

Background: Mice deficient in the platelet receptor CLEC-2 for podoplanin showed impaired blood/lymphatic vessel separation.
Results: Functions of lymphatic endothelial cells are inhibited by platelet releasates and BMP-9, which we identified as a novel releasate.
Conclusion: Granule contents including BMP-9 released upon platelet activation by CLEC-2-podoplanin interaction may contribute to the separation in vivo.
Significance: We proposed a novel mechanism of platelet-mediated blood/lymphatic vessel separation.
Keywords: Bone Morphogenetic Protein (BMP), Endothelial Cell, Lymphangiogenesis, Migration, Platelets, CLEC-2, Podoplanin, Proliferation, Tube Formation
Abstract
The platelet activation receptor CLEC-2 plays crucial roles in thrombosis/hemostasis, tumor metastasis, and lymphangiogenesis, although its role in thrombosis/hemostasis remains controversial. An endogenous ligand for CLEC-2, podoplanin, is expressed in lymphatic endothelial cells (LECs). We and others have reported that CLEC-2-deficiency is lethal at mouse embryonic/neonatal stages associated with blood-filled lymphatics, indicating that CLEC-2 is essential for blood/lymphatic vessel separation. However, its mechanism, and whether CLEC-2 in platelets is necessary for this separation, remains unknown. We found that specific deletion of CLEC-2 from platelets leads to the misconnection of blood/lymphatic vessels. CLEC-2+/+ platelets, but not by CLEC-2−/− platelets, inhibited LEC migration, proliferation, and tube formation but had no effect on human umbilical vein endothelial cells. Additionally, supernatants from activated platelets significantly inhibited these three functions in LECs, suggesting that released granule contents regulate blood/lymphatic vessel separation. Bone morphologic protein-9 (BMP-9), which we found to be present in platelets and released upon activation, appears to play a key role in regulating LEC functions. Only BMP-9 inhibited tube formation, although other releasates including transforming growth factor-β and platelet factor 4 inhibited proliferation and/or migration. We propose that platelets regulate blood/lymphatic vessel separation by inhibiting the proliferation, migration, and tube formation of LECs, mainly because of the release of BMP-9 upon activation by CLEC-2/podoplanin interaction.
Introduction
Previously, we had identified C-type lectin-like receptor 2 (CLEC-2)3 as a receptor for platelet-activating snake venom, rhodocytin (1). CLEC-2 is a novel class of platelet activation receptor that belongs to the C-type lectin superfamily and elicits robust activation signals in conjunction with the Src family kinases, Syk, and phospholipase Cγ2 (PLCγ2). SLP-76 (adapter protein SH2 domain-containing leukocyte protein of 76 kDa) and the linker for activation of T cells are also necessary for the full activation of platelets induced by CLEC-2 (1). We also identified podoplanin as an endogenous ligand for CLEC-2, which was later confirmed by other groups (2, 3). Podoplanin is expressed on the surface of tumor cells and induces platelet aggregation by binding to CLEC-2, which facilitates hematogenous tumor metastasis (2, 4). Podoplanin is also expressed in kidney podocytes (the origin of its nomenclature), type I lung alveolar cells, and lymphatic endothelial cells (LECs) but not in vascular endothelial cells (reviewed in Ref. 5). As podoplanin in these cells cannot interact with CLEC-2 in the bloodstream, roles for CLEC-2/podoplanin interaction in the latter three cell types have remained unknown. Recently, we and others independently generated CLEC-2-deficient mice and reported that CLEC-2-deficient mice show embryonic/neonatal lethality, severe edema, and blood-filled lymphatic vessels because of impaired blood/lymphatic separation (6, 7). CLEC-2-deficient mice show embryonic/neonatal lethality, and almost all of the CLEC-2−/− pups die shortly after birth, probably because of respiratory failure. These studies revealed that the CLEC-2/podoplanin interaction facilitates blood/lymphatic vessel separation in the developmental stages when primary lymph sacs are derived from the cardinal vein. However, because other blood cells, including neutrophils, monocytes, and dendritic cells, express CLEC-2 in mice, it is not known whether this separation requires CLEC-2 expression in platelets. A misconnection phenotype was also observed in mice deficient in podoplanin and the signaling molecules necessary for platelet activation downstream of CLEC-2, including Syk, SLP-76, and PLCγ2 (8, 9). Moreover, platelet aggregate formation, which is an indicator of platelet activation, was observed in the connection between the cardinal vein and primary lymph sacs in normal mice but not in SLP-76- or podoplanin-deficient mice (6, 10). These findings suggest that platelet activation is necessary for blood/lymphatic vessel separation. However, the mechanism by which CLEC-2 in platelets regulates blood/lymphatic vessel separation remains to be elucidated.
In the present study, we demonstrated that CLEC-2 in platelets is required for the blood/lymphatic vessel separation using mice with CLEC-2 specifically deleted from platelets. We found that co-culture of platelets, or supernatants from activated platelets, significantly inhibited cell migration, proliferation, and tube formation of LECs in a manner dependent on CLEC-2. These findings suggest that granule contents released upon platelet activation inhibit lymphangiogenesis. We found that activated platelets released the transforming growth factor-β (TGF-β) family protein bone morphogenetic protein-9 (BMP-9) and that this was involved in platelet-mediated inhibition of lymphangiogenesis. We proved that BMP-9 is an important mediator and that a combination of the platelet releasates (BMP-9, TGF-β, platelet factor 4 (PF4), angiostatin, and endostatin) appears to account for platelet-mediated inhibition of lymphangiogenesis in vivo. We reported some of these results previously in abstract form following the 52nd Annual Meeting of the American Society of Hematology (11).
EXPERIMENTAL PROCEDURES
Reagents
PF4-Cre transgenic mice were kindly donated by Prof. Radek C. Skoda (University Hospital Basel, Switzerland) (12). The glycoprotein VI (GPVI) agonist poly(PHG) was a generous gift from the JNC Corp. (Yokohama, Japan). Lotrafiban was donated by GlaxoSmithKline. Recombinant VEGF165, TGF-β, and BMP-9 were purchased from R&D Systems (Minneapolis). Recombinant PF4 was from Hematologic Technologies, Inc. (Essex Junction, VT). Recombinant angiostatin was purchased from Enzo Life Sciences (Farmingdale, NY). Recombinant endostatin was from Cedarlane Laboratories (Sanford, NC). The recombinant extracellular domain of human CLEC-2 expressed as a dimeric rabbit immunoglobulin Fc domain fusion protein (hCLEC-2-rFc2) was generated as described previously (2). Anti-BMP-9-neutralizing antibody was from R&D Systems. Other reagents were from sources described below or previously (13, 14) .
Generation of Mice
CLEC-2flox/+ mice were generated using a targeting vector designed so that part of exon 1 flanked by two loxP sites could be deleted by expression of Cre protein (7). We crossed a CLEC-2flox/+ mouse with a PF4-Cre transgenic mouse that specifically expresses Cre recombinase in platelets/megakaryocytes to generate PF4-Cre;CLEC-2flox/+ mice. These mice were crossed to generate PF4-Cre;CLEC-2flox/flox mice, which have CLEC-2 specifically deleted from the platelets. Genotypes of CLEC-2 floxed mice and PF4-Cre transgenic mice were analyzed as described previously (7, 12). Because CLEC-2-null mice are embryonic/neonatal lethal, CLEC-2-deficient irradiated chimeric mice were generated as described previously (7). Briefly, adult C57BL/6 male mice were given two irradiations of 500 rads from a 60Co source at 3-h intervals. The mice were then rescued by intravenous injection of 1 × 106 fetal liver cells from CLEC-2−/− (CLEC-2 chimera) or CLEC-2+/+ embryos (WT chimera) at E13.5–E15.5. The reconstituted mice were used for experiments no sooner than 7 weeks following irradiation. This study was approved by the Animal Care and Use Committee at the University of Yamanashi.
Flow Cytometry
Whole blood drawn from mice as described (7) was diluted 15-fold using modified Tyrode's buffer. Twenty-five microliters of the diluted whole blood was incubated with Cy2-labeled anti-mouse CLEC-2 and Cy2-labeled control rabbit IgG, both of which were generated as described previously (7). Reactions were terminated by the addition of 400 μl of PBS, and the samples were analyzed using a FACScan (BD Biosciences) and CellQuest software (BD Biosciences).
Microscopy
Mice were anesthetized by diethyl ether, and the small intestine and mesentery were photographed using an Optio WG-1 digital camera (Pentax-Hoya Corp., Tokyo). The mesentery was removed from euthanized mice, fixed in 3.7% formalin, and embedded in paraffin. Sections were stained with rabbit anti-mouse LYVE-1 antibody (Abcam) using Simple stain MAX peroxidase for mouse tissue and rabbit antibodies, according to the manufacturer's instructions. Immunohistochemical analysis of embryonic back skin was performed as described previously (7).
Cells
Human umbilical vein endothelial cells (HUVECs) and human lymphatic endothelial cells (hLECs) were purchased from Lonza (Basel, Switzerland) and maintained on culture dishes in endothelial growth medium-2 (EGM-2; Lonza) supplemented with 5% FBS and an EGM-2 microvascular set (0.5 ml of human EGF, 0.2 ml of hydrocortone, 25 ml of FBS, 0.5 ml of VEGF, 2 ml of human FGF-B, 0.5 ml of R3-IGF-1, 0.5 ml of ascorbic acid, and 0.5 ml of GA-1000). Cultures were maintained at 37 °C, 5% CO2 and 100% humidity. Conditionally immortalized murine lymphatic endothelial cells (mLECs) were prepared as described previously (15). mLECs were maintained on gelatinized culture dishes in EGM-2 supplemented with 5% FBS and an EGM-2 microvascular set at 33 °C, 5% CO2, and 100% humidity.
Cell Migration Assay
Transwell migration assays were performed in a modified Boyden-type blind well chamber (Neuro Probe, Inc., Gaithersburg, MD) with a 12-μm (for hLECs) or 8-μm (for HUVECs) nucleopore polycarbonate membrane separating the upper compartment from the bottom chamber, which contained serum-free medium (SFM) for endothelial cells (human endothelial SFM, Invitrogen). HUVECs or hLECs were kept in human endothelial SFM for 1 h, harvested, and resuspended in SFM at 3 × 105 cells/ml. One hundred eighty microliters of cells were loaded into the upper compartment with 20 μl of washed platelets or the indicated reagents. Where indicated, the washed platelets were pretreated with 10 μm lotrafiban for 10 min. After incubation for 6 h at 37 °C with 5% CO2, the membrane was fixed for 30 min in 70% ethanol and stained with Wright-Giemsa. Nonmigrated cells from the upper surface of the filter were scraped off with a cotton bud. The membrane was mounted bottom-side-up on glass slides, and the number of migrated cells/well was determined by counting cells in five randomly selected microscopic view fields at ×200 magnification.
For the wound closure assay, a confluent monolayer of HUVECs or hLECs was scratched with a sterile pipette tip across the plate. The plates were washed twice with supplemented EGM-2 medium. After the wounds were marked and photographed, cells were incubated with washed platelets or the indicated reagents for 20 h. Where indicated, washed platelets were pretreated with 10 μm lotrafiban for 10 min. The marked wound was photographed, and the distance of migration was determined by measuring the width of the wound and subtracting this value from the initial width of the wound.
Cell Proliferation Assay
A cell proliferation assay was performed using Click-iTTM EdU flow cytometry assay kits (Invitrogen). Subconfluent HUVECs or hLECs were washed and kept in human endothelial SFM for 2 or 6 h, respectively. After the addition of a thymidine analog, EdU, together with the washed platelets or the indicated reagents, HUVECs and hLECs were incubated for 2 and 4 h, respectively. Where indicated, washed platelets were pretreated with 10 μm lotrafiban for 10 min. The incorporation of EdU into the cells was measured according to the manufacturer's instructions.
Tube Formation Assay
Subconfluent HUVECs, hLECs, or mLECs were prestained with 10 μg/ml DiIC12(3) fluorescent dye (BD Biosciences) at 37 °C for 1 h and then harvested with trypsin/EDTA; detached cells were collected by centrifugation at 1000 rpm for 5 min. Cell suspensions (HUVECs, 5 × 105/ml; hLECs, 4 × 105/ml; mLECs, 1.25 × 105/ml) were then prepared in supplemented EGM-2. Where indicated, cells were pretreated with human washed platelets or the indicated reagents. Then, 50 μl of cell suspension was added to each well of a 96-well BD BioCoat angiogenesis plate (BD Biosciences) that had been precoated with Matrigel matrix. After incubation for 16 h at 37 °C (for mLECs, 33 °C), the tube-like network was visualized by a fluorescent IX71 microscope (Olympus, Tokyo) and photographed using a DP-70 digital camera (Olympus). The tube-like network was traced using a Bamboo tablet (Wacom, Saitama, Japan), and total length was quantified using ImageJ software.
Platelet Preparation
Venous blood from healthy drug-free volunteers was collected into 10% sodium citrate. This study was approved by the Ethical Committee at the University of Yamanashi, and written informed consent was provided according to the Declaration of Helsinki. Wild-type chimeras or CLEC-2 chimeras were killed with diethyl ether, and blood drawn by post-caval puncture was collected into 100 μl of acid-citrate-dextrose. The washed human or murine platelets were obtained by centrifugation as described previously using prostacyclin to prevent activation during the isolation procedure (14). Both types of platelets were resuspended in modified (calcium-free) Tyrode's buffer (14) at the indicated cell densities.
Generation of Platelet-activated Supernatants
The washed human platelets (2 × 109/ml) were stimulated with calcium-free Tyrode's buffer or 1 μg/ml poly(PHG). Poly(PHG) is a synthetic collagen fiber made by polycondensation of Pro-Hyp-Gly, which spontaneously assumes a polymeric structure with a molecular mass greater than 105. Poly(PHG) has been proven to potently stimulate platelets through GPVI (16). After stimulation with poly(PHG) for 10 min without stirring, platelets were removed by centrifugation, and the resultant supernatant was filtered using a 0.22-μm filter to completely remove the platelets from the supernatant. The effect of the supernatant was examined at a final concentration of 10%. For BMP-9 Western blotting, centrifugation was performed in the presence of 0.1 μg/ml prostaglandin I2 and 15% acid-citrate-dextrose.
Western Blotting
Western blotting was performed as described previously (14). Briefly, platelet supernatants, washed human platelets (1 × 109/ml), and recombinant human BMP-9 were dissolved in SDS sample buffer, separated by 4–12% SDS-PAGE, electrotransferred, and Western-blotted with rabbit anti-human BMP-9 antibody (GeneTex).
Statistics
Statistical significance was evaluated by Student's t test. In each case, p values of <0.05 were taken as the minimum indication of statistical significance.
RESULTS
Mice with CLEC-2 Specifically Deleted from Platelets Have Impaired Blood/Lymphatic Vessel Separation
In mice, CLEC-2 expression has been reported in neutrophils, monocytes, and dendritic cells in addition to platelets (17, 18). To examine the role of platelets in blood-lymphatic vascular separation, we generated mice that had tissue-specific, conditional knock-out of CLEC-2 from platelets and megakaryocytes with the use of a PF4-Cre transgene (12). Previously, we had generated CLEC-2flox/flox mice, in which part of the CLEC-2 exon 1 was flanked by two loxP sites (7). We crossed a CLEC-2flox/flox mouse with a mouse that expresses Cre recombinase in megakaryocytes/platelets to generate PF4-Cre;CLEC-2flox/flox mice. In contrast to CLEC-2-null mice, these mice were not embryonic/neonatal lethal. Flow cytometric analysis showed that CLEC-2 was deleted from the platelets (supplemental Fig. 1) but not from neutrophils in PF4-Cre;CLEC-2flox/flox mice (data not shown). Mice with CLEC-2 specifically deleted from the platelets have the macroscopic and histological signs of blood/lymphatic misconnection. We observed blood-filled red lymphatic vessels in the mesentery and small intestines of the conditional knock-out mice but not in the control mice (Fig. 1A). Moreover, the small intestines of the knock-out mice, but not those of the control mice, were severely edematous (Fig. 1A). Histological analysis of the mesentery confirmed blood-filled lymphatic vessels in the absence of CLEC-2 (Fig. 1B).
FIGURE 1.

Blood/lymphatic vessel misconnection in mice with CLEC-2 specifically deleted from platelets. A, mesentery (upper row) and small intestine (lower row) from wild type;CLEC-2flox/+ (control) and PF4-Cre;CLEC-2flox/flox (CLEC-2 conditional knock-out (KO) from platelets/megakaryocytes (plts/megs)). A, V, and L denote artery, vein, and lymphatic vessels, respectively. The arrows in the upper right panel indicate a lymphatic vessel. B, mesenteric sections of 10-week-old WT;CLEC-2flox/flox (left panel (control)) and PF4-Cre;CLEC-2flox/flox mice (right panel (CLEC-2 conditional KO in plts/megs)) stained with LYVE-1. C, whole-mount triple fluorescence confocal microscopy of embryonic back skin was performed with antibodies to PECAM-1 (red), LYVE-1 (green), and TER-119 (blue) at E17.5. Arrows indicate the distended lymphatic vessels containing erythrocytes.
We next performed a whole-mount triple fluorescence confocal microscopic examination of embryonic back skin (E17.5) using antibodies to PECAM-1, LYVE-1, and TER-119 (a molecular marker of erythrocytes). Triple staining revealed that the dilated lymphatic vessels in embryos with CLEC-2-deficient platelets contained erythrocytes, whereas those in control embryos did not. In platelet-specific CLEC-2-deficient embryos, lymphatic vessels stained for LYVE-1 and PECAM-1 exhibited a dilated, tortuous, rugged appearance, whereas lymphatic vessels in control embryos had a narrow, straight, smooth appearance (Fig. 1C). Taken together, these findings indicate that CLEC-2 is required for normal blood/lymphatic separation in platelets but not in other cells.
Platelets Inhibit Cell Migration and Proliferation of hLECs but Not Those of HUVECs in a CLEC-2-dependent Manner
We next investigated the mechanism of how CLEC-2 regulates blood/lymphatic vessel separation. Platelet aggregates have been observed in the connection between the lymph sac and the vein in wild-type mice but not in SLP-76 or podoplanin-deficient mice (6, 10). It is possible that platelet aggregates physically occlude the connection between the lymph sac and the vein. However, it is also possible that platelets have some effect on LEC function close to platelet aggregates. Here, we investigated whether platelets affect LEC migration, proliferation, or tube formation.
In the Boyden chamber, hLECs or HUVECs in the upper chamber randomly migrated and passed through the micropores into the lower chamber without chemoattractant. With a co-culture of human or wild-type mouse platelets, hLEC migration significantly decreased by 30% compared with the platelet-free control (Fig. 2A, i). Interestingly, CLEC-2-deficient platelets did not inhibit hLEC migration (Fig. 2A, i). In contrast to hLEC migration, a co-culture of platelets did not inhibit but rather increased HUVEC migration (Fig. 2A, ii). Migration of both hLECs and HUVECs increased in the presence of VEGF-A (Fig. 2A), indicating that cells were still capable of migration during the experiment.
FIGURE 2.
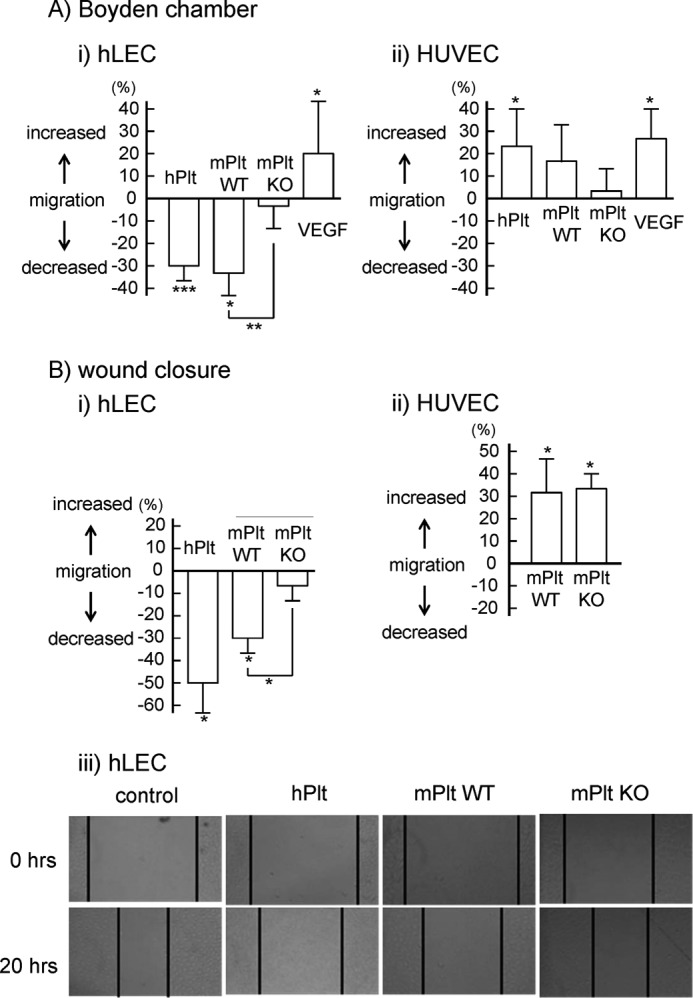
Inhibitory effects of platelets on endothelial cell migration through CLEC-2. A, cell migration of hLEC (i) and HUVEC (ii) in the presence of buffer, human washed platelets (hPlt) (1 × 108/ml), WT murine washed platelets (mPlt) (1 × 108/ml), CLEC-2-deficient murine washed platelets (mPlt KO, 1 × 108/ml), or VEGF (20 ng/ml) was investigated by Boyden-type transwell migration assay. Quantification of the migration was performed as described under “Experimental Procedures.” The graph illustrates percent change ±S.E. from base line (buffer) (n = 10 from four independent experiments). B, cell migration of hLEC (i and iii) and HUVEC (ii) in the presence of buffer, hPlt (1 × 108/ml), mPlt WT (1 × 108/ml), and mPlt KO (1 × 108/ml) was investigated by means of a wound closure assay. Quantification of the migration was performed as described under “Experimental Procedures” (i and ii). The graph illustrates percent change ± S.E. from base line (buffer) (n = 10 from four independent experiments). One, two, and three asterisks denote p < 0.05, p < 0.01, and p < 0.005, respectively. The wound in the hLEC monolayer was photographed (iii). Black lines indicate the front of an hLEC monolayer.
We also investigated migration using a wound closure assay in which migration was measured by a shortening of the scratch width across a confluent monolayer of endothelial cells. Twenty hours after scratches were made, the scratch width decreased as a result of cell migration. Co-culture of human and wild-type mouse platelets significantly inhibited hLEC migration by 50 and 30%, respectively, whereas co-culture of CLEC-2-deficient platelets did not inhibit hLEC migration (Fig. 2B, i and iii). In contrast, co-culture of platelets increased HUVEC migration (Fig. 2B, ii). These findings are consistent with the results from the Boyden chamber, suggesting that platelets inhibit hLEC migration but not HUVEC migration, and that this inhibition is dependent on CLEC-2.
The proliferation of hLECs and HUVECs was investigated by a thymidine analog incorporation assay. Co-culture of human or wild-type mouse platelets significantly inhibited hLEC proliferation by 20% (Figs. 3, A and B). However, co-culture of CLEC-2-deficient platelets did not inhibit hLEC proliferation. Platelets did not affect the proliferation of HUVECs (Figs. 3, A and C). Taken together, these findings indicate that platelets inhibited hLEC migration and proliferation in a CLEC-2-dependent manner.
FIGURE 3.
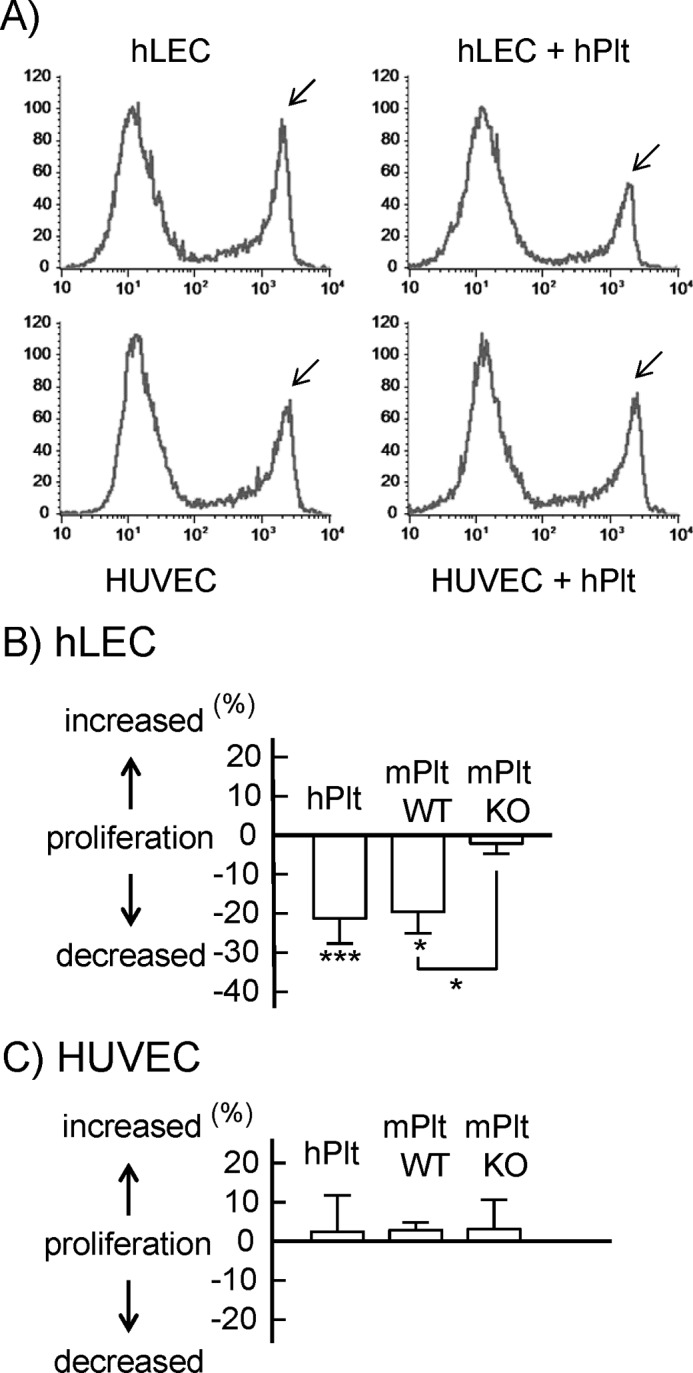
Inhibitory effects of platelets on endothelial cell proliferation. Platelets inhibited hLEC proliferation but not HUVEC proliferation, depending on CLEC-2. A, cell proliferation of hLECs (upper panels) and HUVECs (lower panels) in the presence of buffer (left panels) and hPlt (right panels, 1 × 108/ml) was investigated by thymidine analog incorporation assay. A group of EdU-incorporated cells is indicated by arrows. B and C, cell proliferation of hLECs (B) and HUVECs (C) in the presence of buffer, hPlt (1 × 108/ml), mPlt WT (1 × 108/ml), and CLEC-2-deficient murine washed platelets (mPlt KO, 1 × 108/ml) was investigated by thymidine analog incorporation assay. Quantification of the proliferation was performed as described under “Experimental Procedures.” The graph illustrates percent change ± S.E. from base line (buffer) (n = 10 from four independent experiments).
Platelets Inhibit Tube Formation of LECs but Not Those of HUVECs in a CLEC-2-dependent Manner
Because tube formation is an integrated process of cell migration and proliferation, we next investigated the effects of platelets on the tube formation of LECs. We observed that normal mouse platelets inhibited HUVEC tube formation as well as hLEC tube formation (data not shown), although HUVEC migration and proliferation were not affected (Figs. 2 and 3). We have no explanation for this phenomenon, which will need to be elucidated elsewhere. We utilized mLECs to investigate the effects of CLEC-2-deficient platelets on LEC tube formation.
The co-culture of human platelets significantly inhibited hLEC tube formation by 80% (Fig. 4, A and C). On the other hand, it had no inhibitory effects on HUVEC tube formation (Fig. 4, B and C), although a slight thickening of the tube walls was observed in the presence of platelets (Fig. 4B). Fig. 4, D and E, shows that normal mouse platelets, but not CLEC-2−/− platelets, significantly inhibited tube formation of mLECs, as was the case in hLEC migration and proliferation. Taken together, it is suggested that platelets inhibit the migration, proliferation, and tube formation of LECs, but not of HUVECs, in a CLEC-2-dependent manner.
FIGURE 4.
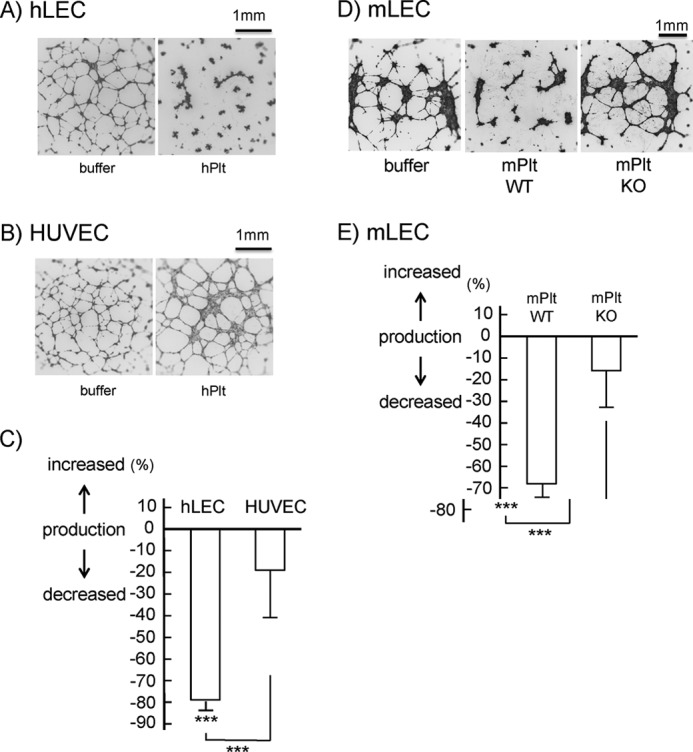
Inhibitory effects of platelets on tube formation of endothelial cells through CLEC-2. A and B, tube formation of hLECs (4 × 105/ml) (A) or HUVECs (5 × 105/ml) (B) in the presence (hPlt) or absence (buffer) of human washed platelets (1 × 108/ml). Images are representative of five different experiments. C, quantification of tube formation. D, tube formation of mLECs (1.25 × 105/ml) in the presence of buffer, wild-type mPlt WT (1 × 107/ml), and CLEC-2-deficient murine washed platelets (mPlt KO, 1 × 107/ml). Images are representative of three different experiments. E, quantification of mLEC tube formation. The graphs in C and E show quantification of tube formation as percent change ± S.E. from base line (buffer) (n = 10–12 from three independent experiments). Three asterisks denote p < 0.005.
There still remains the possibility that podoplanin generates inhibitory signals for LEC migration by binding to CLEC-2, as podoplanin interacts directly with ezrin and increases cell migration and invasiveness in MDCK cells (19). To address this possibility, we investigated the effects of the recombinant extracellular domain of human CLEC-2 expressed as a dimeric rabbit immunoglobulin Fc domain fusion protein (hCLEC-2-rFc2). hCLEC-2-rFc2 significantly inhibited the migration of hLECs but not that of HUVECs (supplemental Fig. 2A). On the other hand, the recombinant CLEC-2 did not affect the proliferation of hLECs/HUVECs (supplemental Fig. 2B) or of hLEC tube formation (supplemental Fig. 2C). These findings suggest that podoplanin cross-linking also regulates LEC function at least in part.
Integrin αIIbβ3 Blocker Did Not Attenuate Platelet-induced Inhibition of hLEC Migration, Proliferation, and Tube Formation, whereas Supernatants from Activated Platelets Inhibited hLEC Migration, Proliferation, and Tube Formation
Previous studies observed the misconnection phenotype in mice deficient in Syk, SLP-76, or PLCγ2, all of which are necessary signaling molecules in CLEC-2-mediated signal transduction (8, 9). Podoplanin-deficient mice also show the same phenotype (10). Moreover, platelet aggregates are observed in the orifice of the lymph sacs in wild-type mice but not in SLP-76- or podoplanin-deficient mice (6, 10). Together, these findings suggest that platelet activation induced by CLEC-2-podoplanin interaction is necessary for blood/lymphatic vessel separation. Platelet activation leads to two events, granular release and platelet aggregate formation. We next sought to clarify which of these events is important for blood/lymphatic vessel separation. To address this question, we utilized an integrin αIIbβ3 blocker, lotrafiban, to inhibit platelet aggregation. The αIIbβ3 blocker itself did not have any effect on the migration, proliferation, and tube formation of hLECs (Fig. 5, A–C, middle bars). Pretreatment of platelets with lotrafiban did not attenuate platelet-induced inhibition of hLEC migration, proliferation, and tube formation (Fig. 5, A–C, compare right and left bars), suggesting that platelet aggregation is not required for lymph/blood vessel formation.
FIGURE 5.
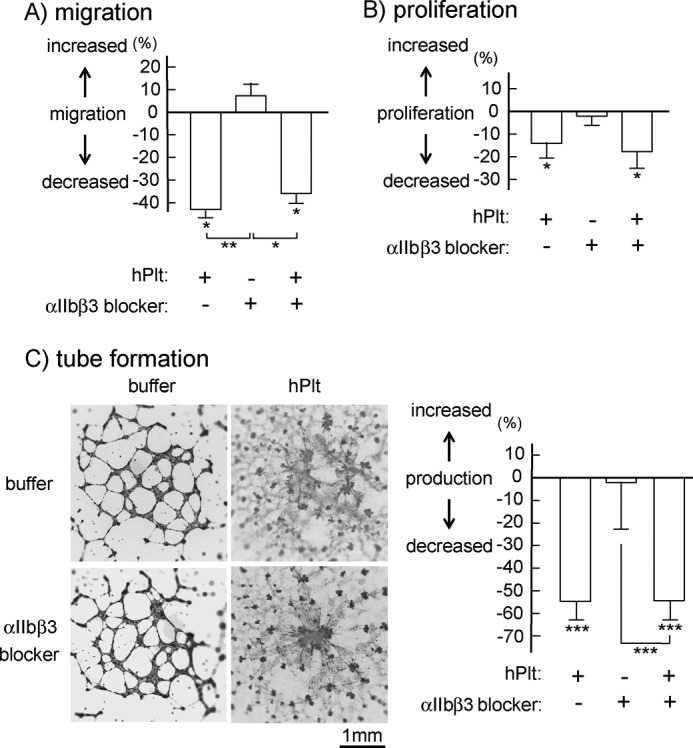
The lack of effect of integrin αIIbβ3 blocker on platelet-induced inhibition of hLEC migration, proliferation, or tube formation. A, cell migration of hLEC in the presence of buffer, hPlt (1 × 108/ml), lotrafiban (αIIbβ3 blocker; final concentration, 1 μm), or human washed platelets pretreated with 10 μm lotrafiban (final concentration, 1 × 108/ml platelets, 1 μm lotrafiban) was investigated by Boyden-type transwell migration assay. Quantification of the migration was performed as described under “Experimental Procedures.” The graph illustrates percent change ± S.E. from base line (buffer) (n = 10 from four independent experiments). B, cell proliferation of hLECs in the presence of buffer, platelets, or lotrafiban as described in A was investigated by thymidine analog incorporation assay. Quantification of the migration was performed as described under “Experimental Procedures.” The graph illustrates percent change ± S.E. from base line (buffer) (n = 10 from four independent experiments). C, tube formation of hLECs in the presence of buffer, platelets, or lotrafiban as described in A was investigated. The images of tube formation were photographed (left images). Quantification of the migration was performed as described under “Experimental Procedures.” The graph on the right illustrates percent change ± S.E. from base line (buffer) (n = 10 from four independent experiments). One, two, and three asterisks indicate p < 0.05, p < 0.01, and p < 0.005, respectively.
As αIIbβ3 blocker did not attenuate platelet-induced inhibition of hLEC migration, proliferation, and tube formation, we next investigated the effects of released granule contents from platelets upon platelet activation. Platelets are activated by the GPVI agonist poly(PHG). After separation of the activated platelets, the resultant supernatants were filtered and added to hLECs. We chose a GPVI agonist to stimulate platelets, because LECs do not express GPVI. The supernatant significantly inhibited the migration, proliferation, and tube formation of hLECs, whereas the agonist alone did not affect migration and proliferation (Fig. 6). These findings suggest that the contents of granules released from activated platelets, but not platelet aggregates, inhibit LEC migration, proliferation, and tube formation.
FIGURE 6.
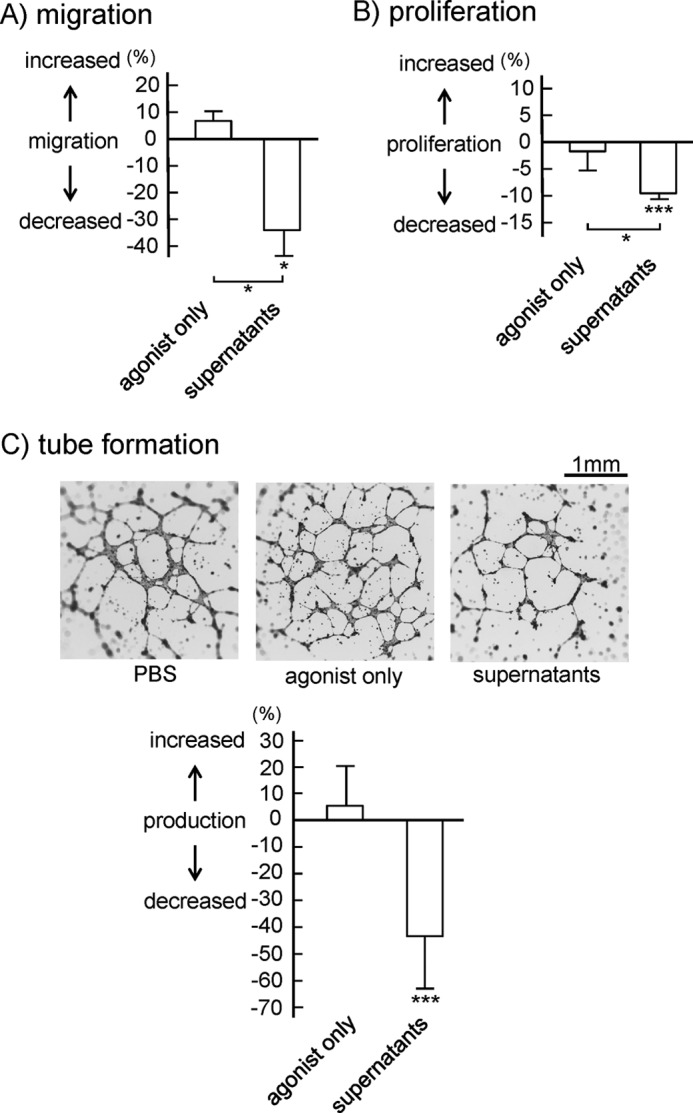
Inhibitory effects of supernatants from activated platelets on hLEC migration, proliferation, and tube formation. A, cell migration of hLECs in the presence of buffer, control supernatants (agonist only), or activated platelet supernatants was investigated by Boyden-type transwell migration assay. Quantification of the migration was performed as described under “Experimental Procedures.” The graph illustrates percent change ± S.E. from base line (buffer) (n = 10 from four independent experiments). B, cell proliferation of hLECs in the presence of buffer, control supernatant, or activated platelet supernatant was investigated by thymidine analog incorporation assay. Quantification of the migration was performed as described under “Experimental Procedures.” The graph illustrates percent change ± S.E. from base line (buffer) (n = 10 from four independent experiments). C, tube formation of hLECs in the presence of buffer (PBS), control supernatant, or activated platelet supernatant was investigated and photographed (upper images). Quantification of tube formation was performed as described under “Experimental Procedures.” The graph (lower panel) illustrates percent change ± S.E. from base line (buffer) (n = 10 from four independent experiments). One and three asterisks indicate p < 0.05 and p < 0.005, respectively.
Platelet Granule Contents Inhibited hLEC Migration, Proliferation, and Tube Formation
To characterize the molecule(s) that is responsible for the inhibition of LEC migration, proliferation, and tube formation, we heated the supernatants from activated platelets to 100 °C and examined the effects of the heated supernatants on hLEC function. Heated supernatants lost their inhibitory effect on hLEC migration (supplemental Fig. 3A) and proliferation (supplemental Fig. 3B). Heated control supernatant (poly(PHG)-containing buffer) also had no effect on migration (supplemental Fig. 3A) and proliferation (data not shown). These findings suggest that proteins present in the activated platelet supernatants, which are capable of being inactivated by heating, are responsible for the inhibition of hLEC function.
We next investigated a molecule responsible for platelet-mediated inhibition of lymphangiogenesis. We identified TGF-β, PF4, angiostatin, and endostatin as probable candidates, because they have been reported to inhibit lymphangiogenesis (20–22), and platelets contain these molecules in α-granules. BMP-9 is a member of the TGF-β family that binds to type I and type II serine-threonine kinase receptors (23). BMP-9 inhibits basic fibroblast growth factor-stimulated migration and proliferation of bovine aortic endothelial cells (24). BMP-9 also inhibits the migration and growth of human dermal microvascular endothelial cells (25), suggesting that BMP-9 has an inhibitory effect on endothelial cells. BMP-9 is highly expressed in the liver (26, 27), but its expression in platelets has not been elucidated to date. Western blotting with an antibody against BMP-9 showed that platelets expressed both BMP prepropeptides of 400–525 amino acids (28, 29) (Fig. 7A, lane c, arrowhead) and mature BMP of 100–140 amino acids (30) (Fig. 7A, lane c, asterisk). The recombinant BMP-9 made in Chinese hamster ovary cells is ∼13 kDa (Fig. 7A, lane d), which is consistent with the molecular mass of mature BMP. In addition to the 50- and 13-kDa bands, platelets have a 25-kDa BMP-9 band (Fig. 7A, lane c, arrow), which appears to be due to human-specific post-translational modifications. The 25-kDa BMP-9 was also detected in supernatants from activated platelets, whereas only a negligible band was detected in supernatants from resting platelets (Fig. 7A, lanes a and b), suggesting that BMP-9 is released from activated platelets.
FIGURE 7.
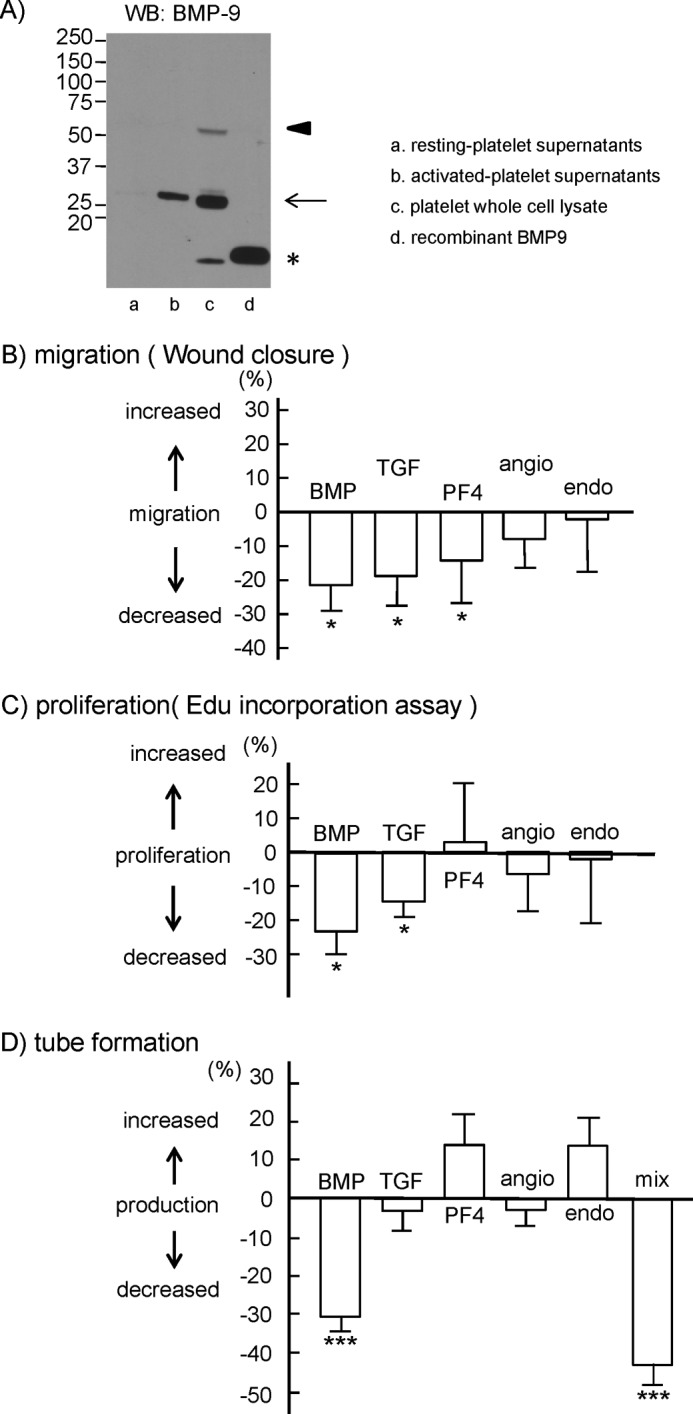
Effects of platelet granule contents on hLEC migration, proliferation, and tube formation. A, Western blotting (WB) with anti-BMP-9 antibody. The asterisk indicates mature BMP-9. The arrow indicates putative glycosylated mature BMP-9. The arrowhead indicates BMP-9 prepropeptides. B, cell migration of hLECs in the presence of buffer, 10 ng/ml BMP-9 (BMP), 10 ng/ml TGF-β (TGF), 1.5 μg/ml PF4, 1 ng/ml angiostatin (angio), or 150 ng/ml endostatin (endo) was investigated by wound closure assay. Quantification of the migration was performed as described under “Experimental Procedures.” The graph illustrates percent change ± S.E. from base line (buffer) (n = 10 from four independent experiments). C, cell proliferation of hLECs in the presence of buffer, 10 ng/ml BMP-9, 10 ng/ml TGF-β, 1.5 μg/ml PF4, 1 ng/ml angiostatin, or 150 ng/ml endostatin was investigated by thymidine analog incorporation assay. Quantification of the proliferation was performed as described under “Experimental Procedures.” The graph illustrates percent change ± S.E. from base line (buffer) (n = 10 from four independent experiments). D, tube formation of hLECs in the presence of 10 ng/ml BMP-9, 10 ng/ml TGF-β, 1.5 μg/ml PF4, 1 ng/ml angiostatin, 150 ng/ml endostatin, or a mixture of these molecules (mix). Quantification of the tube formation was performed as described under “Experimental Procedures.” The graph illustrates percent change ± S.E. from base line (buffer) (n = 10 from four independent experiments). One and three asterisks indicate p < 0.05 and p < 0.005, respectively.
Here, BMP-9, TGF-β, and PF4 significantly inhibited hLEC migration, whereas angiostatin and endostatin did not (Fig. 7B). BMP-9 and TGF-β also significantly inhibited hLEC proliferation, whereas PF4, angiostatin, and endostatin did not (Fig. 7C). As expected, BMP-9, but not angiostatin or endostatin, significantly inhibited the tube formation of hLECs (Fig. 7D). However, although TGF-β inhibited migration and proliferation, it did not inhibit tube formation (Fig. 7D). PF4 also inhibited hLEC migration but did not inhibit tube formation (Fig. 7D). Under in vivo activation conditions, it is most likely that platelets release almost all of the granule contents at the same time. The mixture of TGF-β, BMP-9, PF4, angiostatin, and endostatin significantly inhibited tube formation (Fig. 7D); suggesting that BMP-9 was still able to inhibit tube formation in the presence of PF4 and endostatin. Thus, we propose that podoplanin in LECs activates platelets by binding to CLEC-2 in the connection between the lymph sac and vein during the developmental stages and that released BMP-9, in conjunction with other releasates, inhibits migration, proliferation, and tube formation, which facilitates blood/lymphatic vessel separation.
Anti-BMP-9-neutralizing Antibody Cancelled the Inhibitory Effects of Activated Platelet Supernatants on LEC Migration, Proliferation, and Tube Formation
We next investigated whether BMP-9 is the major secreted mediator for inhibition of hLEC migration, proliferation, and tube formation using an anti-BMP-9 neutralizing antibody. The antibody alone had no significant effects on hLEC migration, proliferation, and tube formation, although it showed a tendency to stimulate these responses (Fig. 8). Inhibition of hLEC migration, proliferation, and tube formation by platelet supernatants was cancelled in the presence of the neutralizing anti-BMP-9 antibody (Fig. 8), suggesting that the inhibitory effects of platelet supernatants are mainly because of BMP-9.
FIGURE 8.
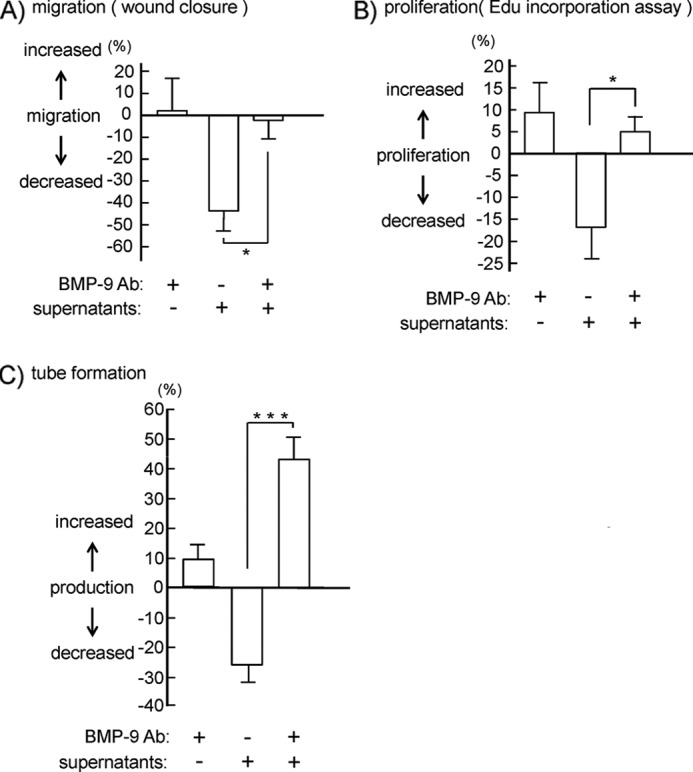
Effects of the anti-BMP-9-neutralizing antibody on inhibition of hLEC migration, proliferation, and tube formation by platelet granule contents. A, cell migration of hLECs in the presence of buffer, 10 μg/ml anti-BMP-9-neutralizing antibody (BMP-9 Ab), supernatants from activated platelets, and supernatants plus 10 μg/ml anti-BMP-9-neutralizing antibody was investigated by wound closure assay. B, cell proliferation of hLECs in the presence of buffer, 1 μg/ml the anti-BMP-9-neutralizing Ab, supernatants from activated platelets, and supernatants plus 1 μg/ml anti-BMP-9-neutralizing antibody was investigated by thymidine analog incorporation assay. C, tube formation of hLECs in the presence of buffer, 1 μg/ml anti-BMP-9-neutralizing Ab, supernatants from activated platelets, and supernatants plus 1 μg/ml anti-BMP-9-neutralizing antibody was investigated. Quantification of migration, proliferation, and tube formation was performed as described under “Experimental Procedures” (migration assay, n = 6 from three independent experiments; proliferation assay, n = 8 from four independent experiments; tube formation assay, n = 16 from three independent experiments). The graph illustrates percent change ± S.E. from base line (buffer). One and three asterisks indicate p < 0.05 and p < 0.005, respectively.
DISCUSSION
In the present study, we have demonstrated that CLEC-2 in platelets, but not in other cells, is required for blood/lymphatic vessel separation. We also have proposed that podoplanin in LECs activates platelets by binding to CLEC-2 in the connection between lymph sacs and veins during the developmental stages and that released BMP-9 from activated platelets, in conjunction with other releasates, inhibits the migration, proliferation, and tube formation of LECs, which facilitates blood/lymphatic vessel separation.
In initial studies, RT-PCR analysis has shown CLEC-2 transcripts in peripheral blood mononuclear cells, bone marrow cells, monocytes, dendritic cells, and granulocytes (31). However, subsequent studies revealed that the surface expression of CLEC-2 is limited to in platelets/megakaryocytes (1, 32) in human blood cells, although it is also expressed in the liver sinusoid (32, 33). In mice, although an analysis of the SAGE (serial analysis of gene expression) library derived from mouse megakaryocytes has revealed that CLEC-2 is one of the most megakaryocyte-specific and abundant molecules in megakaryocytes (34), CLEC-2 is expressed in other blood cells such as neutrophils (17), macrophages (18), and dendritic cells (35) in addition to platelets. Therefore, it is conceivable that CLEC-2 in blood cells other than platelets may play a role in blood/lymphatic vessel separation in mice. To address this issue, we generated mice with CLEC-2 specifically deleted from platelets. Specific deletion of CLEC-2 from platelets resulted in blood-filled, dilated, and tortuous lymphatic vessels (Fig. 1), proving that platelets regulate blood/lymphatic vessel separation through CLEC-2. CLEC-2-deficient mice die at the embryonic/neonatal stage (6, 7), whereas mice that are specifically deleted of CLEC-2 from platelets survive, suggesting that CLEC-2 expressed in cells other than platelets play a crucial role in maintaining life at the embryonic/neonatal stage.
We next investigated the mechanism by which CLEC-2 facilitates lymph/blood vessel separation. We observed that normal platelets, but not CLEC-2-deficient platelets, significantly inhibited the migration, proliferation, and tube formation of LECs but not those of HUVECs (Figs. 2–4), suggesting that platelets inhibit lymphangiogenesis by way of CLEC-2. It is obvious that activation of platelets is required for blood/lymphatic vessel separation, because the lack of Syk and SLP-76, which are necessary in CLEC-2-mediated signal transduction, results in a misconnection between blood and lymphatic vessels (8, 9). Platelet activation is likely to be induced by the association between CLEC-2 in platelets and podoplanin in LECs, because mice deficient in either molecule have the misconnection phenotype (6, 7, 10). Platelet activation leads to two events: release of granule contents and platelet aggregate formation. A blocker of integrin αIIbβ3, lotrafiban, inhibits platelet aggregation. We found that pretreatment of platelets with lotrafiban did not affect platelet-induced inhibition of lymphangiogenesis (Fig. 5). We observed CD62P expression and fibrinogen binding in platelets after seeding on surfaces coated with LECs but not HUVECs; platelet activation and fibrinogen binding were encountered in our in vivo system shown in Fig. 5, although the fibrinogen binding was very weak, probably due to an absence of stirring (data not shown). Integrin αIIbβ3-deficient mice, in which the platelets can release granule contents but cannot form aggregates, reportedly lack the misconnection phenotype, consistent with our results. Hence, it is unlikely that platelet aggregates physically occlude the connection between the lymph sac and the vein. As it is impossible to specifically inhibit granule release without inhibiting aggregation, we utilized activated platelet supernatants, which supposedly contain all of the granule contents released upon platelet activation, in order to investigate the role of granule release. We did not use rhodocytin to obtain activated-platelet supernatants because it also binds to integrin α2β1 (36, 37), which is expressed in LECs and HUVECs (38, 39). We did not use thrombin either, because endothelial cells express thrombin receptors, proteinase-activated receptors 1 and 2 (40). Hence, we used supernatants from platelets stimulated by a GPVI agonist, poly(PHG), as GPVI is not expressed in LECs or HUVECs (41). The supernatants of platelets activated through GPVI significantly inhibited the migration, proliferation, and tube formation of LEC. These findings suggest that the contents released from granules facilitate blood/lymphatic vessel separation and that the inhibitory effect is not restricted to supernatants from platelets activated through CLEC-2. It has been reported that kindlin-3-deficient mice, in which the platelets can release granule contents but cannot form aggregates, have the misconnection phenotype (10, 42), which seems to be at odds with our hypothesis. However, it has recently been demonstrated that kindlin-3 is present in human endothelial cells derived from various anatomical origins and that kindlin-3 knockdown results in the impaired formation of tube-like structures in Matrigel (43), leaving open the possibility that endothelial cell defects can contribute to the misconnection phenotype. We found that recombinant CLEC-2 inhibited hLEC migration but not proliferation and tube formation (supplemental Fig. 2), suggesting that cross-linking of podoplanin by recombinant CLEC-2 generates activation signals for cell migration. It has been reported recently that small interfering RNA depletion of podoplanin expression in human lung microvascular lymphatic endothelial cells (HMVEC-LLy) causes a dramatic reduction in directional migration compared with control siRNA-transfected cells (44). Podoplanin depletion causes Cdc42 activation and RhoA inhibition, leading to a decrease in HMVEC-LLy migration (44). It may be possible that podoplanin clustering by recombinant CLEC-2 regulates the activity of small GTPases and inhibits LEC migration. However, recombinant CLEC-2 did not inhibit the proliferation and tube formation of LEC, suggesting that podoplanin signaling only partly contributes to the inhibition of LEC functions.
While this manuscript was being reviewed, Finney et al. (45) reported that platelets inhibit LEC transmigration depending on CLEC-2, which is consistent with our results (Fig. 2). However, they report that the application of platelet releasate did not affect LEC transmigration and that treatment with an anti-podoplanin plus a secondary cross-linking antibody decreased VEGF-C-induced LEC migration (45). These findings suggest that platelets inhibit LEC migration in vitro by contact-dependent mechanisms. We agree that LEC migration is inhibited partly due to contact-dependent mechanism, as we also observed that recombinant CLEC-2 inhibited LEC migration (supplemental Fig. 2). In our hands, however, platelet releasate clearly inhibited not only LEC migration but also LEC proliferation and tube formation (Fig. 6). Finney et al. (45) added platelet releasate from platelets stimulated with rhodocytin to the transmigration well. However, we used supernatants from platelets stimulated with poly(PHG) but not those from platelets stimulated with rhodocytin, because rhodocytin also binds to integrin α2β1 (36, 37), which is expressed in LECs and HUVECs (38, 39). The different results may be due to some of the effects of rhodocytin in the supernatants on LEC migration in their experiment. Alternatively, it may be possible that different agonists cause different pattern of granule release from platelets, leading to the different results. In the present study, we reported for the first time that BMP-9 is released from activated platelets. The kinetics of BMP-9 release is an issue to be addressed in the future.
The next question is which molecule(s) are required for blood/lymphatic vessel separation. We investigated the effects of TGFβ, PF4, angiostatin, and endostatin on lymphangiogenesis, because these are cytokines released from platelets and have been reported to inhibit lymphangiogenesis (20–22). In addition to these molecules, we identified BMP-9 as a component of platelet releasates that affects LEC function. This finding has not been reported elsewhere to the best of our knowledge. Among all of the molecules, BMP-9 had the most potent inhibitory effects on migration, proliferation, and tube formation of hLECs (Fig. 7). Angiostatin and endostatin had no effect on these functions in hLECs (Fig. 7). On the other hand, TGF-β and PF4 showed inconsistent effects on the hLEC function; TGF-β inhibited migration and proliferation but not tube formation. PF4 inhibited the migration but not the proliferation and tube formation of hLECs. The differential effects of these cytokines on LEC function may have important physiologic implications, which will be future topics for research. The combination of all of these molecules (BMP-9, TGF-β, PF4, angiostatin, and endostatin) strongly inhibited tube formation in hLECs. Their combined administration inhibited hLEC tube formation by 45%, which was almost equivalent to the inhibitory effect of platelet supernatants on tube formation (Figs. 6C and 7D). Although other releasates may also be involved in the regulation of lymphangiogenesis, we suggest that this combination (BMP-9, TGF-β, PF4, angiostatin, and endostatin) plays a key role in platelet-induced inhibition of lymphangiogenesis in vivo. Although our data shown in Fig. 7 suggest that BMP-9 plays a key role in platelet-mediated inhibition of lymphangiogenesis, this is not demonstrated directly. To directly prove a major role of BMP-9 in platelet-mediated inhibition of lymphangiogenesis, we utilized an anti-BMP-9-neutralizing antibody. The anti-BMP-9-blocking antibody cancelled the inhibitory effects of platelet supernatants on hLEC migration, proliferation, and tube formation (Fig. 8). Among these, hLEC tube formation was increased in the presence of supernatants plus antibody (Fig. 8C), proving the major role of BMP-9 in platelet-mediated inhibition of lymphangiogenesis. The generation of BMP-9-deficient mice is awaited to prove the major role of BMP-9 in vivo.
BMP-9 also reportedly inhibits the migration or proliferation of vascular endothelial cells (24). However, we observed that platelet supernatant, including BMP-9 and other releasates, inhibited LEC function (Fig. 6) but not HUVEC function (data not shown). These findings suggest that the inhibitory effects of BMP-9 on HUVECs are cancelled by other releasates from activated platelets. The cancellation of inhibitory effects of BMP-9 on HUVEC migration may also occur in vivo.
Mice deficient in the NF-E2 transcription factor show severe thrombocytopenia but fail to develop the misconnection phenotype (46). This finding apparently contradicts the hypothesis that platelets regulate the separation. However, these mice produce a few platelet-like particles, which can express P-selectin, an α-granule membrane protein, in response to thrombin (47). It is conceivable that a small number of platelets may be enough for blood/lymphatic separation, because the key factor for the separation is granule release, not platelet aggregates, which may require a large number of platelets to physically seal the orifice between lymph sacs and veins. Myeloid ecotropic viral integration site 1 (Meis1)-deficient mice or PF4-Cre;Rosa26R-LacZbpa-DTA transgenic mice, both of which completely lack megakaryocyte/platelets, show the misconnection phenotype (48), suggesting that the notion that a complete lack of platelets is responsible for misconnection.
Normal platelets, but not CLEC-2-deficient platelets, significantly inhibited the migration, proliferation, and tube formation of LECs, whereas normal platelets as well as CLEC-2-deficient platelets did not inhibit those of HUVECs (Figs. 2–4). Platelets and platelet releasates have been shown to have proangiogenic effects in various models of angiogenesis (reviewed in Ref. 49). For example, platelets or platelet releasates stimulate HUVEC proliferation, migration, and tube formation in vitro (50, 51). We also observed that platelets significantly increased HUVEC migration (Fig. 2). Moreover, the slightly thickened walls of HUVEC tubes were observed in the presence of platelets (Fig. 4B), which may be a part of the proangiogenic effect of platelets. HUVEC tube formation was not inhibited either by intact platelets (Fig. 4B) or by supernatants from activated platelets (data not shown), suggesting that LECs and HUVECs respond differently to platelet releasates.
Acknowledgments
We are grateful to Chiaki Komatsu, Hisaichiro Nakazawa, Tsutomu Yuminamochi, and Dr. Kumiko Nakazawa for excellent technical assistance. We are also grateful to Prof. Radek C. Skoda for the kind donation of PF4-Cre mice and to Drs. Naohiro Kodama and Testuro Takehara for help with this study.
This work was supported in part by a grant-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science, and Technology and the Japan Society for the Promotion of Science (JSPS) through the Funding Program for Next Generation World-leading Researchers (NEXT Program) initiated by the Council for Science and Technology Policy (CSTP).

This article contains supplemental Figs. 1–3.
- CLEC-2
- C-type lectin-like receptor 2
- PLCγ2
- phospholipase C γ2
- HUVEC
- human umbilical vein endothelial cell
- LEC
- lymphatic endothelial cell
- hLEC
- human lymphatic endothelial cell
- mLEC
- murine lymphatic endothelial cells
- BMP-9
- bone morphogenetic protein-9
- PF4
- platelet factor 4
- GPVI
- glycoprotein VI
- EGM-2
- endothelial growth medium-2
- SFM
- serum-free medium
- EdU
- 5-ethynyl-2′-deoxyuridine
- hPlt
- human washed platelets
- mPlt
- murine washed platelets.
REFERENCES
- 1. Suzuki-Inoue K., Fuller G. L., García A., Eble J. A., Pöhlmann S., Inoue O., Gartner T. K., Hughan S. C., Pearce A. C., Laing G. D., Theakston R. D., Schweighoffer E., Zitzmann N., Morita T., Tybulewicz V. L., Ozaki Y., Watson S. P. (2006) A novel Syk-dependent mechanism of platelet activation by the C-type lectin receptor CLEC-2. Blood 107, 542–549 [DOI] [PubMed] [Google Scholar]
- 2. Suzuki-Inoue K., Kato Y., Inoue O., Kaneko M. K., Mishima K., Yatomi Y., Yamazaki Y., Narimatsu H., Ozaki Y. (2007) Involvement of the snake toxin receptor CLEC-2, in podoplanin-mediated platelet activation, by cancer cells. J. Biol. Chem. 282, 25993–26001 [DOI] [PubMed] [Google Scholar]
- 3. Christou C. M., Pearce A. C., Watson A. A., Mistry A. R., Pollitt A. Y., Fenton-May A. E., Johnson L. A., Jackson D. G., Watson S. P., O'Callaghan C. A. (2008) Renal cells activate the platelet receptor CLEC-2 through podoplanin. Biochem. J. 411, 133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kato Y., Kaneko M. K., Kunita A., Ito H., Kameyama A., Ogasawara S., Matsuura N., Hasegawa Y., Suzuki-Inoue K., Inoue O., Ozaki Y., Narimatsu H. (2008) Molecular analysis of the pathophysiological binding of the platelet aggregation-inducing factor podoplanin to the C-type lectin-like receptor CLEC-2. Cancer Sci. 99, 54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tsuruo T., Fujita N. (2008) Platelet aggregation in the formation of tumor metastasis. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 84, 189–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bertozzi C. C., Schmaier A. A., Mericko P., Hess P. R., Zou Z., Chen M., Chen C. Y., Xu B., Lu M. M., Zhou D., Sebzda E., Santore M. T., Merianos D. J., Stadtfeld M., Flake A. W., Graf T., Skoda R., Maltzman J. S., Koretzky G. A., Kahn M. L. (2010) Platelets regulate lymphatic vascular development through CLEC-2-SLP-76 signaling. Blood 116, 661–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Suzuki-Inoue K., Inoue O., Ding G., Nishimura S., Hokamura K., Eto K., Kashiwagi H., Tomiyama Y., Yatomi Y., Umemura K., Shin Y., Hirashima M., Ozaki Y. (2010) Essential in vivo roles of the C-type lectin receptor CLEC-2: embryonic/neonatal lethality of CLEC-2-deficient mice by blood/lymphatic misconnections and impaired thrombus formation of CLEC-2-deficient platelets. J. Biol. Chem. 285, 24494–24507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abtahian F., Guerriero A., Sebzda E., Lu M. M., Zhou R., Mocsai A., Myers E. E., Huang B., Jackson D. G., Ferrari V. A., Tybulewicz V., Lowell C. A., Lepore J. J., Koretzky G. A., Kahn M. L. (2003) Regulation of blood and lymphatic vascular separation by signaling proteins SLP-76 and Syk. Science 299, 247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ichise H., Ichise T., Ohtani O., Yoshida N. (2009) Phospholipase Cγ2 is necessary for separation of blood and lymphatic vasculature in mice. Development 136, 191–195 [DOI] [PubMed] [Google Scholar]
- 10. Uhrin P., Zaujec J., Breuss J. M., Olcaydu D., Chrenek P., Stockinger H., Fuertbauer E., Moser M., Haiko P., Fässler R., Alitalo K., Binder B. R., Kerjaschki D. (2010) Novel function for blood platelets and podoplanin in developmental separation of blood and lymphatic circulation. Blood 115, 3997–4005 [DOI] [PubMed] [Google Scholar]
- 11. Osada M., Inoue O., Ding G., Hirashima M., Suzuki-Inoue K., Ozaki Y. (2010) ASH Annual Meeting Abstracts. Blood 116, 160 (abstr.) [Google Scholar]
- 12. Tiedt R., Schomber T., Hao-Shen H., Skoda R. C. (2007) Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood 109, 1503–1506 [DOI] [PubMed] [Google Scholar]
- 13. Suzuki-Inoue K., Tulasne D., Shen Y., Bori-Sanz T., Inoue O., Jung S. M., Moroi M., Andrews R. K., Berndt M. C., Watson S. P. (2002) Association of Fyn and Lyn with the proline-rich domain of glycoprotein VI regulates intracellular signaling. J. Biol. Chem. 277, 21561–21566 [DOI] [PubMed] [Google Scholar]
- 14. Suzuki-Inoue K., Inoue O., Frampton J., Watson S. P. (2003) Murine GPVI stimulates weak integrin activation in PLCγ2−/− platelets: involvement of PLCγ1 and PI3-kinase. Blood 102, 1367–1373 [DOI] [PubMed] [Google Scholar]
- 15. Ichise T., Yoshida N., Ichise H. (2010) H-, N-, and Kras cooperatively regulate lymphatic vessel growth by modulating VEGFR3 expression in lymphatic endothelial cells in mice. Development 137, 1003–1013 [DOI] [PubMed] [Google Scholar]
- 16. Inoue O., Suzuki-Inoue K., Shinoda D., Umeda Y., Uchino M., Takasaki S., Ozaki Y. (2009) Novel synthetic collagen fibers, poly(PHG), stimulate platelet aggregation through glycoprotein VI. FEBS Lett. 583, 81–87 [DOI] [PubMed] [Google Scholar]
- 17. Kerrigan A. M., Dennehy K. M., Mourão-Sá D., Faro-Trindade I., Willment J. A., Taylor P. R., Eble J. A., Reis e Sousa C., Brown G. D. (2009) CLEC-2 is a phagocytic activation receptor expressed on murine peripheral blood neutrophils. J. Immunol. 182, 4150–4157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chang C. H., Chung C. H., Hsu C. C., Huang T. Y., Huang T. F. (2010) A novel mechanism of cytokine release in phagocytes induced by aggretin, a snake venom C-type lectin protein, through CLEC-2 ligation. J. Thromb. Haemost. 8, 2563–2570 [DOI] [PubMed] [Google Scholar]
- 19. Martín-Villar E., Megías D., Castel S., Yurrita M. M., Vilaró S., Quintanilla M. (2006) Podoplanin binds ERM proteins to activate RhoA and promote epithelial-mesenchymal transition. J. Cell Sci. 119, 4541–4553 [DOI] [PubMed] [Google Scholar]
- 20. Oka M., Iwata C., Suzuki H. I., Kiyono K., Morishita Y., Watabe T., Komuro A., Kano M. R., Miyazono K. (2008) Inhibition of endogenous TGF-β signaling enhances lymphangiogenesis. Blood 111, 4571–4579 [DOI] [PubMed] [Google Scholar]
- 21. Shao X. J., Chi X. Y. (2005) Influence of angiostatin and thalidomide on lymphangiogenesis. Lymphology 38, 146–155 [PubMed] [Google Scholar]
- 22. Shao X. J., Xie F. M. (2005) Influence of angiogenesis inhibitors, endostatin and PF-4, on lymphangiogenesis. Lymphology 38, 1–8 [PubMed] [Google Scholar]
- 23. Miyazono K., Kamiya Y., Morikawa M. (2010) Bone morphogenetic protein receptors and signal transduction. J. Biochem. 147, 35–51 [DOI] [PubMed] [Google Scholar]
- 24. Scharpfenecker M., van Dinther M., Liu Z., van Bezooijen R. L., Zhao Q., Pukac L., Löwik C. W., ten Dijke P. (2007) BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J. Cell Sci. 120, 964–972 [DOI] [PubMed] [Google Scholar]
- 25. David L., Mallet C., Mazerbourg S., Feige J. J., Bailly S. (2007) Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 109, 1953–1961 [DOI] [PubMed] [Google Scholar]
- 26. Song J. J., Celeste A. J., Kong F. M., Jirtle R. L., Rosen V., Thies R. S. (1995) Endocrinology 136, 4293–4297 [DOI] [PubMed] [Google Scholar]
- 27. Miller A. F., Harvey S. A., Thies R. S., Olson M. S. (2000) Bone morphogenetic protein-9. An autocrine/paracrine cytokine in the liver. J. Biol. Chem. 275, 17937–17945 [DOI] [PubMed] [Google Scholar]
- 28. Yamashita H., Ten Dijke P., Heldin C. H., Miyazono K. (1996) Bone morphogenetic protein receptors. Bone 19, 569–574 [DOI] [PubMed] [Google Scholar]
- 29. Hogan B. L. (1996) Bone morphogenetic proteins: multifunctional regulators of vertebrate development. Genes Dev. 10, 1580–1594 [DOI] [PubMed] [Google Scholar]
- 30. Wozney J. M. (1992) The bone morphogenetic protein family and osteogenesis. Mol. Reprod. Dev. 32, 160–167 [DOI] [PubMed] [Google Scholar]
- 31. Colonna M., Samaridis J., Angman L. (2000) Molecular characterization of two novel C-type lectin-like receptors, one of which is selectively expressed in human dendritic cells. Eur. J. Immunol. 30, 697–704 [DOI] [PubMed] [Google Scholar]
- 32. Chaipan C., Soilleux E. J., Simpson P., Hofmann H., Gramberg T., Marzi A., Geier M., Stewart E. A., Eisemann J., Steinkasserer A., Suzuki-Inoue K., Fuller G. L., Pearce A. C., Watson S. P., Hoxie J. A., Baribaud F., Pöhlmann S. (2006) DC-SIGN and CLEC-2 mediate human immunodeficiency virus type 1 capture by platelets. J. Virol. 80, 8951–8960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tang T., Li L., Tang J., Li Y., Lin W. Y., Martin F., Grant D., Solloway M., Parker L., Ye W., Forrest W., Ghilardi N., Oravecz T., Platt K. A., Rice D. S., Hansen G. M., Abuin A., Eberhart D. E., Godowski P., Holt K. H., Peterson A., Zambrowicz B. P., de Sauvage F. J. (2010) A mouse knockout library for secreted and transmembrane proteins. Nat. Biotechnol. 28, 749–755 [DOI] [PubMed] [Google Scholar]
- 34. Senis Y. A., Tomlinson M. G., García A., Dumon S., Heath V. L., Herbert J., Cobbold S. P., Spalton J. C., Ayman S., Antrobus R., Zitzmann N., Bicknell R., Frampton J., Authi K. S., Martin A., Wakelam M. J., Watson S. P. (2007) A comprehensive proteomics and genomics analysis reveals novel transmembrane proteins in human platelets and mouse megakaryocytes including G6b-B, a novel immunoreceptor tyrosine-based inhibitory motif protein. Mol. Cell. Proteomics 6, 548–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mourão-Sá D., Robinson M. J., Zelenay S., Sancho D., Chakravarty P., Larsen R., Plantinga M., Van Rooijen N., Soares M. P., Lambrecht B., Reis e Sousa C. (2011) CLEC-2 signaling via Syk in myeloid cells can regulate inflammatory responses. Eur. J. Immunol. 41, 3040–3053 [DOI] [PubMed] [Google Scholar]
- 36. Suzuki-Inoue K., Ozaki Y., Kainoh M., Shin Y., Wu Y., Yatomi Y., Ohmori T., Tanaka T., Satoh K., Morita T. (2001) Rhodocytin induces platelet aggregation by interacting with glycoprotein Ia/IIa (GPIa/IIa, Integrin α2β1). Involvement of GPIa/IIa-associated Src and protein tyrosine phosphorylation. J. Biol. Chem. 276, 1643–1652 [DOI] [PubMed] [Google Scholar]
- 37. Navdaev A., Clemetson J. M., Polgar J., Kehrel B. E., Glauner M., Magnenat E., Wells T. N., Clemetson K. J. (2001) Aggretin, a heterodimeric C-type lectin from Calloselasma rhodostoma (Malayan pit viper), stimulates platelets by binding to α2β1 integrin and glycoprotein Ib, activating Syk and phospholipase Cγ2, but does not involve the glycoprotein VI/Fc receptor γ chain collagen receptor. J. Biol. Chem. 276, 20882–20889 [DOI] [PubMed] [Google Scholar]
- 38. Avraamides C. J., Garmy-Susini B., Varner J. A. (2008) Integrins in angiogenesis and lymphangiogenesis. Nat. Rev. Cancer 8, 604–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chung C. H., Wu W. B., Huang T. F. (2004) Aggretin, a snake venom-derived endothelial integrin α2β1 agonist, induces angiogenesis via expression of vascular endothelial growth factor. Blood 103, 2105–2113 [DOI] [PubMed] [Google Scholar]
- 40. Banfi C., Brioschi M., Barbieri S. S., Eligini S., Barcella S., Tremoli E., Colli S., Mussoni L. (2009) Mitochondrial reactive oxygen species: a common pathway for PAR1- and PAR2-mediated tissue factor induction in human endothelial cells. J. Thromb. Haemost. 7, 206–216 [DOI] [PubMed] [Google Scholar]
- 41. Jandrot-Perrus M., Busfield S., Lagrue A. H., Xiong X., Debili N., Chickering T., Le Couedic J. P., Goodearl A., Dussault B., Fraser C., Vainchenker W., Villeval J. L. (2000) Cloning, characterization, and functional studies of human and mouse glycoprotein VI: a platelet-specific collagen receptor from the immunoglobulin superfamily. Blood 96, 1798–1807 [PubMed] [Google Scholar]
- 42. Moser M., Nieswandt B., Ussar S., Pozgajova M., Fässler R. (2008) Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med. 14, 325–330 [DOI] [PubMed] [Google Scholar]
- 43. Bialkowska K., Ma Y. Q., Bledzka K., Sossey-Alaoui K., Izem L., Zhang X., Malinin N., Qin J., Byzova T., Plow E. F. (2010) The integrin co-activator Kindlin-3 is expressed and functional in a non-hematopoietic cell, the endothelial cell. J. Biol. Chem. 285, 18640–18649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Navarro A., Perez R. E., Rezaiekhaligh M. H., Mabry S. M., Ekekezie I. I. (2011) Polarized migration of lymphatic endothelial cells is critically dependent on podoplanin regulation of Cdc42. Am. J. Physiol. Lung Cell. Mol. Physiol. 300, L32–L42 [DOI] [PubMed] [Google Scholar]
- 45. Finney B. A., Schweighoffer E., Navarro-Núñez L., Bénézech C., Barone F., Hughes C. E., Langan S. A., Lowe K. L., Pollitt A. Y., Mourao-Sa D., Sheardown S., Nash G. B., Smithers N., Reis e Sousa C., Tybulewicz V. L., Watson S. P. (2012) CLEC-2 and Syk in the megakaryocytic/platelet lineage are essential for development. Blood 119, 1747–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shivdasani R. A., Orkin S. H. (1995) Erythropoiesis and globin gene expression in mice lacking the transcription factor NF-E2. Proc. Natl. Acad. Sci. U.S.A. 92, 8690–8694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Levin J., Peng J. P., Baker G. R., Villeval J. L., Lecine P., Burstein S. A., Shivdasani R. A. (1999) Pathophysiology of thrombocytopenia and anemia in mice lacking transcription factor NF-E2. Blood 94, 3037–3047 [PubMed] [Google Scholar]
- 48. Carramolino L., Fuentes J., García-Andrés C., Azcoitia V., Riethmacher D., Torres M. (2010) Platelets play an essential role in separating the blood and lymphatic vasculatures during embryonic angiogenesis. Circ. Res. 106, 1197–1201 [DOI] [PubMed] [Google Scholar]
- 49. Ho-Tin-Noé B., Demers M., Wagner D. D. (2011) How platelets safeguard vascular integrity. J. Thromb. Haemost. 9, Suppl. 1, 56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kandler B., Fischer M. B., Watzek G., Gruber R. (2004) Platelet-released supernatant increases matrix metalloproteinase-2 production, migration, proliferation, and tube formation of human umbilical vascular endothelial cells. J. Periodontol. 75, 1255–1261 [DOI] [PubMed] [Google Scholar]
- 51. Pipili-Synetos E., Papadimitriou E., Maragoudakis M. E. (1998) Evidence that platelets promote tube formation by endothelial cells on Matrigel. Br. J. Pharmacol. 125, 1252–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]