

Background: GMX1778 is an inhibitor of nicotinamide phosphoribosyltransferase for the regeneration of NAD+ from nicotinamide.
Results: GMX1778 increases intracellular ROS in cancer cells but does not induce ROS in normal cells.
Conclusion: Exposure to GMX1778 may be a novel way of inducing ROS selectively in NAPRT1-negative tumor cells.
Significance: Selectively modulating intracellular ROS in cancers by GMX1778 provides a useful therapeutic opportunity.
Keywords: Apoptosis, p53, Reactive Oxygen Species (ROS), Redox Regulation, Small Molecules, GMX1778, Normal Tissue Toxicity
Abstract
Cancer cells undergo mitosis more frequently than normal cells and thus have increased metabolic needs, which in turn lead to higher than normal reactive oxygen species (ROS) production. Higher ROS production increases cancer cell dependence on ROS scavenging systems to balance the increased ROS. Selectively modulating intracellular ROS in cancers by exploiting cancer dependence on ROS scavenging systems provides a useful therapeutic approach. Essential to developing these therapeutic strategies is to maintain physiologically low ROS levels in normal tissues while inducing ROS in cancer cells. GMX1778 is a specific inhibitor of nicotinamide phosphoribosyltransferase, a rate-limiting enzyme required for the regeneration of NAD+ from nicotinamide. We show that GMX1778 increases intracellular ROS in cancer cells by elevating the superoxide level while decreasing the intracellular NAD+ level. Notably, GMX1778 treatment does not induce ROS in normal cells. GMX1778-induced ROS can be diminished by adding nicotinic acid (NA) in a NA phosphoribosyltransferase 1 (NAPRT1)-dependent manner, but NAPRT1 is lost in a high frequency of glioblastomas, neuroblastomas, and sarcomas. In NAPRT1-deficient cancer cells, ROS induced by GMX1778 was not susceptible to treatment with NA. GMX1778-mediated ROS induction is p53-dependent, suggesting that the status of both p53 and NAPRT1 might affect tumor apoptosis, as determined by annexin-V staining. However, as determined by colony formation, GMX1778 long term cytotoxicity in cancer cells was only prevented by the addition of NA to NAPRT1-expressing cells. Exposure to GMX1778 may be a novel way of inducing ROS selectively in NAPRT1-negative tumors without inducing cytotoxic ROS in normal tissue.
Introduction
Reactive oxygen species (ROS)2 play a dual role in a variety of normal physiological and pathological conditions (1). ROS function as redox messengers in intracellular signaling and regulation at physiological low levels. In contrast, at certain pathological conditions, excessive levels of ROS promote cell death by inducing oxidative damage to cellular macromolecules, including lipids, proteins, and DNA (2–4). Increased damage caused by ROS has been shown to determine the cell fate by inducing cell cycle arrest and apoptosis (5) as well as modulating topoisomerase II activity (6, 7). Normal tissues maintain intracellular redox homeostasis by balancing the equilibrium of ROS generation with the elimination of generated ROS (8). A variety of antioxidant processes is involved in maintaining the cellular redox levels and permits the repair of DNA damage lesions induced by excessive ROS levels by cellular DNA repair pathways (9). p53 has a critical role in redox homeostasis (10), and cells containing decreased nuclear p53 have been shown to accumulate oxidative DNA damage (11).
The redox status in tumors is usually different from that of normal tissue counterparts due in part to increased ROS production (12, 13). The adaptive antioxidant capacity in certain cancer cells allows tumors to tightly control ROS levels while maintaining proliferative capacity (14). This oxidative stress phenotype in cancer cells may provide therapeutic opportunities and challenges for eliminating tumors by specifically increasing ROS in tumors while maintaining ROS levels in normal tissues (14–16). Oxidative stress caused by chemotherapeutic drugs is often one of the main reasons for toxicity in normal tissue (17). Thus, a critical parameter for ROS-inducing anticancer drugs is the demonstration of a therapeutic effect while producing little oxidative stress in normal tissues (15–17). In higher eukaryotes, two major biochemical pathways, the salvage and the de novo pathway, are involved in the biosynthesis of NAD+. The salvage pathway operates via the two major pathways using nicotinamide phosphoryltransferase (NAMPT) and nicotinamide phosphoribosyltransferase 1 (NAPRT1), which use nicotinamide and nicotinic acid (NA), respectively, as the substrate for NAD+ recycling (18). Although both pathways are employed to generate NAD+, in cells expressing endogenous NAPRT1, only the NA added in the salvage pathway can increase cellular levels of NAD+ and reduce cytotoxicity by an oxidizing agent (18, 19). GMX1778 (CHS 828; teglarinad) is a potent inhibitor of NAMPT that exerts a cytotoxic effect by decreasing the cellular level of NAD+ (20, 21). GMX1778 has been shown to synergize with ionizing radiation in head and neck cancer tumor models (22). When applied in a metronomic treatment regimen of lower doses at frequent intervals, GMX1777, an intravenously administered pro-drug of GMX1778, regressed neuroblastomas in a preclinical model (23). Furthermore, exogenous addition of NA rescues NAD+ depletion via the NAPRT1-dependent salvage pathway (20, 21). Thus, in tumors deficient in nicotinamide phosphoribosyltransferase, targeting NAMPT with GMX1778 may provide a novel synthetic lethal therapeutic approach by inducing metabolic stress.
Based on the importance of NAD+ in regulating cellular ROS levels (24, 25), we hypothesized that decreasing NAD+ levels by exposure to GMX1778 could increase cellular oxidative stress. Based on the concept of nononcogene addiction (26), we also tested the hypothesis that the ROS stress induced by GMX1778 would be particularly cytotoxic in tumors defective in the NAPRT1-dependent salvage pathway and that normal cells could be protected from ROS induction via activating the NAPRT1-dependent salvage pathway with rescue by nicotinic acid. GMX1778 induced ROS, which could be reversed if NAPRT1 was activated by the addition of NA. Furthermore, p53 expression delayed initial ROS generation but did not suppress the GMX1778-mediated ROS increase more than 72 h. Normal cells were more resistant to GMX1778 because most normal cells have active NAPRT1, p53, and a lower level of ROS requiring less dependence on ROS scavenging systems.
EXPERIMENTAL PROCEDURES
Cell Lines and GMX1778
Human glioblastoma cell line (U251) and brain metastatic derivative of the breast cancer cell line MDA-MB-231 genetically engineered to express luciferase (MDA-MB-231 BR) were obtained from the NCI Frederick Tumor Repository. U251 were grown in RPMI 1640 media (Quality Biological, Gaithersburg, MD) containing 2 mmol/liter l-glutamine and 5% fetal bovine serum. MDA-MB-231 BR cells were grown in high glucose DMEM (Invitrogen) with 10% fetal bovine serum. HCT116 and HCT116 p53−/− cells were obtained from the Vogelstein laboratory (Johns Hopkins University) and grown in high glucose DMEM with 10% fetal bovine serum supplemented with 1× MEM (Invitrogen). Human mammary epithelial cells (HMEC) were purchased from Lonza and maintained in complete mammary epithelial growth media (Lonza, Walkersville, MD). MCF10A cells were maintained in mammary epithelial cell growth medium (Lonza, Walkersville, MD). H1299 cells were obtained from the Prives laboratory (Columbia University) and were grown in complete RPMI 1640 medium supplemented with 10% FBS. H1299 cells are stably transfected with a tetracycline-inducible vector encoding for WT human p53 (27). To suppress p53 expression, H1299 cells were incubated with fresh medium supplemented with 0.5 μg/ml anhydrotetracycline; p53 expression was induced when cells were incubated with fresh medium without anhydrotetracycline for 24 h. Cultures were maintained at 37 °C in an atmosphere of 5% CO2 and 95% room air. GMX1778, provided by Gemin X Pharmaceuticals Inc. (Montreal, Canada), was reconstituted in DMSO (10 mmol/liter) and stored at −80 °C.
Measurement of Intracellular Superoxide Levels
Superoxide levels were measured using the oxidation of the fluorescent dye, dihydroethidine (DHE), obtained from Molecular Probes (Invitrogen). Cells were seeded in 60-mm2 plates and allowed to attach for 24 h prior to 48 h of exposure to GMX1778. Cells were washed once with PBS and then labeled with DHE (10 μm in 0.1% DMSO) by incubation at 37 °C for 40 min in PBS (containing 5 mm pyruvate). Antimycin A (Sigma), a reactive oxygen species (ROS) generator, was prepared in DMSO and used at a 10 μm final concentration as a positive control and added at the time of DHE addition. The cells were suspended by treatment with trypsin on ice and then resuspended in ice-cold PBS. Flow cytometry analysis was performed using a Guava EasyCyte Plus microcapillary flow cytometer (Guava Technologies, Hayward, CA) utilizing laser excitation and emission wavelengths of 488 and 532 nm, respectively. The mean fluorescence intensity of 5000 cells was analyzed in each sample and corrected for autofluorescence from unlabeled cells. The mean fluorescence intensity data were normalized to corresponding untreated control cell levels to yield normalized mean fluorescence intensity.
GSH and GSSG Measurement
GSH and GSSG content in U251 cells was measured using a GSH detection kit (MBL International, Woburn, MA) according to the manufacturer's instructions. Briefly, 4 × 106 cells were homogenized in 100 μl of glutathione assay buffer and clarified by spinning in a microcentrifuge for 5 min at 4 °C. Then 60 μl was collected, and 20 μl of 6 n perchloric acid was added to stabilize GSH and stored at −80 °C. For GSH and GSSG detection, 20 μl of 3 n KOH was added to 40 μl of perchloric acid-treated samples, incubated on ice for 5 min, and then spun for 5 min at 4 °C. Neutralized samples were plated in triplicate in clear 96-well plates and mixed with 90 μl of assay buffer for the detection of GSH or 70 μl of assay buffer with 10 μl of GSH quencher to remove GSH and 10 μl of reducing agent to convert GSSG to GSH for measurement. Standard curves were generated with known amounts of GSH provided by the kit and used to calculate cellular GSH and GSSH concentrations in lysates. The absorbance at 405 nm was read by a microplate reader. Protein content of each sample was determined using protein assay kits (Bio-Rad). GSH and GSSG levels were calculated according to the equivalent protein quantity (mg of protein) per data point. Data were presented as fold change compared with normalized control.
NAD+, NADH, NADP+, and NADPH Measurement
Cellular NAD+, NADH, NADP+, and NADPH levels were measured using NAD+/NADH and NAD+/NADPH quantification assay kits according to manufacturer's instructions (MBL International, Woburn, MA). For NAD+ and NADH measurements, 1 × 106 cells were extracted in 200 μl of extraction buffer by two cycles of freeze thawing as directed in the protocol from MBL International. Total NAD+ and NADH were calculated by adding 50 μl of sample to 100 μl of NAD cycling mix and incubated for 5 min, and then 10 μl of NADH developer was added. NADH only was detected by decomposing NAD+ by heating at 60 °C for 30 min and clarified by centrifugation. Then 50 μl of cooled sample was added to 100 μl of NAD cycling mix and incubated for 5 min, and then 10 μl of NADH developer was added. The absorbance of samples at 405 nm was read by a microplate reader and compared with an NADH standard curve. The NAD+ concentration was determined by measuring NADH concentrations from heat-treated samples in which NAD+ had been decomposed and subtracting NADH concentrations from a duplicate sample that had not been heated and quantified for total NADt (NAD+ and NADH) as described in the NAD+/NADH quantitation kit. For the measurement of NADP+ and NADPH levels in lysates, a similar protocol and method were used with the NADP+/NADPH quantification kit. The concentration of NADPH in samples was calculated using standard curves generated by using known amounts of NADPH included in the kit. NAD+, NADH, NADP+, and NADPH levels were calculated according to the equivalent protein quantity (mg of protein) per data point. Data were presented as fold change compared with normalized control.
Western Blots
Cells were harvested and lysed in buffer containing 50 mm Tris-HCl, pH 7.4, 1 mm EDTA, 1% Nonidet P-40, 150 mm NaCl, and protease inhibitor and phosphatase inhibitor mixtures (Sigma). Lysates were centrifuged at 16,000 × g at 4 °C for 30 min and normalized for protein concentration. Lysates were then subjected to SDS-PAGE, 10% BisTris (Invitrogen), transferred to polyvinylidene fluoride (GE Healthcare), and probed with either NAPRT1 (1:1000) (Proteintech, Chicago), p53 (1:1000), or p21 (1:1000) (Santa Cruz Biotechnology). Equalization of protein loading was assessed using α-actin (1:5000) (Sigma) as the housekeeping protein. Primary antibody incubation was overnight at 4 °C, and the secondary horseradish peroxidase-conjugated anti-mouse or rabbit antibody (1:1000) (Santa Cruz Biotechnology) incubation was for 1 h at room temperature. Blots were visualized by the chemiluminescent substrate Luminol (Santa Cruz Biotechnology) and imaged using a Fuji LAS4000 (Fujifilm Life Science). Western blots shown are representative blots from three or more independent experiments. The following antibodies were used: NAPRT1 (Proteintech, Chicago) and p53 and p21 (Santa Cruz Biotechnology).
Annexin-V Apoptosis Assay
Apoptosis was measured using a Guava EasyCyte Plus as per the manufacturer's protocol.
Briefly, cells were washed twice in PBS, harvested by treatment with a trypsin/EDTA solution, centrifuged, washed with complete media, and suspended in 1× Nexin buffer with annexin V-PE and nexin 7-aminoactinomycin D. The cells were allowed to incubate for 30 min at room temperature and then analyzed using a Guava EasyCyte Plus flow cytometer.
Colony Formation Assay of Cells Treated with GMX1778 and/or Niacin
For determination of the survival of cells treated with GMX1778, exponentially growing cells were seeded at a density of 1200 cells/100-mm2 dish. Cells were allowed to attach overnight before the addition of the varied concentrations (5–50 nm) of freshly prepared GMX1778 and/or 20 μm niacin (Sigma). Plates were incubated for colony formation for 10–14 days. Medium was left unchanged during the duration of the experiment; therefore, the cells were continuously exposed to GMX1778. Colonies were stained with crystal violet and counted (GelCount, Oxford).
RESULTS
GMX1778 Induces ROS and Modulates a Redox Status in Cancer Cells by Reducing NAD+ Biosynthesis
GMX1778 decreases the intracellular NAD+ level by inhibiting the NAD+ biosynthesis enzyme NAMPT. Because of the roles of NADH and NADPH as reducing equivalents in oxidative stress responses for survival of cancer and normal cells, we hypothesized that a decrease in cellular levels of NAD+ by GMX1778 would result in a decrease in the steady state levels of NADH, NADP+, and NADPH, which would increase ROS levels leading to cell damage and finally death. To test this hypothesis, exponentially growing U251 cells defective in NAPRT1 expression were subjected to 5 nm GMX1778 (IC50 for U251 cells). ROS levels were then monitored by measuring intracellular oxidation of dihydroethidine (DHE) at various time points after exposure to GMX1778. DHE is a cell membrane-permeable dye used to monitor superoxide production because DHE fluorescence intensity correlates with the relative intracellular state O2˙̄ levels (13). As a positive control for ROS induction, cells were treated with antimycin A, an electron transport chain blocker known to increase mitochondrial oxygen radicals, and increased levels of ROS were observed (Fig. 1A). In cells treated with GMX1778, there was an increase in ROS level beginning at 16 h from initiation of treatment that reached a peak at 48 h (Fig. 1A). ROS levels reached a plateau after 48 h of GMX1778 exposure (data not shown).
FIGURE 1.

A, increased steady state levels of superoxide demonstrated by increased DHE oxidation in human glioma cells, U251 with 5 nm GMX1778. Cells were plated for 24 h prior to GMX1778 treatment. Cells were exposed to GMX1778 for the times indicated prior to DHE treatment. Cells exposed to antimycin A (AntA, 10 μm) for 40 min during DHE treatment were used as a positive control. Error bars represent ± S.D. of three different separate experiments. (*, significantly different from DHE-only group, p < 0.05, n = 3.) B and C, fold changes in NAD+/NADH and NADP+/NADPH levels were measured after cells were treated with 5 nm GMX1778 for the indicated times. Fold change was calculated as the ratio of NAD+ or NADH and NADP+ or NADPH, respectively, per mg of protein normalized to control. Error bars represent ± S.D. of three different separate experiments. (*, significantly different from control group, p < 0.05, n = 3.) D, GSH and GSSG levels were measured after cells were treated with 5 nm GMX1778 for the indicated times. Fold change was calculated as the ratio of GSH or GSSH per mg of protein normalized to control. Error bars represent ± S.D. of three different separate experiments. (*, significantly different from control group, p < 0.05, n = 3.)
To establish that the ROS induction by GMX1778 was directly due to a change in NAD+, NADH, NADP+, and NADPH levels, U251 cells were subjected to GMX1778 treatment, and intracellular NAD+, NADH, NADP+, and NADPH levels were determined. As shown in Fig. 1, B and C, a decrease in the steady state levels of NAD+ and NADP+ was observed 16 h after U251 cells were treated with GMX1778. Although the extent of NADH and NADPH decrease was less than that of NAD+ and NADP+, the change in the steady state levels of NADH and NADPH was consistent with the decrease in the steady state levels of NAD+ and NADP+. The ratio of glutathione (GSH) and the oxidized form glutathione disulfide (GSSH) has been used as a measure of cellular redox status (13, 28). GSH and GSSH levels were measured in U251 cells to determine the cellular redox status after GMX1778 exposure (Fig. 1D). The intracellular GSH concentration was decreased, and the intracellular GSSH concentration was increased when measured at 24 and 48 h after GMX1778 exposure. ATP levels were also measured in U251 cells after GMX1778 treatment and ROS induction was correlated with the ATP and NAD+ decrease (supplemental material). These data indicate that ROS were induced by GMX1778 under the conditions of these experiments. These results provide the following: 1) GMX1778 induces ROS in the U251 cancer cells by inhibiting NAD+ biosynthesis, thereby reducing intracellular NADP+, NADH, and NADPH levels; and 2) the increase in the steady state ROS levels altered the redox status of the U251 cells.
ROS Are Not Induced by GMX1778 in Normal Cells
In NAPRT1 wild-type cells, cytotoxicity of GMX1778 can be prevented by vitamin B3 (niacin) restoration of NAD+ via NAPRT1 (20, 21). To determine whether niacin could also rescue NAPRT1 wild-type cells by modulating ROS levels, we first tested expression levels of NAPRT1 in various cancer and normal cell lines. As shown in Fig. 2A, lanes 1–3, U251, MDA-MB-231 BR, and U2OS cells have undetectable protein expression levels of NAPRT1, whereas HCT116, HCT116 p53−/−, and H1299 as well as immortalized breast cells (MCF10A), and normal HMEC express NAPRT1 at varying levels (Fig. 2A, lanes 4–8). We investigated whether NAPRT1-negative cell lines are resistant to induced ROS by GMX1778 and whether the presence of niacin would lead to decreased ROS in NAPRT1-negative cell lines by measuring DHE oxidation florescent staining. ROS levels produced by exposure to GMX1778 were not diminished in U251 and MDA-MB-231 BR cells that were deficient in NAPRT1 in the absence or presence of niacin (Fig. 2B, panels A and B). Because the normal tissue damage incurred by induced ROS would reduce the therapeutic potential of GMX1778, we assessed the steady state levels of ROS in normal breast epithelial cells that were treated with GMX1778. Notably, in contrast to the cells that are p53 mutants and do not express NAPRT1, such as U251 and MDA-MB 231 BR cells, the steady state levels of ROS in normal HMEC exposed to GMX1778 were not increased compared with the control even when exposed to a 10-fold higher (50 nm) concentration than U251 cells even without adding NA to activate the NAPRT1-dependent NAD+ biosynthesis pathway that is dependent on NAPRT1 and NA (compare Fig. 2B, panels A and C; Fig. 3, A and E).
FIGURE 2.

A, various cell line lysates were probed for the expression of NAPRT1. Actin was used as a loading control. B, U251, MDA-MB-231BR, HCT116, HCT116 p53−/−, and normal HMEC DHE oxidation in the presence of GMX1778 at 5, 15, 5, 5, and 50 nm, respectively. Green histograms represent untreated cells; red indicates GMX1778-only treated cells; blue indicates GMX1778 with 20 μm niacin, and black indicates niacin only. One representative of three independent experiments is shown.
FIGURE 3.

Increased steady state levels of superoxide measured by increased DHE oxidation in various GMX1778 treated human cancer cells U251 (A), MDA-MB-231 BR (B), HCT116 (C), HCT116 p53−/− (D), and normal HMEC (E). Cells were grown and labeled with 10 μm DHE as described under “Experimental Procedures” and analyzed by flow cytometry. Values are expressed as the ratio of normalized mean fluorescence intensity relative to DHE-only cells. Error bars represent ± S.D. of three separate experiments. (*, significantly different from control, p < 0.05; **, p < 0.005.)
p53 Suppresses Initial ROS Levels Induced by the Depletion of NAD+
p53 plays a pivotal role in regulating cellular ROS levels and cell fate of damaged cells by providing anti-oxidant or pro-oxidant responses depending on ROS levels (29). We observed that in contrast to U251 and MDA-MB-231 BR cells, exposure of HCT116 cells to GMX1778 for 48 h did not induce ROS levels (Figs. 2, B and D, and 3C). However, in HCT116 p53−/− cells, exposure to GMX1778 increased DHE fluorescence without NA indicating that increased ROS occurred and that normal ROS levels were restored upon addition of NA to the cultures (Figs. 2, B and E, and 3D). Both variants of HCT116 cell lines expressed NAPRT1, although the expression level of NAPRT1 was higher in HCT116 p53−/− than in HCT116 (Fig. 2A, lanes 4 and 5). It is plausible to assume that HCT116 p53−/− cells have partially adapted to the absence of p53 by increasing NAPRT1 expression to maintain the physiological levels of ROS for their survival. To determine whether p53 was directly involved in regulating ROS levels induced by GMX1778, a genetically engineered H1299 p53 Tet-Off cell line was used (27). Fig. 4A shows a Western blot for p53 in H1299 p53 Tet-Off cell in the presence (lane 1) and absence of tetracycline (lane 2). The effect of p53 status on cellular levels of ROS upon exposure to varied concentrations of GMX1778 for 48 h was examined by comparing ROS levels in p53-repressed (dark gray bars) and p53-induced type (light gray bars) H1299 cells (Fig. 4B). GMX1778 induced ROS in the p53-repressed H1299 (Tet-On) cells, and the ROS induced was diminished by the exogenously added NA (Fig. 4B, lanes 2–5 and 6–10, dark gray bars). In contrast, in the p53-induced H1299 (Tet-Off), the induction of ROS was much less at all concentrations of GMX1778 (10, 50, 100, and 200 nm) tested (Fig. 4B, lanes 2–5 and 6–10, light gray bars). Slightly induced ROS levels in the presence of p53 were further reduced to control levels when NA was added (Fig. 4B, compare lanes 2–5 and 6–10, light gray bars). Thus, induced p53 in H1299 (Tet-Off) cells suppresses the generation of ROS in response to GMX1778. Also, exogenously added NA that activates the NAPRT1-dependent NAD+ biosynthesis pathway further diminished the ROS levels induced by GMX1778. These results suggest that the p53 status along with NA addition may maintain the redox status of NAPRT1-expressing cells when treated with GMX1778 within 48 h post-treatment.
FIGURE 4.
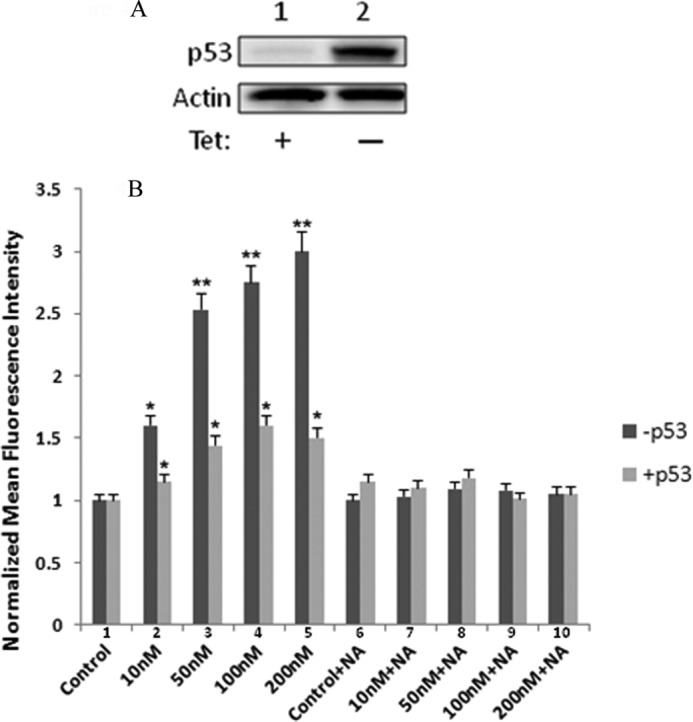
Effects of GMX1778 and niacin in a p53-inducible Tet-Off system. A, in the H1299-Tet-Off system, expression of wild-type p53 is allowed when tetracycline (Tet) is removed from media for 24 h. B, measurement of DHE oxidation with various concentrations of GMX1778 in the absence or presence of 20 μm niacin after 48 h of treatment. −p53 cells media contained 1 μg/ml of tetracycline. (*, significantly different from control, p < 0.05; **, p < 0.005.)
Apoptosis Is Induced by ROS That Are Increased by GMX1778 and Suppressing ROS Induction Protects Cells from Apoptosis
Apoptosis is induced to remove damaged cells when cellular damages by ROS pass the threshold that cells cannot tolerate for their survival (29–32). To determine whether increased ROS levels by GMX1778 are capable of inducing apoptosis in cells that lack NAPRT1, the level of apoptosis was measured in U251 and MDA-MB 231 BR cells treated with GMX1778 either in the absence or presence of NA (Fig. 5A and Table 1). Apoptosis was induced by GMX1778 in more than 80% of U251 and MDA-MB 231 BR cells, and the addition of NA does not significantly rescue these cells from apoptosis. Apoptosis was induced in 46% of HCT116 and 37% of HCT116 p53−/− cells treated with GMX1778 (Table 1). These results suggest that even though the level of induced ROS in HCT116 was less than that of HCT116 p53−/−, the slightly elevated ROS level in both cells was still able to induce apoptosis. Furthermore, in contrast to U251 and MDA-MB-231 BR cells, the induced apoptosis in these two cells was prevented in the presence of niacin. Apoptosis was determined in MCF10A and HMEC cells when treated with GMX1778 (Fig. 5B and Table 1). Importantly, exposure to GMX1778 for 48 h did not induce apoptosis in MCF10A and HMEC cells even in the absence of NA.
FIGURE 5.
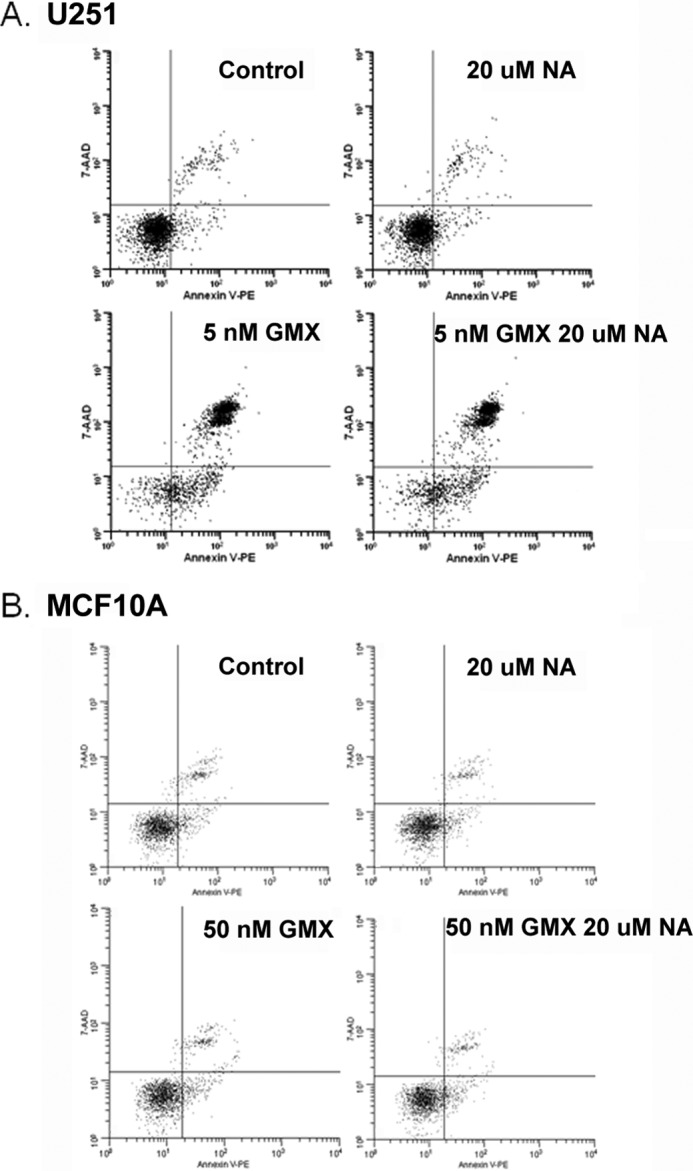
Representative apoptosis results of cells treated with GMX1778 ± 20 μm niacin. Conditions for the above are as labeled. Control, 20 μm niacin, GMX1778, and GMX1778 + 20 μm niacin. The concentration of GMX1778 used for U251 was 5 nm and the immortalized breast cell line. MCF10A cells were treated with 50 nm GMX1778. Cells were treated for 48 h and then stained with the annexin V apoptosis assay. Representatives of triplicate experiments are shown.
TABLE 1.
Apoptosis results of cells treated with GMX1778 repeated in triplicate were used for calculating average ± S.D. as in Fig. 5
GMX1778 concentrations used are in parentheses, and 20 μm NA was added concurrently with GMX1778.
| Cell line | % apoptotic cells (annexin-V-positive) |
||
|---|---|---|---|
| Control | GMX1778 | GMX1778+ NA | |
| U251 (5 nm) | 11.9 ± 3.5 | 86.2 ± 5.3a | 88.7 ± 2.6 |
| HCT116 (5 nm) | 12.8 ± 3.5 | 46.8 ± 6.3a | 11.6 ± 2.4b |
| HCT116 p53−/− (5 nm) | 11.7 ± 3.0 | 37.0 ± 4.5c | 10.5 ± 2.9b |
| MDA-MB-231 BR (15 nm) | 8.0 ± 4.8 | 93.8 ± 4.4a | 90.8 ± 1.2 |
| HMEC (50 nm) | 10.0 ± 0.7 | 14.4 ± 2.5 | 10.8 ± 1.8 |
| MCF10A (50 nm) | 20.3 ± 7.8 | 17.1 ± 7.5 | 17.6 ± 4.2 |
a p ≤ 0.001 compared with control.
b p ≤ 0.001 compared with GMX1778 alone.
c Increased cell death (p ≤ 0.05).
Niacin Rescues GMX1778-induced Cell Death in an NAPRT1-dependent Manner in Colony Formation Assay
We assessed the long term survival of cells that were treated with GMX1778 (5, 15–25, or 50 nm) with or without niacin (20 μm) using a colony formation assay. The data showed a correlation between survival and NAPRT1 status of the cell lines. No colonies were evident when U251 and MDA-MB-231 BR and U2OS cells deficient in NAPRT1 were treated with 5, 15, and 25 nm GMX1778, respectively, indicating a surviving fraction of <0.001 (Fig. 6, A, c, and B). Niacin (20 μm) could not rescue these cells from GMX1778 cytotoxicity (Fig. 6, A, d, and B). Neither HCT116, HCT116 p53−/−, nor H1299 cells survived when treated with 5 or 25 nm GMX1778 alone; however, both cell types survived when niacin (20 μm) was added to the cultures (Fig. 6, A and B). These results suggest that the survival of cells treated with GMX1778 may depend on the NAPRT1 status and may be independent of the p53 status. Finally, the survival of MCF10A and HMEC cells was assessed when treated with GMX1778. When MCF10A and HMEC cells were treated with GMX1778 (50 nm) in the absence of niacin, no colonies were detected. However, both the MCF10A and HMEC cells were rescued when niacin (20 μm) was added exogenously. Together, these results indicate that the survival of cells treated with GMX1778 was dependent on the NAPRT1 expression and the addition of niacin to cultures. These findings are consistent with the dual roles of p53 (pro-survival and pro-death) under continued ROS stress (29).
FIGURE 6.

A, survival of cells treated with GMX1778 in the absence or presence of 20 μm niacin. Conditions for the above are as follows: a, control; b, 20 μm niacin; c, GMX1778; d, GMX1778 + 20 μm niacin. The concentration of GMX1778 used for U251, U2OS, HCT116, and HCT116 p53−/− cell lines was 5 nm. The breast cancer line MDA-MB-231 BR was treated with 15 nm and H1299 with 25 nm GMX11778. Immortalized breast cell MCF10A and normal breast cell HMEC were treated with 50 nm GMX1778. B, graph of percentage of colonies surviving in the presence of vehicle (DMSO), niacin, GMX1778, or GMX1778 with niacin. Concentrations are noted above and data is normalized to cells treated with vehicle.
DISCUSSION
In this report, we present evidence that GMX1778 induces ROS, specifically superoxide radicals evidenced by DHE oxidation, via decreasing the cellular level of NAD+, NADP+, NADH, and NADPH. We employed several approaches to demonstrate the ability of GMX1778 to induce ROS specifically in cancer cells in culture while maintaining nontoxic levels of ROS in normal cells in culture. GMX1778 induced ROS and altered the cellular redox status in various cancer cells by inhibiting NAD+ biosynthesis. We hypothesized that the ROS levels induced by GMX1778 were initially suppressed by p53. Because p53 plays either an anti-oxidant or pro-oxidant role depending on the extent of the ROS level, p53 might initially modulate intracellular ROS induced by GMX1778 by activating anti-oxidant genes for survival (33). However, when the ROS levels that are continually induced by GMX1778 pass the threshold for survival, p53 might induce apoptosis to remove damaged cells. p53 plays a key role in cell fate decisions after sensing various cellular stresses. Some of the molecular mechanisms of how activated p53 determines damaged cell fates, eliciting pro-survival pathways (cell cycle arrest and DNA repair) or pro-death pathways (apoptosis) upon different stresses, have been reported (34–36). ROS induce p53 activation as an upstream signal and trigger apoptosis when activated p53 up-regulates pro-oxidant enzymes that further increase cellular ROS level (29). Our results measuring apoptosis by annexin V staining after treatment with GMX1778 in the cell lines tested are consistent with the dual roles of p53 in response to ROS. In addition to p53, the induced ROS levels by GMX1778 were further suppressed when the NAD+ levels were restored by exogenously adding niacin to NAPRT1-expressing cancer cells tested. Notably, the ROS were not induced by GMX1778 within 48 h of post-GMX1778 exposure in normal (HMEC) or immortalized cells (MCF10A) tested in the absence of NA. We also demonstrate that the extent of apoptosis induction is related to the status of either p53 or NAPRT1, which modulates the extent of apoptosis induction by GMX1778. The induced apoptosis is further prevented when niacin is added to the NAPRT1-expressing cancer or normal cells. Finally, we evaluated the effect of p53 or NAPRT1 on cellular survival using colony formation assay. In contrast to the apoptosis-mediated cell death, when treated with GMX1778, only cells expressing NAPRT1 survived when rescued with exogenously added niacin. Together, these results suggest that although the short term normal tissue protection from apoptosis induced by GMX1778 might be provided by p53 and NAPRT1 with or without niacin, the long term protection of normal tissues, when treated GMX1778, depends on the activation of NAPRT1-dependent NAD+ biosynthesis pathway with the addition of niacin.
The major goal in developing molecularly targeted agents in cancer treatment has been to induce selective killing of tumors with minimal damage to normal tissues by exploiting the Achilles heel of tumors. Recent progress in our knowledge of cancer biology has allowed us to adopt a new strategy, termed the synthetic lethality approach, with various molecularly targeted agents (26, 37). Targeting the replication stresses with poly(ADP-ribose) polymerase inhibitors in tumors with BRCA1/2 mutations highlights the potential of the synthetic lethality approach (38, 39). Based on the observation that NAPRT1 expression was defective in certain cancer cells, the synthetic lethality strategy has been proposed to specifically target tumors that are lacking in NAPRT1 expression. Although this strategy appears attractive and feasible based on the previous report (20, 21), the efficacy of GMX1778 might be compromised because of the possibility that GMX1778 might also induce ROS in normal tissues. Because ROS may be one of the major sources of normal tissue toxicities for approximately half of Food and Drug Administration-approved chemotherapeutics (17), it would be critical to improve a therapeutic index of GMX1778 by minimizing normal tissue damage incurred by ROS.
The results here suggest a novel direction for selective killing of tumors that are defective in NAPRT1 expression while sparing normal tissues from either ROS-mediated short term apoptosis or long term mitotic cell death when treated with GMX1778 and niacin. Furthermore, because it has been reported that cancer cells develop drug or radiation resistance in certain situations by developing anti-oxidant mechanisms (15) and putative cancer stem cells also tightly control the ROS level for their survival by using ROS scavenging systems, compared with normal counterpart cells (40), the ability of GMX1778 together with niacin to induce the higher level of ROS specifically in cancer cells deficient in NAPRT1 provides an attractive option in targeting drug-resistant, ionizing radiation-resistant, or putative cancer stem cells.
Acknowledgments
We thank Drs. Beverly Teicher, Deborah Wilsker, and Eric Bernhard for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant HHSN261200800001E from NCI.

This article contains supplemental Fig. S1.
- ROS
- reactive oxygen species
- NA
- nicotinic acid
- NAMPT
- nicotinamide phosphoribosyltransferase
- HMEC
- human mammary epithelial cell
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- DHE
- dihydroethidine.
REFERENCES
- 1. Circu M. L., Aw T. Y. (2010) Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 48, 749–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Camhi S. L., Lee P., Choi A. M. (1995) The oxidative stress response. New Horiz. 3, 170–182 [PubMed] [Google Scholar]
- 3. Martindale J. L., Holbrook N. J. (2002) Cellular response to oxidative stress. Signaling for suicide and survival. J. Cell. Physiol. 192, 1–15 [DOI] [PubMed] [Google Scholar]
- 4. Thannickal V. J., Fanburg B. L. (2000) Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 279, L1005–L1028 [DOI] [PubMed] [Google Scholar]
- 5. Kuo P. L., Chen C. Y., Hsu Y. L. (2007) Isoobtusilactone A induces cell cycle arrest and apoptosis through reactive oxygen species/apoptosis signal-regulating kinase 1 signaling pathway in human breast cancer cells. Cancer Res. 67, 7406–7420 [DOI] [PubMed] [Google Scholar]
- 6. Meng L. H., Ding J. (2007) Salvicine, a novel topoisomerase II inhibitor, exerts its potent anticancer activity by ROS generation. Acta Pharmacol. Sin. 28, 1460–1465 [DOI] [PubMed] [Google Scholar]
- 7. Li T. K., Chen A. Y., Yu C., Mao Y., Wang H., Liu L. F. (1999) Activation of topoisomerase II-mediated excision of chromosomal DNA loops during oxidative stress. Genes Dev. 13, 1553–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wondrak G. T. (2009) Redox-directed cancer therapeutics. Molecular mechanisms and opportunities. Antioxid. Redox. Signal. 11, 3013–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kryston T. B., Georgiev A. B., Pissis P., Georgakilas A. G. (2011) Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 711, 193–201 [DOI] [PubMed] [Google Scholar]
- 10. Maillet A., Pervaiz S. (2012) Redox regulation of p53, redox effectors regulated by p53. A subtle balance. Antioxid. Redox. Signal. 16, 1285–1294 [DOI] [PubMed] [Google Scholar]
- 11. He X., Ni Y., Wang Y., Romigh T., Eng C. (2011) Naturally occurring germ line and tumor-associated mutations within the ATP-binding motifs of PTEN lead to oxidative damage of DNA associated with decreased nuclear p53. Hum. Mol. Genet. 20, 80–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Szatrowski T. P., Nathan C. F. (1991) Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 51, 794–798 [PubMed] [Google Scholar]
- 13. Aykin-Burns N., Ahmad I. M., Zhu Y., Oberley L. W., Spitz D. R. (2009) Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem. J. 418, 29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shi X., Zhang Y., Zheng J., Pan J. (2012) Reactive oxygen species in cancer stem cells. Antioxid. Redox. Signal. 16, 1215–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trachootham D., Alexandre J., Huang P. (2009) Targeting cancer cells by ROS-mediated mechanisms. A radical therapeutic approach? Nat. Rev. Drug Discov. 8, 579–591 [DOI] [PubMed] [Google Scholar]
- 16. Raj L., Ide T., Gurkar A. U., Foley M., Schenone M., Li X., Tolliday N. J., Golub T. R., Carr S. A., Shamji A. F., Stern A. M., Mandinova A., Schreiber S. L., Lee S. W. (2011) Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 475, 231–234 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17. Chen Y., Jungsuwadee P., Vore M., Butterfield D. A., St Clair D. K. (2007) Collateral damage in cancer chemotherapy: oxidative stress in nontargeted tissues. Mol. Interv. 7, 147–156 [DOI] [PubMed] [Google Scholar]
- 18. Hara N., Yamada K., Shibata T., Osago H., Hashimoto T., Tsuchiya M. (2007) Elevation of cellular NAD levels by nicotinic acid and involvement of nicotinic acid phosphoribosyltransferase in human cells. J. Biol. Chem. 282, 24574–24582 [DOI] [PubMed] [Google Scholar]
- 19. Garten A., Petzold S., Körner A., Imai S., Kiess W. (2009) Nampt. Linking NAD biology, metabolism, and cancer. Trends Endocrinol. Metab. 20, 130–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beauparlant P., Bédard D., Bernier C., Chan H., Gilbert K., Goulet D., Gratton M. O., Lavoie M., Roulston A., Turcotte E., Watson M. (2009) Preclinical development of the nicotinamide phosphoribosyltransferase inhibitor prodrug GMX1777. Anti-Cancer Drugs 20, 346–354 [DOI] [PubMed] [Google Scholar]
- 21. Watson M., Roulston A., Bélec L., Billot X., Marcellus R., Bédard D., Bernier C., Branchaud S., Chan H., Dairi K., Gilbert K., Goulet D., Gratton M. O., Isakau H., Jang A., Khadir A., Koch E., Lavoie M., Lawless M., Nguyen M., Paquette D., Turcotte E., Berger A., Mitchell M., Shore G. C., Beauparlant P. (2009) The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis. Strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol. Cell. Biol. 29, 5872–5888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kato H., Ito E., Shi W., Alajez N. M., Yue S., Lee C., Chan N., Bhogal N., Coackley C. L., Vines D., Green D., Waldron J., Gullane P., Bristow R., Liu F. F. (2010) Efficacy of combining GMX1777 with radiation therapy for human head and neck carcinoma. Clin. Cancer Res. 16, 898–911 [DOI] [PubMed] [Google Scholar]
- 23. Fuchs D., Rodriguez A., Eriksson S., Christofferson R., Sundberg C., Azarbayjani F. (2010) Metronomic administration of the drug GMX1777, a cellular NAD synthesis inhibitor, results in neuroblastoma regression and vessel maturation without inducing drug resistance. Int. J. Cancer 126, 2773–2789 [DOI] [PubMed] [Google Scholar]
- 24. Anastasiou D., Poulogiannis G., Asara J. M., Boxer M. B., Jiang J. K., Shen M., Bellinger G., Sasaki A. T., Locasale J. W., Auld D. S., Thomas C. J., Vander Heiden M. G., Cantley L. C. (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334, 1278–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vander Heiden M. G., Cantley L. C., Thompson C. B. (2009) Understanding the Warburg effect. The metabolic requirements of cell proliferation. Science 324, 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luo J., Solimini N. L., Elledge S. J. (2009) Principles of cancer therapy. Oncogene and nononcogene addiction. Cell 136, 823–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen X., Ko L. J., Jayaraman L., Prives C. (1996) p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev. 10, 2438–2451 [DOI] [PubMed] [Google Scholar]
- 28. Kirsch M., De Groot H. (2001) NAD(P)H, a directly operating antioxidant? FASEB J. 15, 1569–1574 [DOI] [PubMed] [Google Scholar]
- 29. Liu B., Chen Y., St Clair D. K. (2008) ROS and p53. A versatile partnership. Free Radic. Biol. Med. 44, 1529–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Simon H. U., Haj-Yehia A., Levi-Schaffer F. (2000) Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 5, 415–418 [DOI] [PubMed] [Google Scholar]
- 31. Izeradjene K., Douglas L., Tillman D. M., Delaney A. B., Houghton J. A. (2005) Reactive oxygen species regulate caspase activation in tumor necrosis factor-related apoptosis-inducing ligand-resistant human colon carcinoma cell lines. Cancer Res. 65, 7436–7445 [DOI] [PubMed] [Google Scholar]
- 32. Huang H. L., Fang L. W., Lu S. P., Chou C. K., Luh T. Y., Lai M. Z. (2003) DNA-damaging reagents induce apoptosis through reactive oxygen species-dependent Fas aggregation. Oncogene 22, 8168–8177 [DOI] [PubMed] [Google Scholar]
- 33. Sablina A. A., Budanov A. V., Ilyinskaya G. V., Agapova L. S., Kravchenko J. E., Chumakov P. M. (2005) The antioxidant function of the p53 tumor suppressor. Nat. Med. 11, 1306–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Das S., Raj L., Zhao B., Kimura Y., Bernstein A., Aaronson S. A., Lee S. W. (2007) Hzf determines cell survival upon genotoxic stress by modulating p53 transactivation. Cell 130, 624–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sen N., Satija Y. K., Das S. (2011) PGC-1α, a key modulator of p53, promotes cell survival upon metabolic stress. Mol. Cell 44, 621–634 [DOI] [PubMed] [Google Scholar]
- 36. Tanaka T., Ohkubo S., Tatsuno I., Prives C. (2007) hCAS/CSE1L associates with chromatin and regulates expression of select p53 target genes. Cell 130, 638–650 [DOI] [PubMed] [Google Scholar]
- 37. Luo J., Emanuele M. J., Li D., Creighton C. J., Schlabach M. R., Westbrook T. F., Wong K. K., Elledge S. J. (2009) A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 137, 835–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bryant H. E., Schultz N., Thomas H. D., Parker K. M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N. J., Helleday T. (2005) Specific killing of BRCA2-deficient tumors with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 [DOI] [PubMed] [Google Scholar]
- 39. Farmer H., McCabe N., Lord C. J., Tutt A. N., Johnson D. A., Richardson T. B., Santarosa M., Dillon K. J., Hickson I., Knights C., Martin N. M., Jackson S. P., Smith G. C., Ashworth A. (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 [DOI] [PubMed] [Google Scholar]
- 40. Diehn M., Cho R. W., Lobo N. A., Kalisky T., Dorie M. J., Kulp A. N., Qian D., Lam J. S., Ailles L. E., Wong M., Joshua B., Kaplan M. J., Wapnir I., Dirbas F. M., Somlo G., Garberoglio C., Paz B., Shen J., Lau S. K., Quake S. R., Brown J. M., Weissman I. L., Clarke M. F. (2009) Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458, 780–783 [DOI] [PMC free article] [PubMed] [Google Scholar]