

Abstract
Non-technical summary
The sizes of neurons and their synaptic connections are regulated by multiple molecular mechanisms to provide neuronal networks that perform well-defined functions. Deletion of the tumour suppressor phosphatase and tensin homologue (PTEN) during early development leads to a 2- to 3-fold increase in neuronal and synaptic size and abnormalities in synaptic plasticity, the cellular mechanism underlying learning and memory. Whether PTEN deletion affects synaptic plasticity directly or as a consequence of its effect on the neuronal and synaptic size remained unclear. Here we show that deletion of the Pten gene Pten in mice during postnatal development, when the central nervous system is formed, does not affect neuronal or synaptic size but impairs synaptic plasticity. Thus, PTEN affects neuronal structure and synaptic plasticity through independent mechanisms.
Abstract
The tumour suppressor PTEN is the central negative regulator of the phosphatidylinositol 3-kinase (PI3K) signalling pathway, which mediates diverse processes in various tissues. In the nervous system, the PI3K pathway modulates proliferation, migration, cellular size, synaptic transmission and plasticity. In humans, neurological abnormalities such as autism, seizures and ataxia are associated with inherited PTEN mutations. In rodents, Pten loss during early development is associated with extensive deficits in neuronal migration and substantial hypertrophy of neurons and synaptic densities; however, whether its effect on synaptic transmission and plasticity is direct or mediated by structural abnormalities remains unknown. Here we analysed neuronal and synaptic structures and function in Pten-conditional knockout mice in which the gene was deleted from excitatory neurons postnatally. Using two-photon imaging, Golgi staining, immunohistochemistry, electron microscopy, and electrophysiological tools, we determined that Pten loss does not affect hippocampus development, neuronal or synaptic structures, or basal excitatory synaptic transmission. However, it does cause deficits in both major forms of synaptic plasticity, long-term potentiation and long-term depression, of excitatory synaptic transmission. These deficits coincided with impaired spatial memory, as measured in water maze tasks. Deletion of Pdk1, which encodes a positive downstream regulator of the PI3K pathway, rescued Pten-mediated deficits in synaptic plasticity but not in spatial memory. These results suggest that PTEN independently modulates functional and structural properties of hippocampal neurons and is directly involved in mechanisms of synaptic plasticity.
Introduction
The phosphatidylinositol 3-kinase (PI3K) signalling pathway regulates crucial biological processes, including proliferation, growth, migration, metabolism, and neuronal and synaptic plasticity. Activated PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3), resulting in a complex downstream signalling cascade. The serine–threonine kinase PDK1 is an important downstream regulator that is recruited to the membrane by PIP3, where it phosphorylates and contributes to the activation of AKT, a major mediator of downstream PI3K signalling. PTEN is a lipid phosphatase that converts PIP3 to PIP2, thereby negatively regulating the PI3K pathway (Engelman et al. 2006; Chalhoub et al. 2009).
Individuals with PTEN germline mutations are prone to various neurological conditions, including autism, learning delay and mental retardation (Goffin et al. 2001; Waite & Eng, 2002; Butler et al. 2005; Gustafson et al. 2007; Endersby & Baker, 2008). Synaptic mechanisms have been implicated in the pathogenesis of these conditions (Hoeffer & Klann, 2010; Stornetta & Zhu, 2011). During the development of the CNS, the deletion of the Pten gene in mice disrupts multiple processes, including neuronal migration and proliferation (Endersby & Baker, 2008). The hallmark of Pten deletion during development is the spectacular progressive hypertrophy of neuronal somata and processes, including dendrites and dendritic spines, leading to as much as a 250% increase in neuronal and synaptic size (Backman et al. 2001, 2002; Kwon et al. 2001; Fraser et al. 2004, 2008; Jaworski et al. 2005; Kwon et al. 2006; Chow et al. 2009). In addition to these substantial neuronal morphological abnormalities, both major forms of synaptic plasticity, long-term potentiation (LTP) (Fraser et al. 2008) and long-term depression (LTD) (Wang et al. 2006; Jurado et al. 2010), are deficient when Pten is deleted or downregulated in hippocampal neurons, and behavioural abnormalities also occur (Kwon et al. 2006; Zhou et al. 2009). Thus, it is unknown whether the effects on neuronal morphology and synaptic function are independent of each other. Functional synaptic defects in Pten-deficient animals could be secondary to morphological changes that involve hypertrophy of dendritic spines, postsynaptic densities, and presynaptic terminals (Kwon et al. 2001, 2006; Fraser et al. 2008), where crucial components of postsynaptic and presynaptic plasticity are located (Blundon & Zakharenko, 2008). A previous attempt to separate PTEN's effect on synaptic plasticity from its morphological effects was made in developing neurons in organotypic slice cultures (Jurado et al. 2010). However, it is not clear if PTEN has a direct effect on synaptic plasticity in acute preparations and behaviour in vivo.
Here we employed a transgenic mouse line that expressed Cre recombinase (Cre) under the control of an excitatory neuron-specific CaMKIIα promoter to investigate the effect of Pten deletion during postnatal development. The timing of Cre activity bypassed developmental defects in neuronal positioning and allowed assessment of synaptic function prior to the onset of the hypertrophic effects of PTEN deficiency. We determined that postnatal Pten deletion affects LTP and LTD at excitatory hippocampal synapses and hippocampal-dependent spatial memory without affecting morphological characteristics of excitatory neurons in the hippocampus. We also demonstrated that the additional deletion of Pdk1, the gene for a positive downstream effector of PI3K signalling, rescued synaptic phenotypes in Pten-deficient mice. These results suggest that the PI3K pathway independently affects the function and structure of neuronal networks.
Methods
Animals
Mice with a conditional knockout of Pten or Pdk1 in forebrain excitatory neurons were generated by crossing Pten+/fl (Suzuki et al. 2001) or Pdk1+/fl mice (Lawlor et al. 2002) with mice that express Cre under the control of the CamKIIα promoter (Tsien et al. 1996a,b). Mice with the genotype CamKIIα-Cre;Ptenfl/fl are hereafter referred to as Pten−/− mice, and mice with the genotype CamKIIα-Cre;Pdk1fl/fl are referred to as Pdk1−/− mice. Mice with deletions of both Pten and Pdk1 (CamKIIα-Cre;Ptenfl/fl;Pdk1fl/fl) are referred as Pten−/−;Pdk−/− or double-mutant mice. CamKIIa-Cre mice were maintained on the C57BL/6J genetic background for more than 12 generations. Pten+/fl and Pdk1+/fl mice were maintained on the mixed C57BL/6J;FVB/NJ background. To better control for background differences, we compared mutant mice and wild-type (WT) littermates in all experiments. Cre− littermates were used as WT controls. For some single-cell imaging experiments, we produced mice carrying a yellow fluorescent protein (YFP) reporter of Cre activity (CamKIIα-Cre;Ptenfl/fl;R26LSL-EYFP) by crossing Pten−/− mice with R26LSL-EYFP reporter mice (Srinivas et al. 2001). All experiments were done in 7- to 8-week-old mice. Mice were killed by quick decapitation. The care and use of animals were reviewed and approved by the Institutional Animal Care and Use Committee of St Jude Children's Research Hospital. The authors have read, and the experiments comply with the policies and regulations of The Journal of Physiology given by Drummond (2009).
Brain slice preparation
Acute transverse hippocampal slices (400 μm) were prepared as previously described (Bayazitov et al. 2007). Briefly, mouse brains were quickly removed and placed in cold (4°C) dissecting artificial cerebrospinal fluid (ACSF) containing 125 mm choline chloride, 2.5 mm KCl, 0.4 mm CaCl2, 6 mm MgCl2, 1.25 mm NaH2PO4, 26 mm NaHCO3 and 20 mm glucose (285–295 mosmol l-1), under 95% O2/5% CO2. After dissection, slices were incubated for 1 h in ACSF containing 125 mm NaCl, 2.5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 1.25 mm NaH2PO4, 26 mm NaHCO3 and 10 mm glucose (285–295 mosmol l-1), under 95% O2/5% CO2 at room temperature and then transferred into the submerged recording chamber and superfused (2–3 ml min−1) with warm (30–32°C) ACSF.
Field potential recordings
The field recordings were performed using a set-up with eight submerged recording chambers (Campden Instruments, Lafayette, IN, USA). We recorded field excitatory postsynaptic potentials (fEPSPs) from the CA1 stratum radiatum by using an extracellular glass pipette (3–5 MΩ) filled with ACSF. Schaffer collateral/commissural fibres in the stratum radiatum were stimulated with a bipolar tungsten electrode placed 200–300 μm from the recording pipette. Stimulation intensities were chosen to produce an fEPSP with a 1 V s-1 slope based on an input/output curve from stimulations of 0, 25, 50, 100, 150, 200, 250 and 300 μA. Paired-pulse facilitation experiments were performed using a pair of stimuli of the same intensity delivered 20, 50, 100, 200 and 1000 ms apart. LTP at CA3–CA1 synapses was induced by three periods of 200 Hz tetanization delivered every 5 min. Every period of tetanization consisted of 10 trains of 200 Hz stimulation delivered at the same intensity for 200 ms (40 stimulations) every 5 s. A similar protocol was previously used to induce compound (presynaptic and postsynaptic) LTP at CA3–CA1 synapses in the hippocampus (Zakharenko et al. 2001, 2003; Bayazitov et al. 2007). LTD was induced with a train of 900 stimulations delivered at 1 Hz (Zakharenko et al. 2002).
Single-cell electrophysiology
Whole-cell recordings were obtained from the soma of CA1 neurons. For current-clamp recordings, patch pipettes (open pipette resistance, 3–5 MΩ) were filled with an internal solution containing 140 mm KMeSO4, 8 mm NaCl, 1 mm MgCl2, 10 mm Hepes, 5 mm MgATP, 0.4 mm Na2GTP, 300 μm Fluo 5F and 10 to 25 μm Alexa 594 (pH 7.3). For voltage-clamp recordings, we replaced the potassium-based solution with a caesium-based internal solution. Whole-cell recordings were registered using a Multiclamp 700B (Molecular Devices, Sunnyvale, CA, USA), digitized (10 kHz; DigiData 1322A, Molecular Devices), and recorded using pCLAMP 9.0 software (Molecular Devices). Spontaneous miniature excitatory postsynaptic currents (mEPSCs) were recorded at –70 mV holding potential in the presence of picrotoxin (100 μm) and tetrodotoxin (1 μm) in the extracellular solution for at least 1 h. Amplitude and inter-event intervals of mEPSCs were analysed off-line using the Mini-Analysis Program (Synaptosoft Inc., Leonia, NJ, USA). All software-detected events were verified visually. The current ratio of AMPA receptors to NMDA receptors (AMPAR/NMDAR) was calculated from the EPSC traces recorded at +40 mV. The amplitude of stimulation was adjusted to evoke 50 pA EPSCs at –70 mV. The AMPAR current was determined at time points when EPSCs recorded at –70 mV reached their peaks; the NMDAR current was determined 100 ms after the peaks. EPSCs were analysed off-line using Clampfit 10.1 software (Molecular Devices).
Two-photon imaging
Two-photon laser-scanning microscopy (TPLSM) was performed using an Ultima Imaging System (Prairie Technologies, Middleton, WI, USA), a Ti:sapphire Chameleon Ultra femtosecond-pulsed laser (Coherent Inc., Santa Clara, CA, USA), and 60× (0.9 NA) water-immersion IR objectives (Olympus, Centre Valley, PA, USA), as described previously (Earls et al. 2010). Alexa Fluor 594 was included in the internal solution and were excited at 820 nm. Alexa Fluor 594 fluorescence was used to image and reconstruct dendritic and axonal morphology of CA1 neurons. ImageJ was used to analyse dendritic branching and the morphology of dendritic spines.
Electron microscopy
Hippocampal slices were fixed in 2.5% glutaraldehyde in 0.1 m sodium cacodylate buffer, thrice rinsed in the same buffer, and dehydrated in a graded series of alcohol rinses and then propylene oxide washes. The tissue was infiltrated and embedded in eponaraldite and polymerized overnight at 70°C. We cut 70 nm sections on a Leica UC6 ultramicrotome fitted with a Diatome diamond knife and stained with lead citrate and 8% uranyl acetate. Thick sections were trimmed to a region containing the CA1 cell body layer where synapses were identified. The sections were imaged on a JEOL 1200EX11 transmission electron microscope with an AMT XR111 11-megapixel digital camera. Synapses were counted as regions of membrane enclosing synaptic vesicles in close proximity to a postsynaptic density. Vesicle size and number and the length of postsynaptic densities were measured in ImageJ. Vesicles were traced with the line or elliptical selection tools, and the traces were then measured.
Immunohistochemistry and immunofluorescence
Mice were perfused transcardially with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). Tissues were postfixed in 4% PFA overnight, cut sagittally into halves, embedded in paraffin, and then cut into 5 μm sections. Tissues from littermate controls were processed simultaneously with those from the mutant animals. Using microwave antigen retrieval, we probed sections with a primary antibody against PTEN (1:100; no. 9559, Cell Signaling Technology, Inc., Danvers, MA, USA) or p-S6 S235/236 (1:500, no. 2211, Cell Signaling). The primary antibody was detected using biotinylated secondary antibodies (1:200, Vector labs), peroxidase-conjugated avidin (Elite ABC, Vector Laboratories, Inc., Burlingame, CA, USA), and treatment with DAB substrate and hematoxylin counterstain (Vector). For dual-labelling fluorescence immunohistochemistry for PTEN and yellow fluorescent protein (YFP), mice were perfused transcardially with 4% PFA. After dissection, tissues were placed into the 4% PFA solution overnight and then transferred to the PBS solution containing 25% sucrose. Tissues were frozen, sectioned, and probed for YFP using primary antibodies against green fluorescent protein (GFP; 1:200; ab6673, Abcam) and PTEN (1:100; no. 9559, Cell Signaling). The primary antibodies were detected using secondary antibodies conjugated with Alexa Fluor 488 (1:400, A-11055, Invitrogen) or Cy3 (1:400, 111–165-144, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA).
Nuclear size measurement and cell counting
Paraffin-embedded tissue sections were stained for haematoxylin and eosin (H&E), and 20× images were taken of the CA1, CA3 and dentate gyrus soma from sections in approximately 150 μm increments. ImageJ was used to analyse nuclear diameter and cell density in sections collected from three mice of each genotype.
Western blotting
Unilateral hippocampi were dissected and snap frozen. The tissue was lysed using disposable pestles and microtubes (Fisher Scientific) in ice-cold RIPA buffer (50 mm Tris-HCl (pH 7.5), 1% NP-40, 0.5% sodium deoxycholate, 150 mm NaCl, and 0.1% SDS and supplemented with protease and phosphatase inhibitions (Roche, Indianapolis, IN, USA). Protein extract (25 μg) was electrophoresed on 10% SDS-PAGE gels (Invitrogen), and protein was transferred to PVDF (Invitrogen). The primary antibodies used for chemiluminescence detection were pAkt (T308) (1:1000, Cell Signaling, no. 9275), pAkt (S473) (1:1000, Cell Signaling no. 9271), pS6 (Ser235/236) (1:1000, Cell Signaling no. 2211), and mouse anti-β-actin (1:5000, no. A5441, Sigma-Aldrich). Blots were probed with secondary antibody goat anti-rabbit IgG HRP (1:5000, Elite ABC, Vector Laboratories). Western blots were probed with anti-mouse (1:5000) and anti-rabbit (1:5000) secondary antibodies conjugated to IR dye 680 or 800 (LI-COR Biosciences, Lincoln, NE, USA). Blots were imaged and quantified using the Odyssey infrared imaging system (LI-COR Biosciences).
Spatial memory testing
We tested spatial learning and memory in the Morris water maze. We used a circular polypropylene maze (diameter, 120 cm; depth, 60 cm) filled with water (26°C) clouded with white, non-toxic, water-based paint. Compass points labelled along the rim served as trial starting positions. For the spatial task, water levels were raised 0.6 cm above the clear, Plexiglass escape platform. For the nonspatial task, the escape platform was made visible by lowering the water 0.6 cm below the surface of the platform. The water maze environment was full of visual cues whose locations remained fixed throughout the learning protocol. Mouse movements in the maze were recorded using a video camera tracking system (HVS Image, Co., Buckingham, UK) mounted above the pool, and path length was measured. Animals were trained in the standard spatial version of the Morris water maze task for 10 successive days. Each training day, animals were given three trials, one from each quadrant's start location except the training (TRA) quadrant, which contained the escape platform. Each trial had a duration of 60 s with an inter-trial period of 10 min. Between trials, animals were placed into heated holding cages that sat atop thermal water blankets to control for hypothermia. The order of starting locations was counterbalanced each day by using a Latin-square design. A spatial memory (probe) trial was administered the day following the completion of spatial learning. With the escape platform removed, animals received a single 60 s trial in which the animal tried to find the escape platform in the TRA quadrant. This trial originated from the quadrant opposite the TRA quadrant. The overall path length was measured for each mouse, and the relative path length for each quadrant was calculated. The standard non-spatial version of the Morris water maze task was initiated 2 days after completion of the spatial protocol. In this task, the escape platform was visible above the water's surface. Animals were trained for 3 successive days. Each training day, the escape platform was rotated, in a clockwise manner, to the next quadrant with the exception of the TRA quadrant. Each day, animals were given three 60 s trials in the same manner that was used during their spatial training.
Statistics
All data are represented as means ± SEM. Statistical tests for all experiments, except behavioural testing studies, were computed using the non-parametric Mann–Whitney rank sum and Wilcoxon's signed rank tests or Student's t test measured in SigmaStat (Systat Software, Inc., Point Richmond, CA, USA). For behavioural testing, statistical tests were computed using repeated measures ANOVA measured in SPSS Statistics (SPSS Inc., Chicago, IL, USA).
Results
Postnatal Pten deletion does not affect hippocampal structure
To test whether PTEN has a role in synaptic transmission and plasticity that is separate from its developmental effect, we first generated Pten−/− mice in which the deletion of Pten was not only restricted to the forebrain pyramidal neurons but also restricted temporally to postnatal development (see Methods); thus, the deletion of Pten became apparent only after postnatal day (P)14, with increasing proportions of cells showing deletion with increasing age (Tsien et al. 1996b; Zakharenko et al. 2003). To verify the deletion of Pten and the concomitant loss of phosphorylation of downstream components in the PI3K signalling cascade, we performed immunostaining and Western blot analyses on hippocampal protein extracts from 8-week-old Pten−/− mice (Fig. 1A–C). PTEN immunostaining of hippocampal slices from Pten−/− mice showed PTEN− neurons throughout the hippocampus, including the CA1 and CA3 areas (Fig. 1B). The Pten deletion was less dramatic in the CA3 area, where we found fewer neurons were PTEN− (Fig. 1B). These results were consistent with the distribution of Cre in the hippocampus of CamKIIα-Cre mice, which were used to produce Pten−/− mice (Tsien et al. 1996a,b). As expected from deletion of Pten only in a subpopulation of cells in the hippocampus, quantitative Western blot analysis in Pten−/− hippocampi showed a 41% decrease in the level of PTEN protein compared to that of WT hippocampi (P = 0.04, 3 mice per genotype) (Supplemental Fig. 1). We also detected higher p-Akt (S473 and T308) and p-S6 levels in Pten−/− hippocampal extracts, as expected for activated PI3K signalling (Fig. 1C). Despite normal hippocampal development, the lifespan of Pten−/− mice was substantially shorter than that of WT mice. On average, Pten−/− mice died at 11 weeks of age from unidentified causes, and some succumbed as early as 9 weeks. Therefore, we performed our synaptic transmission and plasticity experiments using 8-week-old mice. At this age, Pten−/− mice have no apparent gross abnormalities in the hippocampus. The CA3 and CA1 areas of the hippocampus of Pten−/− mice were normally developed (Fig. 1A and B), which was not the case in mice that have the Pten gene deleted in neurons early during development (Fraser et al. 2004, 2008). Basal synaptic transmission at CA3–CA1 synapses, measured as an input–output relationship between stimulation intensity and fEPSPs in the CA1 area, was also normal in Pten−/− mice compared to that of WT littermates (P > 0.05, 23 slices for WT mice, 42 slices for Pten−/− mice {23–42 slices}), indicating the normal development of CA3 and CA1 synaptic circuits in Pten−/− mice (Fig. 1D). This result differed substantially from decreased basal synaptic transmission in mice with early deletion of Pten (Fraser et al. 2008). Short-term plasticity at CA3–CA1 synapses was also normal in mutant mice. Thus, the paired-pulse ratio was not significantly different (P > 0.05, 23–42 slices) between Pten−/− and WT littermates (Fig. 1E). These results indicate that postnatal deletion of Pten does not affect the development of the hippocampal circuits or basal synaptic properties at excitatory hippocampal synapses.
Figure 1. Postnatal Pten deletion preserves hippocampal architecture and basal synaptic transmission.
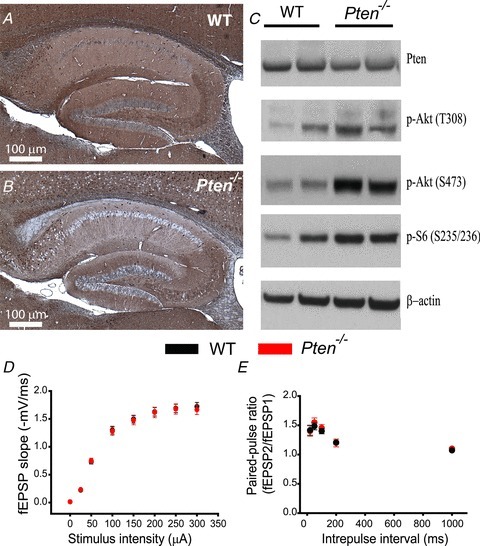
A and B, immunohistochemistry for PTEN in the hippocampus of 8-week-old WT (A) and Pten−/− (B) mice. Note the normal architecture of all hippocampal areas in Pten−/− mice. C, Western blot analysis of protein lysates from hippocampi of WT and Pten−/− littermates. Pten−/− mice show increased phosphorylation of Akt at T308 and S473 and of the downstream substrate S6. D, mean fEPSP slope as a function of stimulation intensity measured at CA3–CA1 synapses in slices from WT or Pten−/− mice shows that basal synaptic transmission is normal in both genotypes. E, mean fEPSP slopes as a function of interpulse interval between the first and second fEPSPs evoked at CA3–CA1 synapses in slices from WT and Pten−/− mice shows that paired-pulse ratio is normal in Pten−/− mice.
Postnatal Pten deletion hinders synaptic plasticity
To test whether the postnatal deletion of Pten affects long-term synaptic plasticity, we measured LTP and LTD of fEPSPs at CA3–CA1 synapses in acute hippocampal slices of Pten−/− and WT littermates. To examine whether the Pten deletion affects LTP at excitatory CA3–CA1 synapses, we recorded fEPSPs before and after the delivery of high-frequency tetanus (200 Hz) to the Schaffer collaterals. LTP was significantly decreased in Pten−/− mice compared to WT littermates (Fig. 2A). The LTP of fEPSPs measured 180 min after tetanization was approximately 55% lower in Pten−/− mice than in WT mice, increasing to 124.1 ± 6.0% over baseline, as compared to 153.6 ± 8.6% in WT littermates (P = 0.022, 23–42 slices). Similarly, LTD was impaired in Pten−/− mice (Fig. 2B). The magnitude of LTD of fEPSPs measured 180 min after delivery of low-frequency train (1 Hz, 900 stimulations) was substantially lower in Pten−/− mice (93.6 ± 4.7% of baseline, 27 slices) compared to WT littermates (79.8 ± 3.5%, 23 slices, P = 0.024). These deficits in LTP and LTD were not caused by the abnormal expression of postsynaptic ionotropic receptors; the AMPAR/NMDAR ratio was not changed in Pten-null (PTEN−) neurons, compared to neurons containing PTEN (PTEN+ neurons, P = 0.77, 7 neurons for each genotype, see below how PTEN− and PTEN+ neurons were identified) (Fig. 2C). Similarly, no differences in spontaneous mEPSCs were detected between neurons of the two genotypes (Fig. 2D and E). Thus, mEPSC amplitudes and intervals between mEPSCs were not significantly different between PTEN− and PTEN+ neurons (P = 0.304 and P = 0.907, respectively; 12–25 neurons). Together, these results indicate that postnatal deletion of the Pten gene affects both major forms of hippocampal synaptic plasticity without affecting the development of the hippocampal circuitry or basal properties of synaptic transmission.
Figure 2. Postnatal deletion of Pten causes deficits in LTP and LTD.
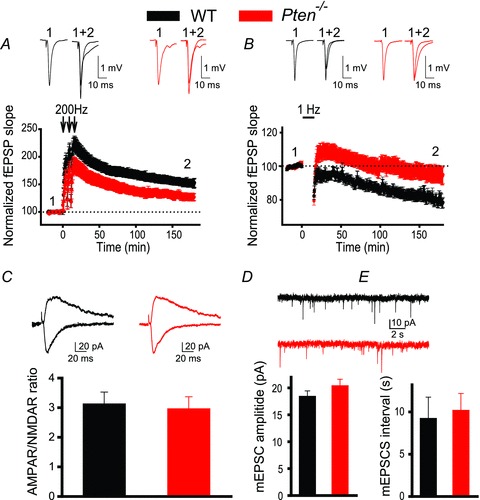
A and B, mean fEPSPs as a function of time before and after induction of LTP (A) or LTD (B) in slices from WT (black) or Pten−/− (red) mice. Insets show representative fEPSP traces before and 180 min after induction of synaptic plasticity. C, mean ratios of AMPAR/NMDAR currents recorded at CA3–CA1 synapses in slices from WT or Pten−/− mice are similar. Insets show representative traces of AMPAR-mediated (recorded at –70 mV, downward traces) and NMDAR-mediated (recorded at 40 mV, upward traces) EPSCs recorded in YFP− (WT) and YFP+ (Pten−/−) neurons. D and E, mean amplitudes (D) and intervals (E) of spontaneous mEPSCs measured in CA1 neurons of WT or Pten−/− mice are comparable. Insets show representative traces of mEPSCs measured in YFP− (WT) and YFP+ (Pten−/−) CA1 neurons.
PTEN-null pyramidal neurons have normal morphology
Because Pten deletion early in development leads to a dramatic hypertrophy in cells and synapses, we tested whether the deficit in synaptic plasticity observed in Pten−/− mice is caused by morphological abnormalities of hippocampal pyramidal neurons. To this end, we took advantage of the mosaic pattern of Pten deletion in these animals. We compared soma size and dendritic morphology between PTEN− and PTEN+ neurons in Pten−/− mice. These experiments were done in 8-week-old mice to match those used in synaptic plasticity experiments. To distinguish these neurons, we crossed Pten−/− mice with YFP-reporter mice that express this fluorescent protein in Cre+ neurons. Labelling of the hippocampal neurons in Pten−/−;R26LSL-EYFP mice with antibodies against PTEN and GFP revealed that the presence of YFP in the cell body is a reliable indicator of the Pten deletion (Fig. 3A). Indeed, approximately 84% of YFP+ pyramidal neurons in the CA1 area were PTEN− (3 mice). This is consistent with the notion that the Cre-lox method does not guarantee deletion of the floxed allele in 100% of intended cells. Although the majority of Cre+ cells will have a deletion of the intended allele, false-positive and false-negative deletions may also occur. Therefore, on the basis of the above results, we assumed that targeted deletion of Pten occurs in 84% of YFP+ neurons in Pten−/−;R26LSL-EYFP mice. We used TPLSM to perform targeted whole-cell electrophysiological recordings and fill YFP+ (PTEN−) or YFP− (PTEN+) neurons with the fluorescent dye Alexa 594 in Pten−/−;R26LSL-EYFP mice. This approach allowed clear visualization of cell bodies, dendrites and dendritic spines of YFP− and YFP+ neurons (YFP visible only in the soma in these cells) and comparison of their morphological and electrophysiological features (Fig. 3B). In contrast to results from previous studies in which Pten was deleted early during development (Fraser et al. 2004, 2008), our results showed no increase in the soma size of YFP+ neurons in which the gene was deleted postnatally (Fig. 3C). Thus, the size of somata did not significantly differ between YFP+ and YFP− neurons (P = 0.79, 14–23 neurons). Similarly, the length of apical dendrites did not differ significantly between the two genotypes (P = 0.247, 5–7 neurons). To verify the effect of the postnatal Pten deletion on the neuronal cell size, we performed Golgi (Fig. 3D) and H&E staining (Fig. 3F) and compared soma and nuclei sizes in CA3 and CA1 neurons in Pten−/− and WT littermates. Both measurements showed that postnatal Pten deletion does not increase cell or nucleus size. Thus, we found no difference in the cell size (P = 0.911, 94–102 neurons) between the genotypes (Fig. 3E) and a slight decrease in the nucleus size in Pten−/− mice (P = 0.002, 6–8 slides, 78–194 cells per slide, 3 mice per genotype) compared to that in WT littermates (Fig. 3G). Next, using TPLSM, we compared morphological characteristics of dendritic spines in PTEN+ and PTEN− CA1 neurons in Pten−/−;R26LSL-EYFP and WT;R26LSL-EYFP mice (Fig. 3H). Similar to other morphological characteristics, the width (P = 0.64) and length (P = 597, 5–8 neurons, 1146–1286 spines) of the dendritic spine were not significantly different between neurons of two genotypes (Fig. 3I). The density of dendritic spines was also similar between PTEN− neurons (1.19 ± 0.26 spines μm−1, 5 neurons) and PTEN+ neurons (1.61 ± 0.29 spines μm−1, 8 neurons, 25–314 spines per neuron, P = 0.101). Further testing using electron microscopy revealed that in the CA1 area of the hippocampus, the presynaptic (P = 0.247, 12–24 regions, 39–68 synapses) and postsynaptic (P = 0.790, 12–24 regions, 39–68 synapses) areas are of similar size in both genotypes (Fig. 3J and K). Furthermore, the length of postsynaptic density, the number of synaptic vesicles per synapse, and the number of active-zone synaptic vesicles did not differ between the genotypes (P > 0.05, 39–68 synapses, data not shown). Together, these results indicate that changes in synaptic plasticity caused by postnatal deletion of the Pten gene are not the result of morphological changes in pyramidal neurons.
Figure 3. Postnatal Pten deletion in pyramidal neurons does not induce neuronal hypertrophy in the CA3 and CA1 areas of the hippocampus.
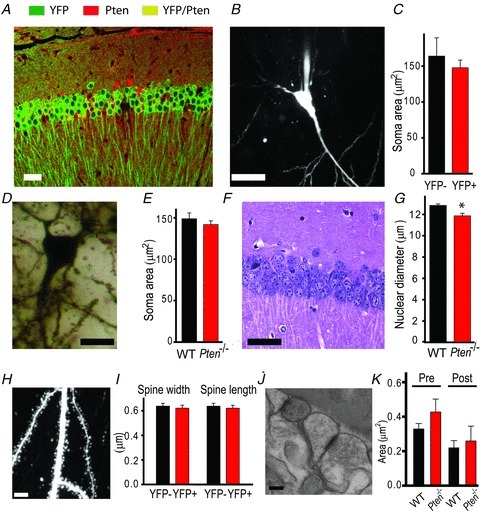
A, double-immunofluorescence staining for PTEN (red) and YFP (green) reveals that YFP is a reliable indicator of Pten deletion in Cre;Ptenfl/fl; R26LSL-EYFP mice, with only rare false-positive cells expressing both YFP and PTEN (yellow). B and C, TPLSM image of a neuron filled with Alexa 594 shows the soma clearly. This method was used to measure mean soma areas in WT (YFP−) or Pten-null (YFP+) neurons (C). D and E, Golgi staining of somata (D) of CA1 neurons was used to measure soma areas in slices from WT and Pten−/− mice (E). F and G, H&E staining of the CA1 area of the hippocampus shows the nuclei of cells. G, the mean nuclear diameter of neurons in H&E-stained slices from Pten−/− mice was smaller than that in WT mice (*P < 0.05). H, a two-photon image of an apical dendrite of a CA1 neuron. Note that the dendritic spines are clearly visible. These images were used to measure the mean width and length of dendritic spines in slices from WT and Pten−/− mice (I). J and K, scanning electron microscopic images of the hippocampus (J) were used to measure the mean presynaptic (Pre) and postsynaptic (Post) areas of synapses in the CA1 areas of WT and Pten−/− mice (K). Scale bars, 40 μm (A), 20 μm (B), 10 μm (D), 30 μm (F), 5 μm (H), and 100 nm (J).
Postnatal Pten loss impairs spatial memory
To determine whether LTP and LTD deficits at hippocampal synapses affect hippocampal-dependent spatial learning and memory, we compared the behaviour of Pten−/− and WT mice in the Morris water maze task. To match synaptic plasticity and morphological experiments, we performed these experiments in 7- to 8-week-old mice. Similar to WT mice, Pten−/− mice learned the spatial task during a 10 day training session (P < 0.001, repeated measures ANOVA, F(9, 360) = 25.862) (Fig. 4A). However, spatial memory was significantly impaired in the mutant mice. In the probe trial that was performed 24 h after the last training session, Pten−/− mice performed substantially worse than WT mice (Fig. 4B and C). Specifically, the mutant mice made fewer counter passes across the platform location during the first 10 s (P = 0.006, 18–19 mice) and during the entire duration of the 60 s trial (P = 0.003, 18–19 mice). However, no difference was seen between the two genotypes during the non-spatial task (P > 0.05, 18–19 mice) (Fig. 4D), suggesting that postnatal deletion of Pten specifically affects hippocampal-dependent spatial memory.
Figure 4. Pten deletion in pyramidal neurons during postnatal development causes a deficit in spatial memory.
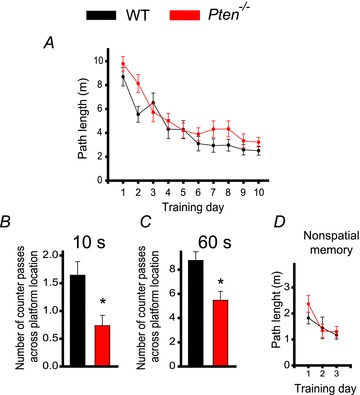
A, spatial learning is not impaired in Pten−/− mice, as indicated by the lack of difference in the mean path lengths of WT (black) or Pten−/− (red) mice swimming in the Morris water maze to find a hidden platform as a function of training days. B and C, long-term memory retention is, however, impaired in Pten−/− mice. The mean number of counter passes across the platform location measured during the first 10 s (B) and during the entire 60 s probe trial (C) in WT and Pten−/− mice differed significantly (*P < 0.05). D, non-spatial memory is normal in Pten−/− mice. Mean path lengths to swim to a visible platform, as a function of training days, was comparable in WT and Pten−/− mice.
Postnatal Pdk1 deletion does not alter synaptic plasticity
PTEN is the central negative regulator of the PI3K pathway, and its deletion causes unrestrained PI3K signalling that leads to a plethora of consequences, including abnormalities in neuronal development and function. Several recent findings have indicated that an additional deletion of a positive regulator of the PI3K pathway, Pdk1, rescues some of these abnormalities (Rintelen et al. 2001; Bayascas et al. 2005; Chalhoub et al. 2009). Therefore, we tested whether the deletion of Pdk1 rescues deficits in hippocampal synaptic plasticity and spatial memory caused by the postnatal deletion of Pten. Pdk1 deletion in brain during early development causes microcephaly and decreases cellular size (Chalhoub et al. 2009). In contrast, we did not find such changes when Pdk1 deletion occurred postnatally. Pdk1−/− mice had no visible anatomic abnormalities and had a normal lifespan (data not shown). Synaptic plasticity at CA3–CA1 hippocampal synapses was also normal in 8-week-old Pdk1−/− mice (Fig. 5). LTP and LTD of fEPSPs were not significantly different in slices from Pdk1−/− mice compared to WT mice (Fig. 5A and B). Thus, 180 min after LTP induction, fEPSPs rose to 129.9 ± 5.6% and 134.4 ± 6.9% of baseline in slices from Pdk−/− and WT mice, respectively (P = 0.67, 28–29 slices). Similarly, 180 min after LTD induction, fEPSPs decreased to 80.3 ± 3.1% and 81.7 ± 2.7% of baseline in slices from Pdk1−/− and WT mice, respectively (P = 0.502, 28–35 slices).
Figure 5. Postnatal deletion of Pdk1 in pyramidal neurons does not induce deficits in synaptic plasticity.
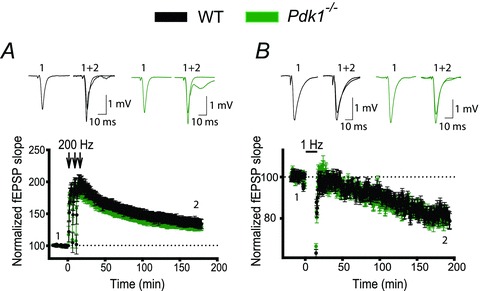
A and B, synaptic plasticity is not perturbed in Pdk1−/− mice. The mean fEPSP slope versus time before and after induction of LTP (A) or LTD (B) in slices from WT and Pdk1−/− mice did not differ. Insets show representative fEPSP traces before and 180 min after induction of synaptic plasticity.
Pdk1 deletion rescues the synaptic plasticity defect in Pten-deficient mice
To determine whether deleting Pdk could rescue the deficits in synaptic plasticity observed in Pten−/− mice, we compared properties of CA3–CA1 synapses in slices from Pten−/−;Pdk1−/− mice and their WT littermates. Double-mutant mice developed normally and had no gross morphological abnormalities (data not shown). Furthermore, in contrast to Pten−/− mice, Pten−/−;Pdk1−/− mice had a normal life span (data not shown). The hippocampi of Pten−/−;Pdk1−/−mice developed normally, and the absence of PTEN was seen clearly in hippocampal pyramidal neurons of these double-mutant mice (Fig. 6A). Also, the density of neurons in the CA1 and CA3 areas was comparable in slices from Pten−/−;Pdk1−/− and WT mice (data not shown), indicating that deletion of Pdk1 did not affect the survival of PTEN− pyramidal neurons. The pattern of Pten deletion (Figs 1B and 6A) and the decrease in PTEN protein compared to WT controls were similar between Pten−/− and Pten−/−;Pdk1−/− mice (Supplemental Fig. 2), suggesting that deletion of Pdk1 did not alter the effectiveness of Pten deletion in double-mutant mice. As expected, the increase in PI3K signalling that we observed in Pten−/− mice was reduced in double-mutant mice. We detected that levels of p-S6 were substantially higher in pyramidal neurons of Pten−/− mice compared to WT mice, but this effect was rescued in Pten−/−;Pdk1−/− mice (Fig. 6B). As in Pten−/− mice, basal synaptic transmission was not changed in the double-mutant mice. The input–output coupling and paired-pulse ratios at CA3–CA1 synapses were normal (P > 0.05, 21–32 slices) in Pten−/−;Pdk1−/− mice compared to WT littermates (Fig. 6C and D). Interestingly, the deletion of Pdk1 rescued deficits in synaptic plasticity caused by the deletion of Pten. In contrast to Pten−/− mice, the double-mutant mice showed normal LTP and LTD (Fig. 6E and F). The fEPSP measured at CA3–CA1 synapses 180 min after induction of LTP increased to 137.5 ± 7.9% (30 slices) of baseline in the double-mutant mice, which was not significantly different from that seen in WT littermates (137.7 ± 7.2%, 23 slices, P = 0.987). Similarly, fEPSPs measured 180 min after LTD induction decreased to 85.9 ± 2.8% and 82.9 ± 2.5% (22–24 slices, P = 0.427) of baseline in slices from double-mutant and WT mice, respectively. Thus, these results indicate that deletion of Pdk1, a positive downstream effector of the PI3K pathway, is sufficient to counterbalance the effect of Pten deletion on synaptic plasticity.
Figure 6. Pdk1 deletion rescues the synaptic plasticity deficit in Pten−/−mice.
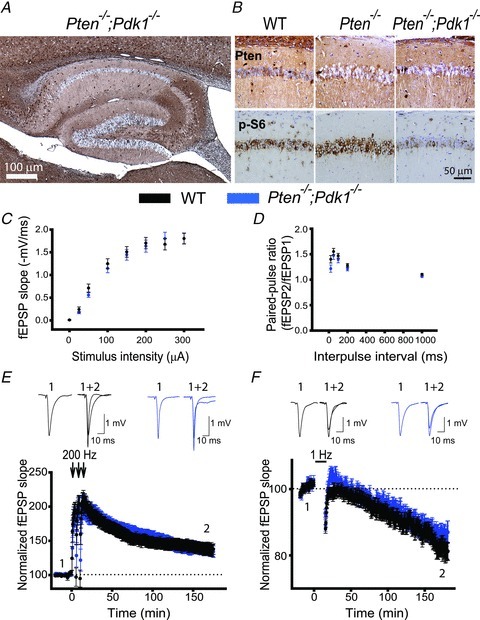
A, a representative image of immunohistochemistry for PTEN in hippocampal slices from Pten−/−;Pdk1−/− mice. As with Pten−/− mice, Pten is effectively deleted in most of the pyramidal neurons from the double-mutant mice, and their hippocampal architecture is normal. B, representative images of double immunostaining for PTEN and p-S6 in hippocampal slices of WT, Pten−/− and Pten−/−;Pdk1−/− littermates. The CA1 areas of the hippocampi are shown. C and D, basal synaptic transmission is not affected in Pten−/−;Pdk1−/− mice. Mean fEPSP slope as a function of stimulation intensity (C) or interpulse interval (D) measured at CA3–CA1 synapses in slices from WT or Pten−/−;Pdk1−/− mice are comparable. E and F, normal synaptic plasticity occurs in double-mutant mice. The mean fEPSP as a function of time before and after induction of LTP (E) and LTD (F) in slices from WT or Pten−/−;Pdk1−/− mice are similar. Insets show representative fEPSP traces before (1) and 180 min after (2) induction of synaptic plasticity.
Pdk1 deletion does not rescue spatial memory deficits in Pten-deficient mice
We next tested whether the deletion of the Pdk1 gene is sufficient to rescue the deficit in spatial memory caused by the postnatal deletion of Pten. To this end, we compared the performance of Pten−/−;Pdk1−/− mice with that of WT littermates in the Morris water maze task. Pten−/−;Pdk1−/− mice and their WT littermates had a somewhat different genetic background than Pten−/− mice and their WT littermates (see Methods). We found that double mutants and their WT littermates performed differently from Pten−/− mice and their WT littermates (Figs 4 and 7). Because genetic background contributes to overall performance in various behavioural tasks (Crawley et al. 1997; Holmes et al. 2002; Wolfer & Lipp, 2000), we assumed that this difference was due to variability in the genetic background between these two mouse strains. Nonetheless, when we compared Pten−/−;Pdk1−/− mice and their WT littermates, we found that, similar to Pten−/− mice, the double-mutant mice effectively learned the spatial task (P < 0.001, repeated measures ANOVA, F(9, 279) = 15.614) (Fig. 7A). However, their spatial memory was significantly impaired compared to their WT littermates. During the probe trial, Pten−/−;Pdk1−/− mice made fewer counter passes over the platform location than did WT mice (Fig. 7B and C). This deficit was apparent during the first 10 s (P = 0.03, 16–17 mice) and during the entire 60 s probe trial (P = 0.011, 16–17 mice). This deficit was specific for spatial memory; Pten−/−;Pdk1−/− and WT mice performed similarly (P > 0.05, 16–17 mice) in the Morris water maze non-spatial task (Fig. 7D).
Figure 7. Spatial memory is impaired in Pten−/−;Pdk1−/− mice.
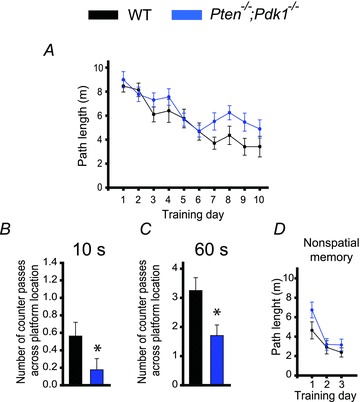
A, spatial learning is normal in double-mutant mice. Mean path length as a function of training days in the Morris water maze task in WT and Pten−/−;Pdk1−/− mice was comparable. B and C, spatial memory, however, is impaired in double-mutant mice. The mean number of counter passes across the platform location during the first 10 s (B) and during the entire (60 s) probe trial (C) for WT or Pten−/−;Pdk1−/−mice were significantly different (*P < 0.05). D, non-spatial memory is normal in Pten−/−;Pdk1−/− mice. The mean path lengths in the Morris water maze as a function of training days during the visual platform task did not differ between WT and Pten−/−;Pdk1−/− mice.
Discussion
Using conditional-knockout mice, we showed that the deletion of the Pten gene from pyramidal neurons during postnatal development impairs hippocampal synaptic plasticity and hippocampal-dependent spatial memory without affecting the developmental positioning or structural integrity of those neurons. These findings implicate PTEN as a direct modulator of synaptic plasticity and show that this functional property is independent of PTEN's role in neuronal development and growth. A direct role of PTEN in synaptic plasticity and memory may also implicate this negative regulator of the PI3K pathway in the pathogenesis of autism. Mutations in the PTEN gene have been implicated in a subset of patients with autism and macrocephaly (Goffin et al. 2001; Butler et al. 2005; Boccone et al. 2006; Buxbaum et al. 2007; Herman et al. 2007; McBride et al. 2010). Although autism-associated macrocephaly may also be associated with PTEN's role in cell size regulation, synaptic plasticity deficits associated with PTEN mutations may underlie cognitive manifestations of autism. The notion of segregated structural and functional properties of PTEN in autism is supported by reports demonstrating that multiple mouse models of autism that are based on mutations in other autism-associated genes (i.e. Shank3, MeCP2 and Fmr1) have substantial deficits in LTP or LTD but no reported macrocephaly (Huber et al. 2002; Hou et al. 2006; Moretti et al. 2006; Bozdagi et al. 2010; Bangash et al. 2011).
The mechanisms of synaptic plasticity modulation by PTEN are currently under investigation. Most likely, PTEN affects LTP and LTD through postsynaptic mechanisms. A recent report suggested that the semiacute blockade of PTEN activity in organotypic hippocampal slice cultures via the disruption of PTEN's interaction with the PDZ domain-containing protein PSD-95 resulted in deficits in LTD (Jurado et al. 2010). PSD-95 is a member of the membrane-associated guanylate kinase family that is highly abundant in the postsynaptic density and is key to postsynaptic mechanisms of LTP (Stein et al. 2003; Ehrlich & Malinow, 2004; Steiner et al. 2008) and LTD (Xu et al. 2008; Bhattacharyya et al. 2009; Sturgill et al. 2009; Jo et al. 2010). Another study reported that PTEN inhibition directly affected postsynaptic glutamatergic receptors by increasing the expression of AMPARs in cultured hippocampal neurons from neonatal rats. As a consequence, the amplitude of mEPSCs increased (Moult et al. 2010). In our study, we used acute slices from young adult Pten−/− mice that lacked Pten preferentially in CA1 pyramidal neurons. We did not detect any changes in mEPSCs recorded in CA1 neurons or in the AMPAR/NMDAR ratio and basal synaptic transmission at CA3–CA1 synapses in these mice, perhaps indicating that PTEN regulates the basal expression of AMPARs in neonatal but not in adult neurons.
Pten-dependent regulation of neuronal morphology may depend on the type of neurons. A recent in vivo study showed that Pten knockdown via short hairpin RNAs (shRNAs) led to the increase in the size of granule neurons, dendritic spines, and mEPSCs in the dentate gyrus of both young and mature mice (Luikart et al. 2011). Notably, the onset of granule neuron morphological abnormalities was significantly delayed when PTEN shRNA was introduced in young adults compared to knockdown in neonatal mice. Thus, the lack of significant morphological changes in CA1 pyramidal neurons in young adult Pten−/− mice in our study may also reflect the age-dependent rate of hypertrophy associated with PTEN deletion at different developmental stages. Differences in the hypertrophic response to PTEN deletion may reflect striking differences between the dentate gyrus and the CA1 area of the hippocampus. Specifically, the CA1 area consists of mostly differentiated cells, and the dentate gyrus is a major site of adult neurogenesis and contains a large fraction of neuronal precursors that give rise to excitatory neurons in both young and adult hippocampi (Kuhn et al. 1996; Eriksson et al. 1998; Gage, 2000). The extent to which the PI3K pathway is stimulated will dramatically influence the magnitude of the growth signal in the absence of PTEN. Thus, differences in the level of PI3K activation between the neurogenic regions of the dentate gyrus compared to the CA1 region may also underlie the rate at which morphological abnormalities are observed following PTEN deletion. In this study, we demonstrated that postnatal deletion of Pten in pyramidal neurons caused abnormal LTP and LTD in the absence of neuronal or synaptic morphological defects, thus separating the PTEN effect on synaptic morphology and function.
Our findings of abnormal synaptic plasticity in Pten−/− mice most likely represent the effect of PTEN on plasticity mechanisms at excitatory synapses. Indeed, we selectively removed PTEN from excitatory neurons by using CaMKIIα-Cre mice that express Cre recombinase in excitatory neurons (Tsien et al. 1996a,b). Thus, the most parsimonious explanation of LTP and LTD defects at CA3–CA1 synapses in Pten−/− mice is a cell-autonomous effect of PTEN on plasticity mechanisms at these excitatory synapses. Alternatively, Pten deletion may have an indirect effect at CA3–CA1 synapses through modulation of CA3 excitatory inputs on inhibitory neurons that, in turn, may affect LTP and LTD recorded in CA1 neurons.
In this study, we showed that LTP and LTD deficits caused by Pten deletion are rescued by the additional deletion of the Pdk1 gene. Double-mutant mice had normal synaptic plasticity at excitatory hippocampal synapses. Previous studies demonstrated that Pdk1 deletion restores some structural abnormalities in PTEN− neurons (Rintelen et al. 2001; Bayascas et al. 2005; Chalhoub et al. 2009). However, this is the first study to show that a similar strategy rescues functional deficits. Deletion of the Pten gene upregulates the PI3K pathway, which results in increased phosphorylation of the downstream effector Akt. Akt is fully activated when phosphorylated by Pdk1 on Thr 308 (T308) and by a second kinase, most commonly the mTorc2 complex, on Ser 473 (S473) (Mora et al. 2004; Sarbassov et al. 2005). Therefore, the block in unregulated signalling through Akt and downstream components in Pten−/−;Pdk1−/− mice was probably sufficient to rescue the LTP and LTD deficits that were observed in Pten−/− mice.
Spatial memory remained impaired in double-mutant mice, suggesting that balancing the deregulated PI3K signalling in Pten-null pyramidal neurons by removal of Pdk1 is sufficient only for certain processes. Indeed, a previous report indicated that Pdk1 deletion rescued cell hypertrophy but failed to correct migration defects of Pten-null neurons (Chalhoub et al. 2009). PTEN signalling is complex. Loss of PTEN lipid phosphatase activity and the resulting increase in PIP3 levels can transduce multiple downstream effects, including Pdk1-independent signals. Furthermore, PTEN has phosphatase-independent functions including roles in the nucleus (Chalhoub et al. 2009; Nardella et al. 2010). Therefore, inhibition of unregulated PI3K signalling downstream of Pdk1 in neurons of Pten−/−;Pdk1−/− mice may rescue only some phenotypes observed in Pten−/− mice. Alternatively, a disconnect between the effect of Pdk1 deletion on deficits in synaptic plasticity at CA3–CA1 synapses and those on the hippocampal-dependent spatial memory in Pten−/− mice may be a reflection of the role of plasticity at other hippocampal synapses in mechanisms of spatial memory. In addition to CA3–CA1 synapses, mossy fibre–CA3, CA3/commissural–CA3, perforant pathway–dentate gyrus, perforant pathway–CA1, CA3–CA2, and other hippocampal synapses may also undergo synaptic plasticity and affect spatial memory (Dudman et al. 2007; Galvan et al. 2008; Nakashiba et al. 2008; Chevaleyre & Siegelbaum, 2010; Malleret et al. 2010), and Pten deletion may affect these synapses differently (Luikart et al. 2011). Furthermore, evidence is emerging to support the theory that although synaptic plasticity mechanisms are a predominant cellular determinant of learning and memory (Milner et al. 1998; Martin et al. 2000; Whitlock et al. 2006), they may not always directly correlate with the behavioural outcome (Migaud et al. 1998; Uetani et al. 2000; Kaksonen et al. 2002; Rutten et al. 2008; Kim et al. 2009; Earls et al. 2010).
In conclusion, we have shown that conditional deletion of the negative regulator of the PI3K pathway, Pten, in pyramidal neurons during postnatal development impairs hippocampal LTP and LTD, as well as spatial memory. These deficits are not mediated by changes in neuronal migration or cellular hypertrophy. Deletion of Pdk1, a positive regulator of the PI3K pathway, rescues synaptic plasticity but not spatial memory deficits in mice with conditional deletion of Pten.
Acknowledgments
We thank the St Jude Cell and Tissue Imaging Shared Resource, Sharon Frase, and Linda Mann for their help with electron microscopy experiments. We thank Tak Mak (University of Toronto) for the Ptenflox mice and Dario Alessi (University of Dundee) for the Pdk1flox mice. This work was supported in part by the National Institute of Mental Health grant R01MH079079 (S.S.Z.), National Cancer Institute grant R01CA135554 (S.J.B.), and the American Lebanese Syrian Associated Charities (ALSAC). The funding sources had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Glossary
Abbreviations
- Cre
Cre recombinase
- fEPSP
field excitatory postsynaptic potential
- GFP
green fluorescent protein
- LTD
long-term depression
- LTP
long-term potentiation
- mEPSCs
miniature excitatory postsynaptic currents
- P
postnatal day
- PFA
paraformaldehyde
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PIP3
phosphatidylinositol 3,4,5-trisphosphate
- PI3K
phosphatidylinositol 3-kinase
- PTEN
phosphatase and tensin homologue
- TPLSM
two-photon laser-scanning microscopy
- TRA
training
- YFP
yellow fluorescent protein
- WT
wild-type
Author contributions
S.S.Z. and S.J.B. designed the experiments. M.S., R.B.B., I.T.B., G.Z., S.J.B. and S.S.Z. collected, analysed and interpreted the data. S.S.Z. wrote the article. All authors gave final approval of the version to be published. All experiments were performed at St Jude Children's Research Hospital.
Supplementary Material
Suppl. Figure 1
Suppl. Figure 2
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Backman S, Stambolic V, Mak T. PTEN function in mammalian cell size regulation. Curr Opin Neurobiol. 2002;12:516–522. doi: 10.1016/s0959-4388(02)00354-9. [DOI] [PubMed] [Google Scholar]
- Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, Tsao MS, Shannon P, Bolon B, Ivy GO, Mak TW. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet. 2001;29:396–403. doi: 10.1038/ng782. [DOI] [PubMed] [Google Scholar]
- Bangash MA, Park JM, Melnikova T, Wang D, Jeon SK, Lee D, Syeda S, Kim J, Kouser M, Schwartz J, Cui Y, Zhao X, Speed HE, Kee SE, Tu JC, Hu JH, Petralia RS, Linden DJ, Powell CM, Savonenko A, Xiao B, Worley PF. Enhanced polyubiquitination of Shank3 and NMDA receptor in a mouse model of autism. Cell. 2011;145:758–772. doi: 10.1016/j.cell.2011.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bayascas JR, Leslie NR, Parsons R, Fleming S, Alessi DR. Hypomorphic mutation of PDK1 suppresses tumorigenesis in PTEN(+/–) mice. Curr Biol. 2005;15:1839–1846. doi: 10.1016/j.cub.2005.08.066. [DOI] [PubMed] [Google Scholar]
- Bayazitov IT, Richardson RJ, Fricke RG, Zakharenko SS. Slow presynaptic and fast postsynaptic components of compound long-term potentiation. J Neurosci. 2007;27:11510–11521. doi: 10.1523/JNEUROSCI.3077-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Biou V, Xu W, Schluter O, Malenka RC. A critical role for PSD-95/AKAP interactions in endocytosis of synaptic AMPA receptors. Nat Neurosci. 2009;12:172–181. doi: 10.1038/nn.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundon JA, Zakharenko SS. Dissecting the components of long-term potentiation. Neuroscientist. 2008;14:598–608. doi: 10.1177/1073858408320643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccone L, Dessi V, Zappu A, Piga S, Piludu MB, Rais M, Massidda C, De Virgiliis S, Cao A, Loudianos G. Bannayan-Riley-Ruvalcaba syndrome with reactive nodular lymphoid hyperplasia and autism and a PTEN mutation. Am J Med Genet A. 2006;140:1965–1969. doi: 10.1002/ajmg.a.31396. [DOI] [PubMed] [Google Scholar]
- Bozdagi O, Sakurai T, Papapetrou D, Wang X, Dickstein DL, Takahashi N, Kajiwara Y, Yang M, Katz AM, Scattoni ML, Harris MJ, Saxena R, Silverman JL, Crawley JN, Zhou Q, Hof PR, Buxbaum JD. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol Autism. 2010;1:15. doi: 10.1186/2040-2392-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, Eng C. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Cai G, Chaste P, Nygren G, Goldsmith J, Reichert J, Anckarsater H, Rastam M, Smith CJ, Silverman JM, Hollander E, Leboyer M, Gillberg C, Verloes A, Betancur C. Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:484–491. doi: 10.1002/ajmg.b.30493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalhoub N, Zhu G, Zhu X, Baker SJ. Cell type specificity of PI3K signaling in Pdk1- and Pten-deficient brains. Genes Dev. 2009;23:1619–1624. doi: 10.1101/gad.1799609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Siegelbaum SA. Strong CA2 pyramidal neuron synapses define a powerful disynaptic cortico-hippocampal loop. Neuron. 2010;66:560–572. doi: 10.1016/j.neuron.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow DK, Groszer M, Pribadi M, Machniki M, Carmichael ST, Liu X, Trachtenberg JT. Laminar and compartmental regulation of dendritic growth in mature cortex. Nat Neurosci. 2009;12:116–118. doi: 10.1038/nn.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley JN, Belknap JK, Collins A, Crabbe JC, Frankel W, Henderson N, Hitzemann RJ, Maxson SC, Miner LL, Silva AJ, Wehner JM, Wynshaw-Boris A, Paylor R. Behavioral phenotypes of inbred mouse strains: implications and recommendations for molecular studies. Psychopharmacology (Berl) 1997;132:107–124. doi: 10.1007/s002130050327. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudman JT, Tsay D, Siegelbaum SA. A role for synaptic inputs at distal dendrites: instructive signals for hippocampal long-term plasticity. Neuron. 2007;56:866–879. doi: 10.1016/j.neuron.2007.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earls LR, Bayazitov IT, Fricke RG, Berry RB, Illingworth E, Mittleman G, Zakharenko SS. Dysregulation of presynaptic calcium and synaptic plasticity in a mouse model of 22q11 deletion syndrome. J Neurosci. 2010;30:15843–15855. doi: 10.1523/JNEUROSCI.1425-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Malinow R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci. 2004;24:916–927. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endersby R, Baker SJ. PTEN signaling in brain: neuropathology and tumorigenesis. Oncogene. 2008;27:5416–5430. doi: 10.1038/onc.2008.239. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- Fraser MM, Bayazitov IT, Zakharenko SS, Baker SJ. Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience. 2008;151:476–488. doi: 10.1016/j.neuroscience.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser MM, Zhu X, Kwon CH, Uhlmann EJ, Gutmann DH, Baker SJ. Pten loss causes hypertrophy and increased proliferation of astrocytes in vivo. Cancer Res. 2004;64:7773–7779. doi: 10.1158/0008-5472.CAN-04-2487. [DOI] [PubMed] [Google Scholar]
- Gage FH( Mammalian neural stem cells. Science. 2000;287:1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- Galvan EJ, Calixto E, Barrionuevo G. Bidirectional Hebbian plasticity at hippocampal mossy fiber synapses on CA3 interneurons. J Neurosci. 2008;28:14042–14055. doi: 10.1523/JNEUROSCI.4848-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin A, Hoefsloot LH, Bosgoed E, Swillen A, Fryns JP. PTEN mutation in a family with Cowden syndrome and autism. Am J Med Genet. 2001;105:521–524. doi: 10.1002/ajmg.1477. [DOI] [PubMed] [Google Scholar]
- Gustafson S, Zbuk KM, Scacheri C, Eng C. Cowden syndrome. Semin Oncol. 2007;34:428–434. doi: 10.1053/j.seminoncol.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Herman GE, Butter E, Enrile B, Pastore M, Prior TW, Sommer A. Increasing knowledge of PTEN germline mutations: Two additional patients with autism and macrocephaly. Am J Med Genet A. 2007;143:589–593. doi: 10.1002/ajmg.a.31619. [DOI] [PubMed] [Google Scholar]
- Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes A, Wrenn CC, Harris AP, Thayer KE, Crawley JN. Behavioral profiles of inbred strains on novel olfactory, spatial and emotional tests for reference memory in mice. Genes Brain Behav. 2002;1:55–69. doi: 10.1046/j.1601-1848.2001.00005.x. [DOI] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci. 2005;25:11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo J, Son GH, Winters BL, Kim MJ, Whitcomb DJ, Dickinson BA, Lee YB, Futai K, Amici M, Sheng M, Collingridge GL, Cho K. Muscarinic receptors induce LTD of NMDAR EPSCs via a mechanism involving hippocalcin, AP2 and PSD-95. Nat Neurosci. 2010;13:1216–1224. doi: 10.1038/nn.2636. [DOI] [PubMed] [Google Scholar]
- Jurado S, Benoist M, Lario A, Knafo S, Petrok CN, Esteban JA. PTEN is recruited to the postsynaptic terminal for NMDA receptor-dependent long-term depression. Embo J. 2010;29:2827–2840. doi: 10.1038/emboj.2010.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaksonen M, Pavlov I, Voikar V, Lauri SE, Hienola A, Riekki R, Lakso M, Taira T, Rauvala H. Syndecan-3-deficient mice exhibit enhanced LTP and impaired hippocampus-dependent memory. Mol Cell Neurosci. 2002;21:158–172. doi: 10.1006/mcne.2002.1167. [DOI] [PubMed] [Google Scholar]
- Kim MH, Choi J, Yang J, Chung W, Kim JH, Paik SK, Kim K, Han S, Won H, Bae YS, Cho SH, Seo J, Bae YC, Choi SY, Kim E. Enhanced NMDA receptor-mediated synaptic transmission, enhanced long-term potentiation, and impaired learning and memory in mice lacking IRSp53. J Neurosci. 2009;29:1586–1595. doi: 10.1523/JNEUROSCI.4306-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 1996;16:2027–2033. doi: 10.1523/JNEUROSCI.16-06-02027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet. 2001;29:404–411. doi: 10.1038/ng781. [DOI] [PubMed] [Google Scholar]
- Lawlor MA, Mora A, Ashby PR, Williams MR, Murray-Tait V, Malone L, Prescott AR, Lucocq JM, Alessi DR. Essential role of PDK1 in regulating cell size and development in mice. EMBO J. 2002;21:3728–3738. doi: 10.1093/emboj/cdf387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart BW, Schnell E, Washburn EK, Bensen AL, Tovar KR, Westbrook GL. Pten knockdown in vivo increases excitatory drive onto dentate granule cells. J Neurosci. 2011;31:4345–4354. doi: 10.1523/JNEUROSCI.0061-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malleret G, Alarcon JM, Martel G, Takizawa S, Vronskaya S, Yin D, Chen IZ, Kandel ER, Shumyatsky GP. Bidirectional regulation of hippocampal long-term synaptic plasticity and its influence on opposing forms of memory. J Neurosci. 2010;30:3813–3825. doi: 10.1523/JNEUROSCI.1330-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- McBride KL, Varga EA, Pastore MT, Prior TW, Manickam K, Atkin JF, Herman GE. Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res. 2010;3:137–141. doi: 10.1002/aur.132. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O’Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Milner B, Squire LR, Kandel ER. Cognitive neuroscience and the study of memory. Neuron. 1998;20:445–468. doi: 10.1016/s0896-6273(00)80987-3. [DOI] [PubMed] [Google Scholar]
- Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15:161–170. doi: 10.1016/j.semcdb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006;26:319–327. doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moult PR, Cross A, Santos SD, Carvalho AL, Lindsay Y, Connolly CN, Irving AJ, Leslie NR, Harvey J. Leptin regulates AMPA receptor trafficking via PTEN inhibition. J Neurosci. 2010;30:4088–4101. doi: 10.1523/JNEUROSCI.3614-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashiba T, Young JZ, McHugh TJ, Buhl DL, Tonegawa S. Transgenic inhibition of synaptic transmission reveals role of CA3 output in hippocampal learning. Science. 2008;319:1260–1264. doi: 10.1126/science.1151120. [DOI] [PubMed] [Google Scholar]
- Nardella C, Carracedo A, Salmena L, Pandolfi PP. Faithfull modeling of PTEN loss driven diseases in the mouse. Curr Top Microbiol Immunol. 2010;347:135–168. doi: 10.1007/82_2010_62. [DOI] [PubMed] [Google Scholar]
- Rintelen F, Stocker H, Thomas G, Hafen E. PDK1 regulates growth through Akt and S6K in Drosophila. Proc Natl Acad Sci U S A. 2001;98:15020–15025. doi: 10.1073/pnas.011318098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutten K, Misner DL, Works M, Blokland A, Novak TJ, Santarelli L, Wallace TL. Enhanced long-term potentiation and impaired learning in phosphodiesterase 4D-knockout (PDE4D) mice. Eur J Neurosci. 2008;28:625–632. doi: 10.1111/j.1460-9568.2008.06349.x. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein V, House DR, Bredt DS, Nicoll RA. Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J Neurosci. 2003;23:5503–5506. doi: 10.1523/JNEUROSCI.23-13-05503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner P, Higley MJ, Xu W, Czervionke BL, Malenka RC, Sabatini BL. Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron. 2008;60:788–802. doi: 10.1016/j.neuron.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stornetta RL, Zhu JJ. Ras and Rap signaling in synaptic plasticity and mental disorders. Neuroscientist. 2011;17:54–78. doi: 10.1177/1073858410365562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgill JF, Steiner P, Czervionke BL, Sabatini BL. Distinct domains within PSD-95 mediate synaptic incorporation, stabilization, and activity-dependent trafficking. J Neurosci. 2009;29:12845–12854. doi: 10.1523/JNEUROSCI.1841-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Yamaguchi MT, Ohteki T, Sasaki T, Kaisho T, Kimura Y, Yoshida R, Wakeham A, Higuchi T, Fukumoto M, Tsubata T, Ohashi PS, Koyasu S, Penninger JM, Nakano T, Mak TW. T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity. 2001;14:523–534. doi: 10.1016/s1074-7613(01)00134-0. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996a;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996b;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- Uetani N, Kato K, Ogura H, Mizuno K, Kawano K, Mikoshiba K, Yakura H, Asano M, Iwakura Y. Impaired learning with enhanced hippocampal long-term potentiation in PTPdelta-deficient mice. EMBO J. 2000;19:2775–2785. doi: 10.1093/emboj/19.12.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite KA, Eng C. Protean PTEN: form and function. Am J Hum Genet. 2002;70:829–844. doi: 10.1086/340026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cheng A, Mattson MP. The PTEN phosphatase is essential for long-term depression of hippocampal synapses. Neuromolecular Med. 2006;8:329–336. doi: 10.1385/NMM:8:3:329. [DOI] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Wolfer DP, Lipp HP. Dissecting the behaviour of transgenic mice: is it the mutation, the genetic background, or the environment? Exp Physiol. 2000;85:627–634. [PubMed] [Google Scholar]
- Xu W, Schluter OM, Steiner P, Czervionke BL, Sabatini B, Malenka RC. Molecular dissociation of the role of PSD-95 in regulating synaptic strength and LTD. Neuron. 2008;57:248–262. doi: 10.1016/j.neuron.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharenko SS, Patterson SL, Dragatsis I, Zeitlin SO, Siegelbaum SA, Kandel ER, Morozov A. Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses. Neuron. 2003;39:975–990. doi: 10.1016/s0896-6273(03)00543-9. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA. Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat Neurosci. 2001;4:711–717. doi: 10.1038/89498. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA. Altered presynaptic vesicle release and cycling diring mGluR-dependent LTD. Neuron. 2002;35:1099–1110. doi: 10.1016/s0896-6273(02)00898-x. [DOI] [PubMed] [Google Scholar]
- Zhou J, Blundell J, Ogawa S, Kwon CH, Zhang W, Sinton C, Powell CM, Parada LF. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci. 2009;29:1773–1783. doi: 10.1523/JNEUROSCI.5685-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.