobilization triggered by the chemokine CXCL12 regulates migration in wounded intestinal epithelial monolayers. J. Biol. Chem. 285, 16066–16075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen W. C., Obrink B. (1991) Cell-cell contacts mediated by E-cadherin (uvomorulin) restrict invasive behavior of L-cells. J. Cell Biol. 114, 319–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martin P., Parkhurst S. M. (2004) Parallels between tissue repair and embryo morphogenesis. Development 131, 3021–3034 [DOI] [PubMed] [Google Scholar]
- 24. Baumgart D. C., Sandborn W. J. (2007) Inflammatory bowel disease. Clinical aspects and established and evolving therapies. Lancet 369, 1641–1657 [DOI] [PubMed] [Google Scholar]
- 25. Boussadia O., Kutsch S., Hierholzer A., Delmas V., Kemler R. (2002) E-cadherin is a survival factor for the lactating mouse mammary gland. Mech. Dev. 115, 53–62 [DOI] [PubMed] [Google Scholar]
- 26. Madison B. B., Dunbar L., Qiao X. T., Braunstein K., Braunstein E., Gumucio D. L. (2002) Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J. Biol. Chem. 277, 33275–33283 [DOI] [PubMed] [Google Scholar]
- 27. Dwinell M. B., Eckmann L., Leopard J. D., Varki N. M., Kagnoff M. F. (1999) Chemokine receptor expression by human intestinal epithelial cells. Gastroenterology 117, 359–367 [DOI] [PubMed] [Google Scholar]
- 28. Zimmerman N. P., Vongsa R. A., Faherty S. L., Salzman N. H., Dwinell M. B. (2011) Targeted intestinal epithelial deletion of the chemokine receptor CXCR4 reveals important roles for extracellular-regulated kinase-1/2 in restitution. Lab. Invest. 91, 1040–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Starodub O. T., Demitrack E. S., Baumgartner H. K., Montrose M. H. (2008) Disruption of the Cox-1 gene slows repair of microscopic lesions in the mouse gastric epithelium. Am. J. Physiol. Cell Physiol. 294, C223–32 [DOI] [PubMed] [Google Scholar]
- 30. Marchiando A. M., Shen L., Graham W. V., Weber C. R., Schwarz B. T., Austin J. R., 2nd, Raleigh D. R., Guan Y., Watson A. J., Montrose M. H., Turner J. R. (2010) Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J. Cell Biol. 189, 111–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kartenbeck J., Schmelz M., Franke W. W., Geiger B. (1991) Endocytosis of junctional cadherins in bovine kidney epithelial (MDBK) cells cultured in low Ca2+ ion medium. J. Cell Biol. 113, 881–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dürer U., Hartig R., Bang S., Thim L., Hoffmann W. (2007) TFF3 and EGF induce different migration patterns of intestinal epithelial cells in vitro and trigger increased internalization of E-cadherin. Cell Physiol. Biochem. 20, 329–346 [DOI] [PubMed] [Google Scholar]
- 33. Oft M., Heider K. H., Beug H. (1998) TGFβ signaling is necessary for carcinoma cell invasiveness and metastasis. Curr. Biol. 8, 1243–1252 [DOI] [PubMed] [Google Scholar]
- 34. Bhowmick N. A., Ghiassi M., Bakin A., Aakre M., Lundquist C. A., Engel M. E., Arteaga C. L., Moses H. L. (2001) Transforming growth factor-β1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 12, 27–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roura S., Miravet S., Piedra J., García de Herreros A., Duñach M. (1999) Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J. Biol. Chem. 274, 36734–36740 [DOI] [PubMed] [Google Scholar]
- 36. Fujita Y., Krause G., Scheffner M., Zechner D., Leddy H. E., Behrens J., Sommer T., Birchmeier W. (2002) Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell Biol. 4, 222–231 [DOI] [PubMed] [Google Scholar]
- 37. Zhao M., Discipio R. G., Wimmer A. G., Schraufstatter I. U. (2006) Regulation of CXCR4-mediated nuclear translocation of extracellular signal-related kinases 1 and 2. Mol. Pharmacol. 69, 66–75 [DOI] [PubMed] [Google Scholar]
- 38. Okabe S., Fukuda S., Broxmeyer H. E. (2002) Src kinase, but not the src kinase family member p56lck, mediates stromal cell-derived factor 1α/CXCL12-induced chemotaxis of a T cell line. J. Hematother. Stem Cell Res. 11, 923–928 [DOI] [PubMed] [Google Scholar]
- 39. Uchida D., Begum N. M., Almofti A., Nakashiro K., Kawamata H., Tateishi Y., Hamakawa H., Yoshida H., Sato M. (2003) Possible role of stromal-cell-derived factor-1/CXCR4 signaling on lymph node metastasis of oral squamous cell carcinoma. Exp. Cell Res. 290, 289–302 [DOI] [PubMed] [Google Scholar]
- 40. Chinni S. R., Yamamoto H., Dong Z., Sabbota A., Bonfil R. D., Cher M. L. (2008) CXCL12/CXCR4 transactivates HER2 in lipid rafts of prostate cancer cells and promotes growth of metastatic deposits in bone. Mol. Cancer Res. 6, 446–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tominaga T., Sahai E., Chardin P., McCormick F., Courtneidge S. A., Alberts A. S. (2000) Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol. Cell 5, 13–25 [DOI] [PubMed] [Google Scholar]
- 42. Hannemann S., Madrid R., Stastna J., Kitzing T., Gasteier J., Schönichen A., Bouchet J., Jimenez A., Geyer M., Grosse R., Benichou S., Fackler O. T. (2008) The Diaphanous-related Formin FHOD1 associates with ROCK1 and promotes Src-dependent plasma membrane blebbing. J. Biol. Chem. 283, 27891–27903 [DOI] [PubMed] [Google Scholar]
- 43. Kuroiwa M., Oneyama C., Nada S., Okada M. (2011) The guanine nucleotide exchange factor Arhgef5 plays crucial roles in Src-induced podosome formation. J. Cell Sci. 124, 1726–1738 [DOI] [PubMed] [Google Scholar]
- 44. Chu Y. S., Thomas W. A., Eder O., Pincet F., Perez E., Thiery J. P., Dufour S. (2004) Force measurements in E-cadherin-mediated cell doublets reveal rapid adhesion strengthened by actin cytoskeleton remodeling through Rac and Cdc42. J. Cell Biol. 167, 1183–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Adams C. L., Nelson W. J., Smith S. J. (1996) Quantitative analysis of cadherin-catenin-actin reorganization during development of cell-cell adhesion. J. Cell Biol. 135, 1899–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Adams C. L., Nelson W. J. (1998) Cytomechanics of cadherin-mediated cell-cell adhesion. Curr. Opin. Cell Biol. 10, 572–577 [DOI] [PubMed] [Google Scholar]
- 47. Vasioukhin V., Bauer C., Yin M., Fuchs E. (2000) Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell 100, 209–219 [DOI] [PubMed] [Google Scholar]
- 48. Moriyama N., Ishihara S., Hirose M., Watanabe S., Sato N., Kinoshita Y. (2001) E-cadherin is essential for gastric epithelial restitution in vitro. A study using the normal rat gastric mucosal cell line RGM1. J. Lab. Clin. Med. 138, 236–242 [DOI] [PubMed] [Google Scholar]
- 49. Hermiston M. L., Gordon J. I. (1995) Inflammatory bowel disease and adenomas in mice expressing a dominant negative N-cadherin. Science 270, 1203–1207 [DOI] [PubMed] [Google Scholar]
- 50. Smalley-Freed W. G., Efimov A., Burnett P. E., Short S. P., Davis M. A., Gumucio D. L., Washington M. K., Coffey R. J., Reynolds A. B. (2010) p120-catenin is essential for maintenance of barrier function and intestinal homeostasis in mice. J. Clin. Invest. 120, 1824–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bjerknes M., Cheng H. (1999) Colossal crypts bordering colon adenomas in Apc(Min) mice express full-length Apc. Am. J. Pathol. 154, 1831–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schneider M. R., Dahlhoff M., Horst D., Hirschi B., Trülzsch K., Müller-Höcker J., Vogelmann R., Allgäuer M., Gerhard M., Steininger S., Wolf E., Kolligs F. T. (2010) A key role for E-cadherin in intestinal homeostasis and Paneth cell maturation. PLoS ONE 5, e14325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nigam A. K., Savage F. J., Boulos P. B., Stamp G. W., Liu D., Pignatelli M. (1993) Loss of cell-cell and cell-matrix adhesion molecules in colorectal cancer. Br. J. Cancer 68, 507–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hay E. D., Zuk A. (1995) Transformations between epithelium and mesenchyme. Normal, pathological, and experimentally induced. Am. J. Kidney Dis. 26, 678–690 [DOI] [PubMed] [Google Scholar]
- 55. Savagner P., Kusewitt D. F., Carver E. A., Magnino F., Choi C., Gridley T., Hudson L. G. (2005) Developmental transcription factor slug is required for effective re-epithelialization by adult keratinocytes. J. Cell Physiol. 202, 858–866 [DOI] [PubMed] [Google Scholar]
- 56. Uttamsingh S., Bao X., Nguyen K. T., Bhanot M., Gong J., Chan J. L., Liu F., Chu T. T., Wang L. H. (2008) Synergistic effect between EGF and TGF-β1 in inducing oncogenic properties of intestinal epithelial cells. Oncogene 27, 2626–2634 [DOI] [PubMed] [Google Scholar]
- 57. Wheelock M. J., Shintani Y., Maeda M., Fukumoto Y., Johnson K. R. (2008) Cadherin switching. J. Cell Sci. 121, 727–735 [DOI] [PubMed] [Google Scholar]
- 58. Shapiro L., Weis W. I. (2009) Structure and biochemistry of cadherins and catenins. Cold Spring Harb. Perspect. Biol. 1, a003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yamada S., Pokutta S., Drees F., Weis W. I., Nelson W. J. (2005) Deconstructing the cadherin-catenin-actin complex. Cell 123, 889–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Abe K., Takeichi M. (2008) EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt. Proc. Natl. Acad. Sci. U.S.A. 105, 13–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kobielak A., Fuchs E. (2004) α-catenin. At the junction of intercellular adhesion and actin dynamics. Nat. Rev. Mol. Cell Biol. 5, 614–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Angres B., Barth A., Nelson W. J. (1996) Mechanism for transition from initial to stable cell-cell adhesion. Kinetic analysis of E-cadherin-mediated adhesion using a quantitative adhesion assay. J. Cell Biol. 134, 549–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hirano S., Nose A., Hatta K., Kawakami A., Takeichi M. (1987) Calcium-dependent cell-cell adhesion molecules (cadherins). Subclass specificities and possible involvement of actin bundles. J. Cell Biol. 105, 2501–2510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yamada S., Nelson W. J. (2007) Localized zones of Rho and Rac activities drive initiation and expansion of epithelial cell-cell adhesion. J. Cell Biol. 178, 517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nelson C. M., Pirone D. M., Tan J. L., Chen C. S. (2004) Vascular endothelial-cadherin regulates cytoskeletal tension, cell spreading, and focal adhesions by stimulating RhoA. Mol. Biol. Cell 15, 2943–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Herzog D., Loetscher P., van Hengel J., Knüsel S., Brakebusch C., Taylor V., Suter U., Relvas J. B. (2011) The small GTPase RhoA is required to maintain spinal cord neuroepithelium organization and the neural stem cell pool. J. Neurosci. 31, 5120–5130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lin C. W., Yen S. T., Chang H. T., Chen S. J., Lai S. L., Liu Y. C., Chan T. H., Liao W. L., Lee S. J. (2010) Loss of cofilin 1 disturbs actin dynamics, adhesion between enveloping and deep cell layers and cell movements during gastrulation in zebrafish. PLoS ONE 5, e15331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nguyen T. N., Uemura A., Shih W., Yamada S. (2010) Zyxin-mediated actin assembly is required for efficient wound closure. J. Biol. Chem. 285, 35439–35445 [DOI] [PMC free article] [PubMed] [Google Scholar]