

In this issue of Circulation Research, Marsboom et al (1) describe a novel involvement of a mitochondrial fission regulatory pathway which is likely to be of great importance in pulmonary arterial hypertension (PAH)-associated smooth muscle remodeling. Data in the study provide evidence that this process appears to be controlled by activation of hypoxia inducible factor 1α (HIF-1α). As illustrated in the model in Figure 1, the stabilization or activation of HIF-1α in PAH is shown to promote mitochondrial fission by Cyclin B1/CDK1-dependent phosphorylation of dynamin-related protein-1 (DRP1) at Serine-616. CDK1-cyclin B dependent regulation of tubular mitochondria fragmentation by DRP1 was identified in previous studies as a regulator of the even distribution of mitochondria during mitosis in HeLa cells (2). Data in the study of Marsboom et al (1) document how this process appears to be essential for the in vitro proliferation of pulmonary arterial smooth muscle cells from human hypertensive patients. In addition, evidence is provided for these mechanisms appearing to be a prominent contributing factor to pulmonary arterial remodeling in several different models of PAH.
Fig. 1.
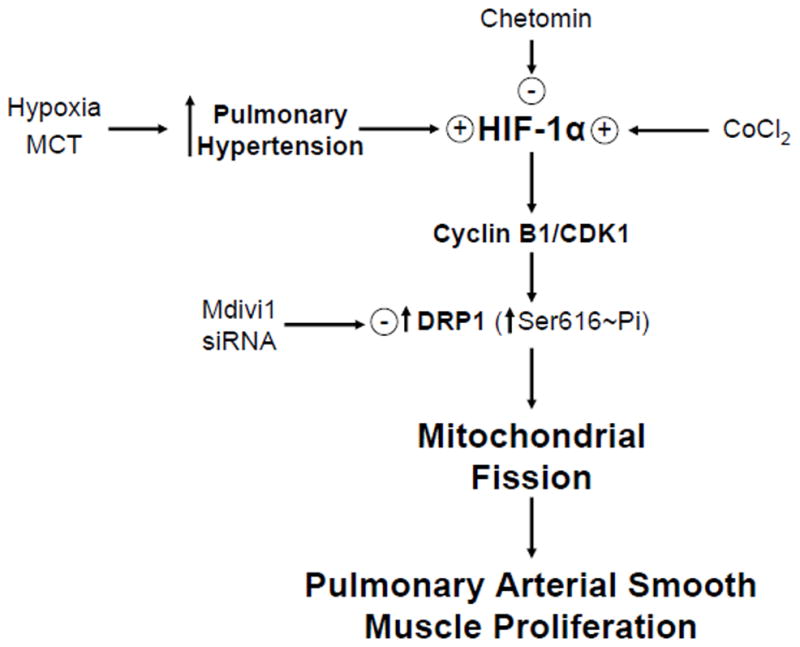
Model showing how conditions associated with pulmonary hypertension development promote HIF-1α regulation of pulmonary arterial smooth proliferative remodeling that appears to be controlled by a Cyclin B1/CDK1-mediated phosphorylation of DRP1, which causes mitochondrial fission needed for their even distribution of during mitosis. The model also highlights approaches described in the text which were used to elucidate mechanisms.
As described in the article of Marsboom et al (1), PAH is disorder in which small pulmonary arteries are obstructed, constricted and inflamed in a manner which increases pulmonary vascular resistance, and this promotes right ventricular hypertrophy and eventually failure. Previous work from Archer and colleagues identified many of components of processes that evolved into the current new mechanism. For example, their studies provided evidence that an activation of HIF-1α under aerobic conditions in PAH was associated with a shifting from aerobic mitochondrial energy metabolism to a cancer cell-type emphasis on using aerobic glycolysis and mitochondrial fragmentation (3). Interestingly, promoting aerobic mitochondrial energy metabolism with the pyruvate dehydrogenase kinase inhibitor dichloroacetate has substantial protective effects against PAH associated with restoring a hypoxic sensing mechanism associated with mitochondrial-derived peroxide regulating voltage-gated potassium (Kv1.5) channels and decreasing HIF-1α activation (3). Dichloroacetate also prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis (4). These previous observations suggest shifting to aerobic glycolysis associated-energy metabolism is potentially an important factor in promoting the remodeling-associated pulmonary arterial smooth muscle proliferation.
The article of Marsboom et al (1) provides an extensive amount of impressive data to support the novel concepts in this paper. Properties of this regulatory system were studied in lung tissue and pulmonary arterial smooth muscle cells (PASMC) from humans with PAH for comparison with PASMC from non-hypertensive controls. In addition, rat monocrotaline (MCT) and chronic hypoxia models were studied, as well as a new model of pulmonary hypertension was developed based cobalt chloride inducing HIF-1α in rats and PASMC. Human PASMC studies indicated that HIF-1α promoted a cyclin B1/CDK1-dependent phosphorylation of DRP1 at Serine-616 in the mechanism of mitochondrial fission regulation, employing inhibitors of HIF-1α (chetomin) and DRP1 (Mdivi-1 and RO-3306). The PAH PASMC had elevated levels of HIF-1α under aerobic conditions and studies with Mdiv-1 and siRNA for DRP1 resulted in evidence that cell cycle arrest occurred in the G2/M phase. The impact of PAH conditions influencing mitochondrial fusion in PASMC was visualized in a movie showing the time-dependent spreading of a mitochondrial matrix fluorescent protein (online supplement). These cells also have properties suggesting there was a shift toward the use of aerobic glycolysis for energy metabolism in PAH. Activating HIF with CoCl2 and/or desferrioxamine promoted DRP1 Serine-616 phosphorylation and rapid mitochondrial fragmentation, in a manner attenuated by HIF-1α siRNA and Mdiv-1. Treatment of PASMC with CoCl2 was also shown to cause a shift in ion channel regulation associated with a reduction in Kv channels, depolarization and increased intracellular calcium that are similar to changes seen in PAH. The mitochondrial fragmentation mechanism appears to be an active process in the remodeling observed in lung tissue from idiopathic PAH patients based on detection by imaging of elevated levels of HIF-1α, increased nuclear localization of Cyclin B1 and increased Serine-616 phosphorylation of DRP1 in pulmonary blood vessels with an external diameter below 150μm that have underwent PASMC proliferation or “muscularization”. A chronic treatment of rats with CoCl2 for 4-weeks was observed to cause PAH associated with a reduction in mitochondrial size. The DRP1 inhibitor Mdivi-1 prevented the development of PAH in both this CoCl2 model and in a chronic hypoxia (4 weeks of 10% oxygen) rat model of PAH. Mdivi-1 also seemed to reverse indicators of hypertension when the therapy was started after three weeks in the rat monocrotaline model of PAH. Thus, data are provided showing the importance of the HIF-1α mitochondrial fission regulatory pathway across multiple animal and human models of PAH.
Recent studies are providing evidence that mitochondrial dynamics associated with proteins promoting fission and fusion are regulated by signaling mechanisms associated with adaptive physiological and pathophysiological processes (5). In the current study (1), the expression of mRNA for mitochondrial fusion protein Mitofusin-2 (MFN2) was decreased, whereas, mitochondrial fusion proteins Mitofusin-1 (MFN1) and Optic atrophy-1 (OPA1) were not different when pulmonary arterial smooth muscle cells from humans with PAH were compared to cells from control humans who were not hypertensive. In contrast, the pro-fusion protein MFN2 was decreased in human PAH PASMCs under conditions where increased phosphorylation on Serine616 of the fission protein DRP1 was observed. Interestingly, it was observed in the present study that inhibition of DRP1 with Mdivi-1 reversed the fragmented phenotype and increased the MFN2. Increased MFN2 has previously been associated with promoting apoptosis in vascular smooth muscle cells (6). Recent studies in mice having conditional cardiac specific ablation of MFN1 and/or MFN2 have provided insight into the metabolic and functional impact of mitochondrial fission promoted as a result of these proteins (7–9). The cardiac MFN1 deficiency results in small spherical mitochondria, however, the mice show rather normal cardiac and mitochondrial respiratory function (7). A cardiac MFN2 deficiency was actually associated with enlarged spherical mitochondria and some cardiac and mitochondrial respiratory dysfunction (8). Interestingly both of these deficiencies were associated with a resistance to oxidant exposure promoting a mitochondrial transition pore response. In contrast, a combined ablation of cardiac myocyte MFN1 and MFN2 and its associated mitochondrial fragmentation resulted in mitochondrial respiratory dysfunction, and rapid progression into a fatal dilated cardiomyopathy (9). Thus, mitochondrial fission and fusion are not specifically linked to a disruption of mitochondrial energy metabolism or highly energy-dependent processes like cardiac function. However, activation of DRP-1 and perhaps decreased MFN2 appear to contribute to promoting proliferative remodeling of vascular smooth muscle.
The HIF-1α controlled mitochondrial fission regulatory pathway illustrated in Figure 1, which is reported in the paper by Marsboom et al (1) in the current issue of Circulation Research, is likely to be of great importance in physiological and pathophysiological processes involving vascular smooth muscle remodeling. Hypoxia sensing and oxidant mechanisms regulating HIF-1α (10) provide pathways for its regulation by many types of stress-type conditions existing in vivo. Regulation of mitochondrial fission and other processes such as promoting aerobic glycolysis (3,4) appear to enable the progression through mitosis that is needed for the stem cell-associated remodeling seen in pulmonary hypertension. Interestingly, Archer and colleagues have also recently reported (11) a 18F-fluorodeoxyglucose positron emission tomography tracer diagnostic method for imaging the enhanced glycolytic metabolism and its therapeutic reversal by dichloroacetate associated with controlling the hyperproliferative changes seen in small pulmonary arteries in PAH. Thus, the DRP1-regulated mechanism controlling mitochondrial fusion and proliferation described in the current issue of Circulation Research (1) opens up a new metabolism-associated target for therapies to treat pulmonary hypertension and perhaps other vascular remodeling associated disease processes.
Acknowledgments
Funding: The Author’s lab has been funded by NIH grants HL031069, HL043023 and HL066331.
Non-standard Abbreviations and Acronyms
- CDK1
Cyclin-dependent kinase 1
- DRP1
Dynamin-related protein-1
- HIF-1α
Hypoxia-inducible factor-1α
- Kv
Voltage-gated K+ channel
- MCT
monocrotaline
- Mdivi-1
Mitochondrial division inhibitor-1
- MFN1
Mitofusin-1
- MFN2
Mitofusin-2
- OPA1
Optic atrophy-1
- PAH
Pulmonary arterial hypertension
- PASMC
Pulmonary artery smooth muscle cell
Footnotes
Conflicts of interest: None
References
- 1.Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang Y-H, Thenappan T, Piao L, Zhang HZ, Pogoriler J, Chen Y, Morrow E, Weir EK, Rehman J, Archer SL. Dynamin-related protein 1 (DRP1)-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res. 2012;110:xxx–xxx. doi: 10.1161/CIRCRESAHA.111.263848. (In this issue; printer, please update) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related gtpase drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 3.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thebaud B, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK, Archer SL. An abnormal mitochondrial-hypoxia inducible factor-1alpha-kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation. 2006;113:2630–2641. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 4.McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, Michelakis ED. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. 2004;95:830–840. doi: 10.1161/01.RES.0000145360.16770.9f. [DOI] [PubMed] [Google Scholar]
- 5.Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- 6.Guo X, Chen K-H, Guo Y, Liao H, Tang J, Xiao R-P. Mitofusin 2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway. Circ Res. 2007;101:1113–1122. doi: 10.1161/CIRCRESAHA.107.157644. [DOI] [PubMed] [Google Scholar]
- 7.Papanicolaou KN, Ngoh GA, Dabkowski ER, O’Connell KA, Ribeiro RF, Stanley WC, Walsh K. Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am J Physiol Heart Circ Physiol. 2012;302:H167–H179. doi: 10.1152/ajpheart.00833.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O’Shea KM, Riley DD, Lugus JJ, Colucci WS, Lederer WJ, Stanley WC, Walsh K. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011;31:1309–1328. doi: 10.1128/MCB.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y, Liu Y, Dorn GW. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. 2011;109:1327–1331. doi: 10.1161/CIRCRESAHA.111.258723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marsboom G, Wietholt C, Haney CR, Toth PT, Ryan JJ, Morrow E, Thenappan T, Bache-Wiig P, Piao L, Paul J, Chen CT, Archer SL. Lung 18F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am J Resp Crit Care Med. 2012;185:670–679. doi: 10.1164/rccm.201108-1562OC. [DOI] [PMC free article] [PubMed] [Google Scholar]