

Abstract
Membrane proteins function in the diverse environment of the lipid bilayer. Experimental evidence suggests that some lipid molecules bind tightly to specific sites on the membrane protein surface. These lipid molecules often act as co-factors and play important functional roles. In this study, we have assessed the evolutionary selection pressure experienced at lipid-binding sites in a set of α-helical and β-barrel membrane proteins using posterior probability analysis of the ratio of synonymous vs. nonsynonymous substitutions (ω-ratio). We have also carried out a geometric analysis of the membrane protein structures to identify residues in close contact with co-crystallized lipids. We found that residues forming cholesterol-binding sites in both β2-adrenergic receptor and Na+-K+-ATPase exhibit strong conservation, which can be characterized by an expanded cholesterol consensus motif for GPCRs. Our results suggest the functional importance of aromatic stacking interactions and interhelical hydrogen bonds in facilitating protein-cholesterol interactions, which is now reflected in the expanded motif. We also find that residues forming the cardiolipin-binding site in formate dehydrogenase-N γ-subunit and the phosphatidylglycerol binding site in KcsA are under strong purifying selection pressure. Although the lipopolysaccharide (LPS)-binding site in ferric hydroxamate uptake receptor (FhuA) is only weakly conserved, we show using a statistical mechanical model that LPS binds to the least stable FhuA β-strand and protects it from the bulk lipid. Our results suggest that specific lipid binding may be a general mechanism employed by β-barrel membrane proteins to stabilize weakly stable regions. Overall, we find that the residues forming specific lipid binding sites on the surfaces of membrane proteins often experience strong purifying selection pressure.
Keywords: Lipid binding, ω-ratio, cholesterol binding motif, lipopolysaccharide
1. Introduction
Biological membranes are an indispensable component of the living cells. They create intercellular and intracellular permeability barriers and incorporate proteins that play important roles in cell communications, protein and solute transport, photosynthesis, motility, and many other vital physiological functions. A fluid mosaic model of the biological membrane was first introduced in 1972 [1], in which a biological membrane was represented as a random two-dimensional liquid crystal, sparsely populated by freely diffusing proteins. Since then, accumulated experimental data has significantly expanded our understanding of the biological membrane. Current models emphasize a membrane with variable patchiness and thickness and a higher content of integrated membrane proteins [2]. Additionally, a multitude of experimental results shifted a general perception of phospholipids as membrane building blocks that provide an appropriate environment for integral membrane proteins, to molecules that play important regulatory roles in modulating membrane protein function, such as directing membrane protein topology, folding, and assembly [3-7].
Due to the physico-chemical constraints imposed by the membrane, membrane proteins have a limited repertoire of residues that can face various regions of the phospholipid bilayer. Computational studies of membrane protein structures revealed that their lipid-facing surfaces are enriched with hydrophobic side chains of Ile, Leu, Val, and Phe residues in the hydrocarbon core region (facing the acyl chains of phospholipids), and with the side chains of Lys, Arg, Trp, Phe, and Leu residues in the interface regions (facing the lipid polar head-groups and glycerol backbones) [8]. These residues interact with boundary (a.k.a. annular) lipids, the majority of which have restricted molecular motions [9, 10], but are still exchangeable with the bulk of membrane lipids. The boundary lipids help to maintain an electrochemically sealed barrier of diffusion and provide a tight integration of the proteins into the membrane [10-12]. Additionally, there are lipids (non-annular) that bind tightly and specifically to the protein surface. They exchange slowly with the surrounding phospholipids, and can directly affect the function of membrane proteins [9, 11, 12]. Palsdottir & Hunte [11] thoroughly analyzed several high-resolution protein-lipid complexes available at the time of their publications, and found that bound lipids are stabilized by multiple noncovalent interactions between protein residues and lipid head-groups, as well as protein and lipid hydrophobic acyl chains. Furthermore, the binding locations of tightly bound lipids are often reproduced in the x-ray structures of the same membrane protein obtained from different species. For example, a comparison of the x-ray structures of the cytochrome c oxidase from R. sphaeroides, P. denitrificans, and B. taurus revealed a remarkable correspondence of the positions occupied by the alkyl chains of the co-crystallized phospholipids in all structures [13]. Further study of the conservation of the lipid-binding site residues in cytochrome c oxidase suggested a higher conservation of the amino acid residues interacting with the alkyl chains, rather than with the phospholipid head-groups [14].
The growing number of high-resolution 3D structures of membrane proteins provides an improved basis for detailed and quantitative studies that can elucidate the interplay between lipids and membrane proteins. In this work, we analyze evolutionary conservation of amino acid residues forming lipid-binding sites by assessing the evolutionary selection pressure acting at each amino acid residue site. We estimated the site-specific ratio of synonymous vs. non-synonymous substitution (called ω-ratio) of the underlying DNA sequences, which can uncover residues important for biological function and structural stability. This approach is well developed [15-19] and was previously used to assess natural selection of the residues forming protein folding nuclei [20] and to discover important protein-protein interactions in the GABAC receptor [21]. Here, we assess the evolutionary conservation of the lipid-facing residues and compare the average conservation of the residues forming a lipid binding site with that of the residues forming the rest of the lipid-facing surface. We observed a statistically significant conservation for cholesterol-binding sites in β2-adrenergic receptor and Na+-K+-ATPase. We also found strong conservation of the residues forming a cardiolipin binding site in formate dehydrogenase-N (γ-subunit), and the phosphatidylglycerol (PG) binding site in the KcsA potassium channel. Two patches of residues experiencing strong purifying selection pressure were found in the lipopolysaccharide-binding site in FhuA β-barrel membrane protein, although when all residues in close contact with the LPS are taken into account, the LPS site is only weakly conserved due to the strict criterion of the synonymous vs. non-synonymous substitution. Further analysis suggested that non-synonymous substitutions within a lipid-binding site occur mostly between residues with similar physicochemical properties, and LPS binding provides significant stability to FhuA. Our analysis shows that residues interacting with co-crystallized lipids often experience stronger purifying selection pressure than residues forming the rest of the membrane-facing protein surface, indicating the importance of lipid binding sites on membrane proteins.
2. Methods
2.1. Calculation of lipid-accessible amino acid residues
Seven membrane protein structures containing lipid and detergent molecules that bind in well-defined sites on the protein surface were chosen for this study (Table 1). We included only one structure containing bound detergent (rhomboid protease from E. coli, PDB ID: 2IC8), as detergent is likely to bind membrane proteins non-specifically. This structure was chosen because the homologous structure from H. influenza (PDB ID: 2NR9) contains detergents bound to the same site, indicating that this is likely a specific binding. In general, there is some uncertainty about whether a bound lipid is a true non-annular lipid that specifically binds to the membrane protein, or an annular lipid that happened to co-crystallize with the protein. We chose to be conservative and regard all structures of the same protein containing different bound lipids and detergents as interacting non-specifically with annular lipids and excluded them from assessment of binding site conservation. In addition, the experimental purification procedures used for protein crystallization may partially strip non-annular phospholipids from the protein surfaces, thus often revealing an incomplete picture of the protein-lipid interactions. As it is very difficult to discriminate these non-annular lipid-binding sites, we again conservatively count all such cases as “lipid-free”, which would lead to underestimation of the significance of observed conservation of lipid binding sites. We included structures containing lipids at the protein-protein interfaces, because endogenous, functionally important lipids often bind at such interfaces, and we can clearly define and take into account residues at the protein-protein interfaces.
Table 1.
Results of the evolutionary conservation calculations
| Protein | PDB | NTM | Nseq | Na | Nna | Mωa | Mωna | p-value |
|---|---|---|---|---|---|---|---|---|
| β2-adrenergic R. | 2RH1:A | 201 | 13 | 100 | 15 | 0.1999 | 0.0979 | 4.2×10−2 |
| Na+-K+-ATPase | 2ZXE:A | 277 | 26 | 125 | 7 | 0.0420 | 0.0051 | 1.0×10−2 |
| Na+-K+-ATPase | 2ZXE:B | 31 | 19 | 24 | 4 | 0.0408 | 0.0165 | 4.7×10−2 |
| Formate dehyd.-N | 1KQF:B | 29 | 14 | 16 | 8 | 0.0773 | 0.0529 | NA |
| Formate dehyd.-N | 1KQF:C | 109 | 15 | 60 | 14 | 0.0662 | 0.0259 | 1.6×10−2 |
| FhuA | 2FCP:A | 325 | 15 | 132 | 24 | 0.0341 | 0.0260 | 8.8×10−2 |
| KcsA | 1K4C:A | 74 | 4 | 35 | 13 | 0.1189 | 0.0998 | 3.4×10−2 |
| GlpG | 2IC8 | 119 | 11 | 79 | 14 | 0.0926 | 0.0421 | 1.0×10−3 |
| ADP/ATP carrier | 2C3E | 164 | 28 | 62 | 78 | 0.0367 | 0.0331 | 1.4×10−1 |
NTM: total number of residues in the transmembrane region (for FhuA, it is the total number of residues in the beta strands, including the non-membrane segments interacting with the LPS head-group).
Nseq: number of orthologous sequences used in ω-ratio calculations.
Na: number of residues interacting with annular lipids.
Nna: number of residues interacting with a bound lipid.
Mωa: mean ω-ratio of the residues interacting with annular lipids.
Mωna: mean ω-ratio of the residues interacting with the non-annular lipid.
Transmembrane helices were determined with the help of the OPM (Orientation of Proteins in Membranes) database [22] as well as visual inspection, using resolved lipid molecules as a reference. The VOLBL [23] program with the probe radius set to 1.9Å has been used to compute lipid-accessible surfaces of membrane proteins as described previously [8]. VOLBL uses weighted Delaunay triangulation and alpha shape to compute metric properties of molecules. The Delaunay triangulation of membrane proteins is computed using the DELCX program [24, 25], and the alpha shape is computed using the MKALF program [24, 26]. The van der Waals radii of protein atoms are from Tsai et al [27]. Residues interacting with the co-crystallized phospholipids were determined using the INTERFACE program with the probe radius set to 0.5Å as described previously [28].
2.2. Conservation analysis
We studied eight chains from six high-resolution membrane protein structures containing co-crystallized lipids and one protein with co-crystallized detergent molecules. The selected sequences from BLAST searches all had e-values of less than 10−25. We manually inspected the selected sequences and their annotations, ensuring that they are true orthologous sequences of the same protein carrying out the same function in different species. We found previously that 10-15 orthologous sequences from sufficiently divergent species are well-suited for the calculation of ω-ratios.
Next, the selected protein sequences were aligned using CLUSTALW [29], followed by manual adjustment with PFAAT [30] when necessary. We also retrieved cDNA sequences for each selected protein sequence, which were aligned with the TRANALIGN program from the EMBOSS software package [31] using multiple protein sequence alignments as a guide. Phylogenetic trees were constructed by a maximum likelihood method as implemented in the PROML package [32]. To identify residues under purifying evolutionary selection pressure, we carried out a posterior probability analysis of evolutionary selection pressure at the individual amino acid residue sites using a maximum likelihood estimator [33]. The evolutionary selection pressure was calculated as the ratio of synonymous vs. non-synonymous substitutions, termed the ω-ratio [15], which measures selection pressure at each amino acid residue position. We use the PAML package, including the codeml module for such analysis [33].
2.3 Randomization tests and statistical analysis
Randomization tests and statistical analysis were performed following the approach described by Tseng and Liang [20]. The mean ω-ratio of the lipid-binding site was tested against the distribution of the mean ω-ratios from 105 random samples containing the same number of amino acid residues as the binding site, but drawn from the pool of lipid-facing residues identified by VOLBL and different from those found at the protein-lipid interface.
2.4 Calculation of energy values
The calculation of energy values of β-strands was performed as described in Naveed et al [34]. We estimate the stability of a strand based on its native as well as non-native conformations. In a non-native conformation, the neighboring strands can slide up or down along the z-axis as many as 7 positions of strand registrations, for a total of 15 different registrations for the two strands. Each conformation will have completely different hydrogen bond patterns between the strands [34]. We enumerate all possible configurations of TM strands of FhuA using this model, with a total of 7×7=49 possible registrations for each strand with its 2 neighbors. We calculate strand energy using an empirical potential function, the development of which is based on extensive combinatorial analysis of known β-barrel membrane protein structures [35-37]. The energy for each residue consists of two components. First, each residue contributes to the energy based on its depth in the lipid bilayer and the orientation of its side-chain. This is termed the “single body propensity”. Second, each residue interacts with two residues on (separate) neighboring strands through strong backbone H-bond interaction, side-chain interactions and weak H-bond interactions, which collectively make up the two-body energy term. Strand energy for a conformation is the summation of both single body and two-body energy terms over all residues in the strand. The summation of the native and non-native conformations weighted by the Boltzmann factor gives the final expected energy for the strand.
3. Results
3.1 Evolutionary selection pressure in the lipid-binding sites
Membrane proteins in the dataset contain a variety of lipids, including cholesterol (as found in β2-adrenergic receptor and Na+-K+-ATPase), lipopolysaccharide (FhuA ferric hydroxamate uptake receptor), cardiolipin (formate dehydrogenase-N and ADP/ATP carrier), phosphatidylglycerol modeled as diacyl glycerol (KcsA K+ channel), as well as detergent molecules in rhomboid intramembrane protease. We have identified lipid-facing residues in each protein, as described in Methods, and separated residues interacting with the bound, co-crystallized lipid/detergent (lipid-binding residues) from those that have no apparent contact with any lipid in the x-ray structure (lipid-free residues).
For each protein chain, we have estimated the mean ω-ratios for the sets of lipid-binding and lipid-free residues. The results are summarized in Table 1, which lists the proteins used in the study, the total number of the lipid-facing residues, the number of the sequences in the phylogenetic tree, mean ω-ratios of the residues in lipid-binding and lipid-free sets of residues, and the p-values for statistical significance. P-values were obtained from randomization tests by recalculating mean ω-ratios of two randomly obtained sets (one was equal in size to the set of lipid-binding residues, while the other corresponded to the set of lipid-free residues). This was repeated 105 times and the fraction where the calculated mean ω-ratios of the lipid-free set were lower than in the lipid-binding set was used as the p-value. The protein and DNA sequence alignments are available as Supplementary Data.
Our results show that the mean ω-ratios of the lipid-binding residues are collectively smaller than the mean ω-ratios of the lipid-free residues, indicating that the lipid-binding sites are generally under stronger purifying evolutionary selection pressure. By the strict criterion of the ω-ratio, in which any non-synonymous substitution changes the encoded amino acid residue - even though it may be of similar physicochemical properties - we found statistically significant conservation of the cholesterol-binding sites in both β2-adrenergic receptor and in Na+-K+ - ATPase. Additionally, the cardiolipin-binding site in formate dehydrogenase-N γ-subunit and the phosphatidylglycerol binding site in KcsA, show robust conservations. The detergent-binding site in E.coli rhomboid proteinase is also strongly conserved. The LPS-binding site of FhuA and three CL-binding sites in ADP/ATP carrier appear to be only weakly conserved by this stringent criterion.
3.2 Cholesterol-binding site in β2-adrenergic receptor, Na+-K+-ATPase and a cholesterol binding motif in GPCRs
Two high-resolution structures of the human β2-adrenergic receptor have recently been reported (PDB ID: 2RH1 and 3D4S). They feature three and two bound cholesterol molecules, respectively. In the 2RH1 structure, two receptors are crystallized as a parallel dimer, in which protein-protein interactions are mediated by six ordered cholesterol and two palmitic acid molecules [38]. In the 3D4S structure, the receptors are crystallized as an antiparallel dimer, with two cholesterol molecules bound at the same binding site as in 2RH1 structure, although in a slightly different conformation. Additionally, a part of another lipid molecule was resolved in the third cholesterol-binding site, demonstrating a strong affinity of this site to retain and bind lipids. The bound cholesterol in 3D4S structure is not at the site of the crystal molecular contacts, providing a clear indication of the physiologically relevant cholesterol-binding sites [39].
The bound cholesterol molecules are found in a shallow surface depression formed by the segments of helices I, II, III, and IV, thus covering a significant part of the membrane-facing surface on the cytoplasmic side of the receptor. We have obtained a complete list of residues interacting with cholesterol in 1RH1 and 3D4S structures using geometric calculations. Comparison of cholesterol-binding residues showed that although cholesterol molecules occupy essentially the same binding sites in both structures (Fig. 1A, B), the sets of cholesterol-binding residues are not identical, even though there is a significant overlap between lipid-binding sites from both structures. This suggests that the cholesterol-binding sites allow a certain degree of binding flexibility that may depend on several factors. The combined results of evolutionary and structural calculations from these two structures are summarized in Table 2. Here, residues under the strongest purifying selection are shown in bold, while “+” or “−“ signs indicate whether the residue interacts (“+”) or does not interact (“−“) with cholesterol in the respective structure. The highly conserved F49, which interacts with cholesterol in the 2RH1 structure, and R151, which interacts with cholesterol in the 3D4S structure, define the cholesterol-binding site boundaries along the z-axis of the phospholipid bilayer.
Fig. 1.

Cholesterol-binding sites in β2-adrenergic receptor shown in (A) 2RH1 and (B) 3D4S structures. Conserved residues are shown in pink, non-conserved residues are in orange. Cholesterol molecules bind in a shallow groove formed by the segments of helices I, II, III, and IV in similar, but not identical locations in both structures. The cholesterol-binding sites in both structures share five residues under strong purifying selection (shown in pink): I55, S74, C77, L80, L84, and W158. (C) The interhelical hydrogen bond between S74 from helix II and W158 from helix IV helps to maintain the optimal conformation of W158 side chain, which interacts with the sterol ring of cholesterol through the stacking interaction.
Table 2.
Cholesterol-binding residues in β2-adrenergic receptor structures (PDB ID: 2RH1 and 3D4S).
| Residue | 2RH1 | 3D4S | ω-ratio |
|---|---|---|---|
| Val 48 | + | − | 0.024 |
| Phe 49 | + | − | 0.019 |
| Val 52 | + | − | 0.122 |
| Ile 55 | + | + | 0.022 |
| Ala 59 | − | + | 0.334 |
| Tyr 70* | − | + | 0.424 |
| Thr 73 | − | + | 0.114 |
| Ser 74 | − | + | 0.026 |
| Cys 77 | + | + | 0.022 |
| Leu 80 | + | + | 0.032 |
| Val 81 | + | + | 0.117 |
| Leu 84 | + | + | 0.030 |
| Ala 85 | − | + | 0.621 |
| Phe 108 | + | − | 0.019 |
| Ile 112 | + | + | 0.528 |
| Leu 115 | − | + | 0.029 |
| Arg 151* | − | + | 0.134 |
| Ile 154* | + | + | 0.112 |
| Leu 155 | + | − | 0.169 |
| Trp 158* | + | + | 0.024 |
| Leu 339 | + | − | 0.114 |
Residues in the cholesterol consensus motif (CCM)
Residues under the strongest purifying selection are shown in bold; “+” or “−“ signs indicate whether the residue interacts (“+”) or does not interact (“−“) with cholesterol.
The cholesterol-binding sites in both structures share five residues under strong purifying selection: I55, S74, C77, L80, L84, and W158. Here, W158 is nearly universally conserved among class A GPCRs and interacts with the sterol ring of cholesterol 1 (following the numbering of cholesterol molecules by Cherezov et al. [38]) through the stacking interactions [39]. To facilitate this interaction, the side chain of tryptophan residue likely maintains a conformation where its aromatic ring is parallel to the lipid-facing protein surface. Analysis of the structures with HBPLUS [40] revealed an interhelical hydrogen bond between the hydroxyl group of S74 (helix II) and the NH group of W158 indole ring (helix IV), as shown in Fig. 1C. Sequence alignments of class A GPCRs, which are regulated by the cholesterol content of the biological membrane, showed that the serine residue at position 74 is highly conserved, with only two out of 25 aligned sequences containing asparagine residues at this position (data not shown). Asparagine is a polar residue and would likely preserve a hydrogen bond with the tryptophan side chain from the neighboring helix. An identical interhelical hydrogen bond is observed in the structure of the human A2A adenosine receptor [41] between S47 and W129. Evolutionary calculations performed on the set of 16 sequences of A2A receptors confirmed that both residues are under strong purifying selection pressure in this receptor as well.
Based on the spatial distribution of the conserved residues that are important for cholesterol binding in β2-adrenergic receptor, a cholesterol consensus motif (CCM) for membrane proteins was proposed [39] using Ballesteros-Weinstein numbering scheme as follows: [4.39-4.43(R,K)]-[4.50(W,Y)]-[4.46(I,V,L)]-[2.41(F,Y)], where position 4.50 corresponds to W158 in β2-adrenergic receptor and can be occupied by either a tryptophan or tyrosine residue. We propose to expand this CCM by adding a position corresponding to S74 due to functional importance of the S74-W158 interhelical H-bond, and the observed high conservation of both residues. The new CCM is now: [4.39-4.43(R,K)]-[4.50(W,Y)]-[2.45(S)]-[4.46(I,V,L)]-[2.41(F,Y)].
Sodium-potassium ATPase is an ATP-powered ion pump that establishes concentration gradient for Na+ and K+ ions across the plasma membrane in all animal cells by pumping Na+ from the cytoplasm and K+ from the extracellular medium [42]. Na+-K+-ATPase is functionally dependent on cholesterol content [5] and plays active role in the intracellular cholesterol distribution [43].
In Na+-K+- ATPase structure, cholesterol binds in a shallow groove between α- and β-subunits (Fig. 2A). The ω-ratio calculations show that all residues (except T788 from the α-subunit) interacting with cholesterol are under strong purifying selection pressure. Analysis of the cholesterol-binding site revealed a conserved Y40 residue on the β-subunit that, similar to W158 in β2-adrenergic receptor, is in a stacking interaction with the aromatic rings of cholesterol and forms an intersubunit hydrogen bond with conserved S851 from the α-subunit (Fig. 2B). This interaction may also play an important role in facilitating protein-protein interactions between α and β subunits of Na+-K+-ATPase.
Fig. 2.

Cholesterol-binding site in Na+-K+-ATPase. (A) Protein surfaces formed by the residues under strong purifying selection pressure from α- and β-subunits are shown in pink. Non-conserved T788 from α-subunit is shown in orange. (B) Intersubunit hydrogen bond between Y40 (β-subunit, the helix is shown in pale green) and S851 (α-subunit) in the cholesterol-binding site, which promotes a stacking interaction with the bound cholesterol.
3.3 Lipopolysacharide-binding site in ferric hydroxamate uptake receptor
The x-ray structure of the E. coli β-barrel membrane protein ferric hydroxamate uptake receptor (FhuA) contains a bound lipopolysaccharide [44], which is a complex lipoglycan found exclusively in the outer membrane of Gram-negative bacteria. LPS is known to be a potent activator of the innate immune system in higher organisms [45, 46]. LPS has an extensive binding site on the FhuA protein surface, formed by the short discontinuous segments found on β-strands 7-11 as shown in Fig. 3A, burying an accessible surface area of 1800Å [47]. Using a recently developed statistical mechanical model of β-barrel membrane proteins [34], we have calculated the thermodynamic properties of the transmembrane region of FhuA. Figure 4 summarizes the results of individual β-strand energy calculations of this receptor. Based on these calculations, we found that strands 7 - 9 form the most unstable region in the protein, and that β-strand 8, which runs through the middle of the LPS-binding site, has the highest energy. This is usually characteristic of the β-strand at the protein-protein oligomerization interface [34].
Fig. 3.

Lipopolysaccharide binding site in ferric hydroxamate uptake receptor. (A) The LPS-binding surface, where 11 residues are under strong purifying selection pressure, are shown in pink. The conserved residues cluster into two regions: one is a cluster of residues interacting with the polar saccharide head-group, and another cluster of residues interacting with the acyl chains in the hydrophobic core of the outer membrane. (B) Salt bridge interactions between conserved charged arginines and the LPS head-group. (C) Interstrand hydrogen bond between conserved Q298 and Y284, which may help to define the surface interacting with the acyl chain of phospholipid and provide additional interstrand stability.
Fig. 4.

Iindividual empirical β-strand energy calculations of the FhuA receptor. Strands 7 - 9 form the most unstable region in the protein, and β-strand 8, which runs through the middle of the LPS-binding site, has the highest energy.
We have identified 24 residues in close contact with the bound LPS in FhuA structure. The evolutionary analysis performed on 15 sequences of FhuA receptors showed that 11 out of 24 interacting residues are under strong purifying selection pressure. These residues are shown in pink on the FhuA LPS-binding surface in Fig. 3A. The randomization test (Table 1) shows that the LPS-binding site in FhuA is only weakly conserved with the strict criterion of ω-ratio, with a p-value of 8.8×10−2. The conserved residues cluster into two regions of the binding site: The first is a cluster of residues interacting with the polar saccharide head-group of LPS, and the second is a cluster of residues interacting with the acyl chains of LPS in the hydrophobic core of the outer membrane. In the head-group region, the conserved polar charged arginine residues R382, R384 (β-strand 9), and R472 (β-strand 11) together with D386, are mostly surrounded by polar non-conserved lysine residues K280, K351, and K439, as well as L437 and E304, forming an outer boundary of the binding site. Here, the conserved R382 and R384 represent an R×R intrastrand antimotif, which was shown to be generally unfavorable in the sequences of membrane β-barrels with the odds ratio of 0.42 and p-value of 3.5×10−2 [48]. This strongly suggests that the energetically unfavorable R×R motif has been selected throughout evolution as a part of a functionally important specific binding site of LPS, as its stability will be counterbalanced by interactions with the LPS molecule. Our structural calculations show that these polar residues facing the LPS head-group region play important functional roles by forming multiple salt bridges and hydrogen bonds with the polar sugar groups and the negatively charged phosphates of LPS.
Additionally, stability calculations identified E304, K351, and Q353, which form multiple hydrogen bonds and salt bridges with the LPS head-group, as destabilizing for the protein native conformation if they do not come into contact with LPS. They contribute significantly to the high energies of strands 7 and 8, as shown in Fig. 4. Although these residues are not under strong purifying selection, the polar side chains are all preserved in these positions in the FhuA multiple sequence alignment. These side chains likely form hydrogen bonds or salt bridges with the LPS.
Specifically, in the 15 sequences used in this study, K351 aligns with arginine, histidine, glutamine, and aspartic acid residues in other proteins, all of which are polar and capable of forming hydrogen bonds or salt bridges with the LPS head-group. Q353 is more buried by the LPS and is less variable in comparison with K351. Q353 position can be substituted by glutamine, asparagine, or a single serine amino acid residue in the aligned sequences. Position of E304 is mostly occupied by glutamic acid residues, although other polar residues such as lysine, arginine, and glutamine are occasionally found in the aligned sequences.
The hydrophobic acyl chains of LPS interact with a number of hydrophobic and non-charged polar residues in the membrane hydrocarbon core. The side-chains of these conserved surface residues appear in the middle of the TM region and interact mostly with the crystallographically resolved ends of the acyl chains. Here, the highly conserved residues Y284 and Q298 form an interstrand hydrogen bond, which may help to define the surface interacting with the acyl chain of phospholipid and provide an additional interstrand stability (Fig. 3C). Since glutamine has low propensity for facing phospholipids in the hydrocarbon core region of β-barrel membrane proteins [37], the conserved Q298 may be playing a functional role.
3.4 Cardiolipin in formate dehydrogenase-N structure
Formate dehydrogenase-N (Fdh-N) is a major component of E. coli nitrate respiration pathway. It is a trimer of heterotrimers that contain a periplasmic α subunit and two subunits with transmembrane domains (β and γ), where β subunit contains one TM helix, and γ subunit contains 4 TM helices. The Fdh-N structure features three well-resolved cardiolipin (CL) molecules tightly bound to each heterotrimer in the transmembrane region. Fig. 5A illustrates how cardiolipin binds to Fdh-N. Each cardiolipin interacts with the β and γ subunits of one heterotrimer and with the γ subunit of the neighboring heterotrimer.
Fig. 5.

Cardiolipin at the protein-protein interface in formate dehydrogenase N (Fdh-N). (A) Fragments of two Fdh-N heterotrimers are shown, where cardiolipin binds to the β and γ-subunits of one heterotrimer (shown in green and blue, respectively), and the γ–subunit from the adjacent heterotrimer (magenta). The bound heme molecules are also shown. Only one β-subunit is shown for clarity (colored in green). Here, cardiolipin (CL) acyl chain A fills in a tunnel leading to the heme-binding site, acyl chain B interacts with the neighboring γ-subunit, and acyl chain C interacts with β-subunit. (B) There are two CL-binding sites on the γ-subunit (shown in cyan and pink). In the first binding site, the acyl chain fills in the tunnel leading to the heme, and the majority of the tunnel residues are conserved (shown in pink). The second binding site is at the protein-protein interface with a neighboring γ-subunit. This site is the most conserved with the lowest ω-ratio, where all but one residue are under strong purifying selection. (C) CL-binding site on the β-subunit, which is shown as a green surface. The periplasmic loop residues N15, S16, and I16 form hydrogen bonds with the CL head-group, although they are not conserved. A263 and I266 that form a groove on the surface of TM helix are also not conserved.
Cardiolipin is a dimeric phospholipid containing four acyl chains, and is often found filling the cavities at the protein-protein interfaces as well as stabilizing interactions between protein subunits [49]. Only three out of four cardiolipin acyl chains are well resolved in the Fdh-N structure 1KQF. These are marked with letters A through C in Fig. 5A; where acyl chain A fills in a tunnel leading to the heme-binding site of the γ-subunit, acyl chain B interacts with the neighboring γ-subunit, and acyl chain C interacts with β subunit.
Residues interacting with cardiolipin acyl chains in γ-subunit are significantly conserved (Table 1 and Fig. 5B). The acyl chains interacting with γ-subunit are likely to play important role in the trimer formation, as they interact with γ-subunits from two adjacent heterotrimers. Additionally, the position of the acyl chain A (Fig. 5B) inside the tunnel leads to the Heme bc, suggesting that cardilipin is involved in the process of electron transfer, thus making it a part of the Fdh-N catalytic process.
The CL-binding surface for the acyl chain C is shown in Fig. 5C. Our structural calculations showed that cardiolipin is in close contact with 8 residues on the β-subunit transmembrane helix, as well as three residues from the periplasmic loop, two of which, N15 and S16, form hydrogen bonds with the head-group as was determined by HBPLUS [40]. Overall, the binding site on the β-subunit is only slightly more conserved when compared with the rest of the lipid-facing residues from the same subunit. It should be noted that many conserved probe-accessible residues (a.k.a. lipid-accessible residues) on the β-subunit are found at the protein-protein interface with a γ-subunit. Consequently, it would be difficult to separate selection pressures due to the lipid binding from those due to the important role they play in protein-protein interaction.
3.5. Cardiolipin in the ADP/ATP carrier
The bovine heart ADP/ATP carrier (AAC) contains six transmembrane domains that fold into a structure with three-fold pseudosymmetry formed by three internal repeats, where each internal repeat binds a molecule of cardiolipin [50]. Experimental data show that cardiolipin is critically important for AAC folding and function [51], and can also mediate dimerization at the protein-protein interfaces [52]. Although the cardiolipins in the AAC complex are only partially resolved [52], our structural calculations revealed that ~44% of lipid-facing residues in the AAC form contacts with the resolved groups of cardiolipins (Table 1). Figs. 6A-C show three CL-binding sites in which conserved and non-conserved residues are shown as gray and red surfaces respectively. The overall conservation varies from site to site. For example, CDL800 (Fig. 6A, average ω=0.032) and CDL802 (Fig. 6C, average ω=0.023) exhibit binding sites with large conserved areas, and patches of non-conserved residues mainly found at the membrane interfaces. In contrast, CL801 binding site (Fig. 6B, average ω=0.049) contains only a small number of conserved residues mainly interacting with the CL head-group, while the residues interacting with the acyl chains are non-conserved. Overall, we find that the mean ω-ratio of all residues interacting with bound CL in all three sites (ω = 0.033) is similar to the mean ω-ratio of the residues that have no contacts with the resolved cardiolipin atoms (ω = 0.037), implying that there is no strong conservation of the CL-binding sites in the AAC.
Fig. 6.

Cardiolipin (CL) binding sites in ADP/ATP carrier. The conserved and non-conserved surfaces are shown in gray and red, respectively. (A, D) A front view (A) of the binding surface for cardiolipin CDL800 (the numbering is from 2C3E structure), mean ω = 0.033, and a view of the conserved tunnel (D) on the surface interacting with the CL head-group. The backbone carboxyl from G72 interacts with the phosphate group of the CL. (B, E) A front view (B) of the binding surface for cardiolipin CDL801, mean ω = 0.049, and a view of the conserved tunnel (E) interacting with the CL head-group. The backbone carboxyl from G272 interacts with the phosphate group. (C, F) A front view (C) of the binding surface for cardiolipin CDL802, mean ω = 0.022, and a view of the conserved tunnel (F) interacting with the CL head-group. The backbone carboxyl from G175 interacts with the phosphate group. (G) A sequence alignment of the five consecutive residues forming the bottom of the conserved depressions interacting with the cardiolipin head-groups. Each sequence contains a conserved aromatic residue (W70, Y173, and F270), and a conserved glycine (G72, G175, and G272). There are polar residues between the aromatics residues and the glycines: R71, Q174, and K271.
The cardiolipin-binding sites in the AAC contain a few interesting features. First, each head-group occupies a groove-like depression that is roughly parallel to the membrane plane (Fig. 6A-C and 6D-F). These head-group binding depressions have similar shapes, and are composed of the highly conserved residues. There is a close contact of one of the CL phosphates with an amide group (shown in blue in Figs. 6A-F) of the conserved glycine residues (G72, G175, and G272) at the bottom of the depression. Additionally, the aromatic residues involved in protein-lipid interactions e.g., W70, Y173, and F270 [52], are all highly conserved and under strong evolutionary selection pressure. In all cases, the phosphate group interacts with five consecutive residues from the second helix of each repeat, i.e. helices H2, H4, and H6. Figure 6G shows a sequence alignment of these residues from each internal repeat.
3.6. Phosphatidylglycerol-binding site in KcsA
In the high-resolution crystal structure of the bacterial KcsA channel, a lipid molecule fills the groove between adjacent subunits (Fig. 7) [4]. Experimental studies identified the bound phospholipid as a phosphatidylglycerol (PG), although its structure was not completely resolved and modeled as diacyl glycerol [4]. In E. coli membranes, phosphatidylethanol (PE) is more abundant than PG [53], and the fact that KcsA experimentally purifies with PG instead of PE suggests that this site has specificity for phosphatidylglycerol binding. The resolved segment of PG is bound on the extracellular side of the intersubunit interface and interacts with 13 residues from the adjacent subunits. Of these, 9 residues are under strong purifying selection, showing significant overall conservation of this lipid-binding site (p-value 3.4×10−2, Table 1). There are three conserved residues W67, T85, and R89 at the intersubunit interfaces, which in addition to forming a lipid-binding site, may play important roles in protein-protein interactions. The remaining six conserved residues are on the lipid-facing surfaces of every subunit and are shown in pink in Fig. 7. The non-conserved residues Y45, Y62, L86, and V93 are at the boundaries of the binding site and are shown in orange. R64 (shown in blue), which was proposed [4] to interact with the negatively charged PG phosphate, is not identified by our structural calculations as interacting with the PG, likely due to the missing phosphate. Although this position is not conserved according to the strict criteria of ω-ratio calculations, we did identify a lysine at this position, capable for forming polar interactions with the negatively charged phosphate.
Fig. 7.
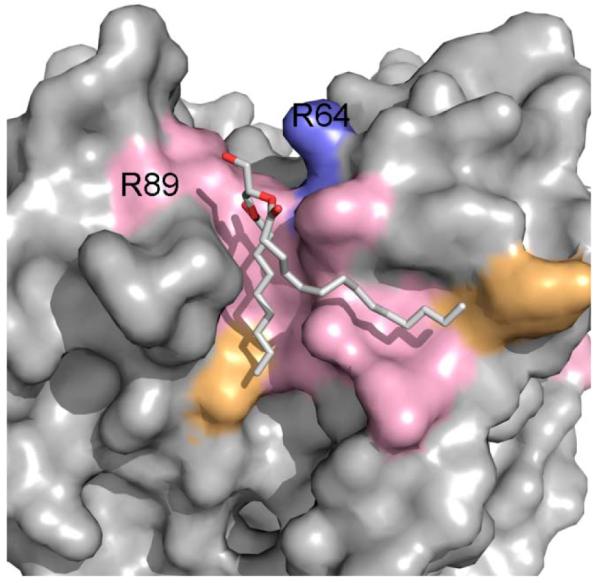
A phosphatidylglycerol (PG) binding site in KcsA potassium channel. The PG binding site is highly conserved with 9 out of 13 lipid-binding residues under strong purifying selection (shown in pink). R64, which was proposed [4] to interact with the negatively charged PG phosphate, is colored in blue.
3.7. Lipid-binding site in rhomboid family intramembrane protease
Intramembrane rhomboid proteases, such as E.coli GlpG, have a core catalytic domain of six transmembrane helices. They cleave type 1 transmembrane substrates a few residues inside of the membrane from the extracellular side [54]. There are crystal structures of the rhomboid protease from E. coli (PDB: 2IC8) and from H. influenzae (PDB: 2NR9). Structures of proteases from both organisms contain several resolved detergent molecules and detergent molecule fragments. Inspection of the lipid-facing surfaces revealed a cleft between transmembrane segments S2 and S5, which is occupied by the detergent molecules in both structures. We have identified residues interacting with the detergent in E. coli structure and carried out evolutionary calculations (Table 1). Our results showed that this site is highly conserved (p-value 10−3) with 10 out of 14 (71%) detergent-interacting residues under strong evolutionary selection pressure.
4. Discussion
The importance of interactions between membrane proteins and their lipid environment is increasingly recognized [6, 55]. Experimental data showed that some phospholipids transiently interact with membrane proteins, while others bind tightly to the grooves on the protein surface [10]. These tightly bound lipids are often resolved in membrane protein structures [11, 12, 14]. In this work, we combined evolutionary and structural analysis to quantitatively assess conservation of the lipid-binding residues. We used the ratio of non-synonymous to synonymous substitutions (Ka/Ks ratio or ω-ratio) to measure purifying selection pressure at individual amino acid residue sites. The estimation of ω-ratio is based on analysis at the DNA sequence level, where the evolutionary relationships among the coding sequences and the underlying stochastic processes [17] were modeled explicitly by a continuous time Markov process [15]. This explicit evolutionary model describes in probabilistic terms how codons evolve along a phylogenetic tree, and has yielded significant insight about the history of molecular evolution [16, 17, 56-58]. An important advantage of this approach is that it distinguishes mutations fixed by evolution from those fixed by chance. In addition, it explicitly takes into account the extent of divergence among sequences, bias in codon frequency, as well as bias favoring transition over transversion [15]. This approach has advantages over other approaches such as entropy-based calculations, because it is more accurate in accounting for bias due to differences in evolutionary history between species.
Evolutionary analysis using ω-ratio is more elaborate and significantly more time-consuming in comparison with other methods such as calculation of residue frequencies or information entropy. Depending on the length and the number of sequences, a calculation may take up to several hours. Evolutionary analysis requires carefully aligned DNA sequences and an accurate phylogenetic tree. However, it works far more effectively, providing a better estimate of selection pressure when conservation is difficult to capture with protein sequence-based methods, or when there are only a limited number of sequences separated by uneven evolutionary time. Originally developed by Nei and Gojobori [18], the ω-ratio approach has been widely used for detecting subtle evolutionary changes in myxovirus resistance genes in mammals [59], beet vein necrotic virus [60], human fetuin-A [61], mammalian lactoferrin [62], tandem-repetitive early-stage histone H3 gene in brooding sea stars [63], 5-HT receptors [64], and for the evolutionary analysis of matrix extracellular phosphoglycoprotein (MEPE) [65]. Several new prediction methodologies were developed based on ω-ratio calculations. For example, the Core-Rim Ka/Ks ratio, or CRK method, uses ω-ratio to predict biologically relevant interfaces in x-ray structures [66]. The method of evolutionary patterning (EP) uses ω-ratio to identify codons under the most intense purifying selection and has been used to predict drug target sites in malaria parasite in an effort to minimize the emergence of parasite resistance [67].
We found statistically significant purifying selection pressure on the cholesterol in β-adrenergic receptor (PDB: 2RH1), and Na+-K+-ATPase (PDB: 2ZXE), cardiolipin in Fhu-N (PDB 1KQF), and phosphatidylglycerol in KcsA (PDB: 1K4C) binding sites, as well as on the surface patches of the ferric hydroxamate receptor interacting with lipopolysaccharide molecule (PDB: 2FCP), although not all lipid-binding sites exhibit strongly significant conservation. This may be attributed to a number of factors, such as incompleteness of data on protein-lipid interactions, e.g., poorly resolved acyl chains, accidental co-crystallization of the annular lipid in a non-specific site, or the dissociation of the specifically bound lipid during purification and crystallization procedures. All these will affect the delineation of “lipid-free” and “lipid-binding” residue sets and, ultimately, the p-values.
Similar problems hinder development of a prediction method based on identification of conserved residue patches on the membrane protein surface. When residues under strong evolutionary selection pressure are mapped to the protein surfaces, they often form continuous patches of conserved residues with sizes appropriate for lipid binding. However, in many cases there are no lipid molecules resolved in structures that are in contact with these surface patches. The functional significance of these lipid-free conserved patches is difficult to assess without additional information, since x-ray structures often do not resolve bound lipid molecules at experimental conditions necessary for crystallography. For example, the structure of β2-adrenergic receptor with PDB ID 3D4S contains only two bound cholesterol molecules, while the structure of the same protein with PDB ID 2RH1 provides coordinates for three bound cholesterol molecules and a palmitic acid. The binding site for the third cholesterol contains many conserved residues, and palmitic acid also interacts with two conserved residues. If the structure of 2RH1 were not solved, we would not know that the surface patch with these conserved residues is indeed the binding site of another cholesterol molecule.
Additionally, inherently flexible nature of the phospholipid acyl chains allows many degrees of conformational freedom. Comparison of conformations of bound lipids shows that acyl chains adopt different conformations to accommodate different geometrical and electrostatic environments of the protein surfaces. This inherent flexibility leads to multiple conformations of the same type of lipid molecule bound to different protein surfaces. For example, conformation of cardiolipins bound to ADP/ATP carrier differs significantly from that of cardiolipins bound to Fdh-N, or to cytochrome bc1 complex. Consequently, the overall shape and size of the lipid-binding sites vary from protein to protein, even for the same lipid molecule. This makes the task of predicting a lipid binding site very difficult.
Overall, we believe that without additional experimental information, one could only conclude that patches of residues under strong evolutionary selection pressure are likely candidate sites for lipid binding. Ultimately, the predictions should be tested by additional targeted experimental studies such as scanning mutagenesis, which would provide useful feedback for assessing the strengths and weaknesses of the approach described in this study, and enable further improvement in identifying specific lipid binding surfaces.
4.1 Sterol binding sites in membrane proteins
Sterols and related compounds play essential roles in the physiology of eukaryotic organisms. For example, cholesterol is an essential component of eukaryotic membranes and plays an important role in membrane organization, dynamics and function. Cholesterol is implicated in the stabilization and function of many membrane proteins [68], among which are class A GPCRs [69], Na+-K+- ATPase [5] and Ca2+-Mg2+-ATPase [70] and Kir channels [71]. Specific protein-sterol interactions are often critical for proteins to function [71]. For example, activities of some class A GPCRs have been demonstrated to be sensitive to cholesterol concentration [72]. Specifically, in β2 – adrenergic receptor, stability against denaturation is increased with the presence of cholesterol [39]. A recent long-timescale molecular dynamics study of A2A receptor in the phospholipid bilayer with and without cholesterol by Lyman et al. [73] clearly demonstrated the crucial role of cholesterol in stabilizing A2A receptor. Specific cholesterol binding sites in GPCRs were reviewed and discussed by Paila et al. [74]. It is, therefore, of great current interest to quantitatively assess the conservation and identify possible binding motifs for cholesterol.
The common theme that emerged from our studies of the cholesterol-binding sites is the requirement of a specific orientation of the aromatic residue that would facilitate stacking interaction with sterol. For example, in the β1- and β2-adrenergic receptor structures [38, 75], this orientation is achieved by interhelical hydrogen bonding of the tryptophan indole amine with the serine hydroxyl from the adjacent helix. A similar interhelical interaction between conserved S47 and W129 that align with S74 and W158 in β2-adrenergic receptor, is found in adenosine A2A receptor [41], a class A GPCR that requires cholesterol to stabilize the functional state of the protein [76]. Although the crystal structure of the A2A receptor contains detergent molecules only [41], the conserved interhelical structural motif may suggest the location of the putative cholesterol-binding site. Similar intersubunit interhelical interaction is found in bovine cytochrome c oxidase, in which the cholic acid binds to the side-chain of W275 from subunit 1, and G22 from subunit 6A2 (following numbering of [77]). In this case, a hydrogen bond is formed between the W275 indole amine and the backbone carboxyl of G22. In Na+-K+- ATPase, a cholesterol molecule is bound at the protein-protein interface and stacks against a hydrogen-bonded tyrosine-serine pair of conserved residues (Fig. 2B). Overall, we found strong conservation of cholesterol-binding residues and high specificity of cholesterol binding.
4.2 Lipopolysacharide binding
In this work, we examined conservation of residues forming the LPS binding site in the x-ray structure of the 22-strand integral membrane protein FhuA (ferric hydroxamate uptake receptor). Using empirical potential function, we determined that strand 8 is the least stable in FhuA (Fig. 4). This strand is in the middle of the LPS binding site in FhuA, where it is shielded from the lipid bulk by the tightly bound LPS. Such high energy strands are typically found at the protein-protein oligomerization interfaces [34]. However, FhuA is a monomer in phospholipid bilayer [78], although it may transiently form dimers and trimers in detergent solution [79].
Previously, we have discussed three general mechanisms by which the weakly stable regions are stabilized in β-barrel membrane proteins, namely out-clamps, in-plugs, and oligomerization [34]. Based on the results of this study, we propose a fourth mechanism employed by the β-barrel membrane proteins to stabilize weakly stable regions using specific binding of lipids. There are several features of the LPS binding site that demonstrate its specificity towards LPS binding, including a patch of highly conserved polar residues in the extracellular cap region, as well as the patch of conserved residues in the hydrocarbon core region (Fig. 4A). The tendency of FhuA to oligomerize in LPS-free detergent solution [79] argues in favor of this general mechanism, where the weakly stable regions are stabilized by shielding from the bulk lipids either via oligomerization, or LPS binding.
4.3 Phosphatidylglycerol in KcsA K+ channel
KcsA requires an anionic lipid, phosphatidylglycerol (PG), for ion channel function, which is a partially resolved PG in the crystal structure [4]. Measurements of KcsA channel conductance showed that the probability of channel opening increases proportionally with the presence of anionic lipids in the membrane, e.g. the open probability is 2% in 25 mol% of POPG and 27% in bilayers of pure POPG [80]. Valiyaveetil et al [4] concluded that the anionic phospholipid is required for the opening of the channel. In agreement with the experimental data, our calculations showed evolutionary conservation of the residues forming this highly specific PG-binding site, which is necessary for the optimal physiological functioning of KcsA in membranes.
4.4. Cardiolipin (CL) binding
We have analyzed surface conservation pattern in two membrane protein structures with bound cardiolipins: a formate dehydrogenase (Fdh-N) and an ADP/ATP carrier (AAC). We found that the CL-binding sites have different distributions of conserved residues and the conservation patterns strongly depend on the functional role of the bound lipid.
A visual comparison of structural complexes of Fdh-N (PDB ID: 1KQF) and AAC (PDB ID: 2C3E) shows that the CL molecules bound to these proteins adopt different conformations, which are pronounced for the acyl chains. The acyl chains in Fdh-N are tightly bound to the grooves on the protein surface and extend to the neighboring subunits, maximizing intra- and inter-subunit van der Waals interactions. Unlike residues interacting with the CL head-groups, residues in the grooves interacting with the acyl chains experience strong evolutionary selection pressure. Stronger conservation of the groove residues is likely due to the importance of the bound CL for the stability and function of the Fdh-N heterotrimeric complex.
The opposite pattern of residue conservation was found for the CL-binding sites in AAC, where residues interacting with the CL head-groups are more conserved than residues interacting with the acyl chains, as the latter are often not completely resolved. Here, one of the two phosphate groups from each CL head-group is tightly bound to the depression on the AAC surface lined with highly conserved residues shown in red in Figs. 6A-F. Fig. 6G shows the sequence alignment of five consecutive residues interacting with one of the two CL phosphates and with the adjacent ester group. The pattern of strong conservation is clearly seen in Fig 6G: each of these sequences contains a conserved aromatic residue as well as a conserved glycine found at the beginnings of the TM helices H2, H4, and H6. For AAC, it is likely that lipid interaction with the head-group is functionally more important, while interactions with the acyl chain play a lesser role. Overall, the difference in conservation patterns between Fdh-N and AAC can be attributed to the different functional roles of bound cardiolipins.
Conclusions
We have quantitatively assessed the evolutionary selection pressure of residues on the lipid-facing surfaces of membrane proteins, which specifically interact with a variety of bound lipids. We found that in general, residues interacting with bound lipids are under stronger purifying selection than the rest of the lipid-facing surface. We also found that the extent of selection pressure varies from site to site and depends on the functional role of the bound lipid. Residues forming functionally important lipid binding sites are under stronger purifying selection, e.g., cholesterol-binding site in β2-adrenergic receptor and Na+-K+-ATPase, and cardiolipin binding site in γ-subunit of Fdh-N, where cardiolipin is involved in trimer formation and possibly in catalytic function of the protein. On the other hand, residues of β-subunit of Fdh-N interacting with the acyl chain of cardiolipin for which no functional importance can be assigned are no more significantly conserved than the rest of the β-subunit transmembrane helix under the stringent criterion of ω-ratio.
Our survey of sterol-binding sites in membrane proteins revealed an important structural motif, which appears in most structures where sterol molecules bind to the protein surface. This structural motif contains an aromatic residue that forms a hydrogen bond with a side chain of Ser or carbonyl oxygen of Gly, securing the orientation of the aromatic side chain to optimize a stacking interaction with the sterol rings. Based on this finding, we propose an expanded cholesterol binding motif in GPCRs that includes an acceptor of H-bond (Ser).
Based on the results of the protein energy calculations together with the structural and evolutionary analysis, we found that specific lipid binding may be employed by the β-barrel membrane proteins as a general mechanism to stabilize weakly stable regions. This is in addition to the previously discussed stabilization mechanisms of out-clamps, in-plugs, and oligomerization.
Overall, our results suggest that specific lipid binding sites are common in membrane proteins. Our study showed that strong evolutionary selection pressure played important role in shaping up the mutual interactions between membrane proteins and lipids, and that the detection of such selection pressure can provide useful information for identifying candidate lipid binding sites on membrane proteins.
Supplementary Material
Acknowledgements
We thank Dr. William Dothan for helpful discussion. We thank Michael Montesano and Vastly Kosynkin for help with proofreading the manuscript. This work was supported by NIH grants GM079804, GM081682, GM086145, NSF grants DBI-0646035 and DMS-0800257, and ONR N00014-09-1-0028.
REFERENCES
- [1].Singer SJ, Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science. 1972;175:720–731. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- [2].Engelman DM. Membranes are more mosaic than fluid. Nature. 2005;438:578–580. doi: 10.1038/nature04394. [DOI] [PubMed] [Google Scholar]
- [3].Dowhan W, Bogdanov M. Lipid-dependent membrane protein topogenesis. J. Ann. Rev. Biochem. 2009;78:515–540. doi: 10.1146/annurev.biochem.77.060806.091251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Valiyaveetil FI, Zhou YF, Mackinnon R. Lipids in the structure, folding, and function of the KcsA K+ channel. Biochemistry. 2002;41:10771–10777. doi: 10.1021/bi026215y. [DOI] [PubMed] [Google Scholar]
- [5].Cornelius F, Turner N, Christensen HRZ. Modulation of Na,K-ATPase by phospholipids and cholesterol. II. steady-state and presteady-state kinetics. Biochemistry. 2003;42:8541–8549. doi: 10.1021/bi034532e. [DOI] [PubMed] [Google Scholar]
- [6].Lee AG. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta. 2004;1666:62–87. doi: 10.1016/j.bbamem.2004.05.012. [DOI] [PubMed] [Google Scholar]
- [7].Divito CB, Amara SG. Close encounters of the oily kind: regulation of transporters by lipids. Mol. Interv. 2009;9:252–262. doi: 10.1124/mi.9.5.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Adamian L, Nanda V, DeGrado WF, Liang J. Empirical lipid propensities of amino acid residues in multispan alpha helical membrane proteins. Proteins. 2005;59:496–509. doi: 10.1002/prot.20456. [DOI] [PubMed] [Google Scholar]
- [9].Epand RM, Epand RF, Berno B, Pelosi L, Brandolin G. Association of phosphatidic acid with the bovine mitochondrial ADP/ATP carrier. Biochemistry. 2009;48:12358–12364. doi: 10.1021/bi901769r. [DOI] [PubMed] [Google Scholar]
- [10].Marsh D. Protein modulation of lipids, and vice-versa, in membranes. Biochim. Biophys. Acta. 2008;1778:1545–1575. doi: 10.1016/j.bbamem.2008.01.015. [DOI] [PubMed] [Google Scholar]
- [11].Palsdottir H, Hunte C. Lipids in membrane protein structures. Biochim. Biophys. Acta. 2004;1666:2–18. doi: 10.1016/j.bbamem.2004.06.012. [DOI] [PubMed] [Google Scholar]
- [12].Hunte C, Richers S. Lipids and membrane protein structures. Curr. Opin. Struct. Biol. 2008;18:406–411. doi: 10.1016/j.sbi.2008.03.008. [DOI] [PubMed] [Google Scholar]
- [13].Qin L, Hiser C, Mulichak A, Garavito RM, Ferguson-Miller S. Identification of conserved lipid/detergent-binding sites in a high-resolution structure of the membrane protein cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 2006;103:16117–16122. doi: 10.1073/pnas.0606149103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Qin L, Sharpe M, Garavito R, Ferguson-Miller S. Conserved lipid-binding sites in membrane proteins: a focus on cytochrome c oxidase. Curr. Opin. Struct. Biol. 2007;17:444–450. doi: 10.1016/j.sbi.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yang Z. Maximum-likelihood estimation of phylogeny from DNA sequences when substitution rates differ over sites. Mol. Biol. Evol. 1993;10:1396–1401. doi: 10.1093/oxfordjournals.molbev.a040082. [DOI] [PubMed] [Google Scholar]
- [16].Delport W, Scheffler K, Botha G, Gravenor MB, Muse SV, Pond SLK. CodonTest: modeling amino acid substitution preferences in coding sequences. PLoS Comput. Biol. 2010;6:17. doi: 10.1371/journal.pcbi.1000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Goldman N, Yang ZH. Codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 1994;11:725–736. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- [18].Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- [19].Tseng YY, Liang J. Estimation of amino acid residue substitution rates at local spatial regions and application in protein function inference: a Bayesian Monte Carlo approach. Mol. Biol. Evol. 2006;23:421–436. doi: 10.1093/molbev/msj048. [DOI] [PubMed] [Google Scholar]
- [20].Tseng YY, Liang J. Are residues in a protein folding nucleus evolutionarily conserved? J. Mol. Biol. 2004;335:869–880. doi: 10.1016/j.jmb.2003.11.007. [DOI] [PubMed] [Google Scholar]
- [21].Adamian L, Gussin HA, Tseng YY, Muni NJ, Feng F, Qian HH, Pepperberg DR, Liang J. Structural model of rho 1 GABA(C) receptor based on evolutionary analysis: testing of predicted protein-protein interactions involved in receptor assembly and function. Protein Sci. 2009;18:2371–2383. doi: 10.1002/pro.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: Orientations of proteins in membranes database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- [23].Edelsbrunner H, Facello M, Fu P, Liang J. Measuring pockets and voids in proteins. Proc. 28th Annu. Hawaii Int. Conf. Syst. Sci. 1995;5:256–264. [Google Scholar]
- [24].Edelsbrunner H, Mucke EP. Three-dimensional alpha shapes. ACM Trans. Graphics. 1994;13:43–72. [Google Scholar]
- [25].Edelsbrunner H, Shah NR. Incremental topological flipping works for regular triangulations. Algorithmica. 1996;15 [Google Scholar]
- [26].Facello MA. Implementation of a randomized algorithm for Delaunay and regular triangulations in 3 dimensions. Comput. Aided Geom. Des. 1995;12:349–370. [Google Scholar]
- [27].Tsai J, Taylor R, Chothia C, Gerstein M. The packing density in proteins: Standard radii and volumes. J. Mol. Biol. 1999;290:253–266. doi: 10.1006/jmbi.1999.2829. [DOI] [PubMed] [Google Scholar]
- [28].Adamian L, Liang J. Helix-helix packing and interfacial pairwise interactions of residues in membrane proteins. J. Mol. Biol. 2001;311:891–907. doi: 10.1006/jmbi.2001.4908. [DOI] [PubMed] [Google Scholar]
- [29].Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- [30].Caffrey DR, Dana PH, Mathur V, Ocano M, Hong EJ, Wang YE, Somaroo S, Caffrey BE, Potluri S, Huang ES. PFAAT version 2.0: A tool for editing, annotating, and analyzing multiple sequence alignments. BMC Bioinformatics. 2007;8:7. doi: 10.1186/1471-2105-8-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rice P, Longden I, Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- [32].Felsenstein J, Churchill GA. A hidden Markov Model approach to variation among sites in rate of evolution. Mol. Biol. Evol. 1996;13:93–104. doi: 10.1093/oxfordjournals.molbev.a025575. [DOI] [PubMed] [Google Scholar]
- [33].Yang Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- [34].Naveed H, Jackups R, Liang J. Predicting weakly stable regions, oligomerization state, and protein-protein interfaces in transmembrane domains of outer membrane proteins. Proc. Natl. Acad. Sci. U. S. A. 2009;106:12735–12740. doi: 10.1073/pnas.0902169106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jackups R, Liang J. Combinatorial analysis for sequence and spatial motif discovery in short sequence fragments. IEEE/ACM Trans. Comput. Biol. Bioinform. 7:524–536. doi: 10.1109/TCBB.2008.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jackups R, Cheng S, Liang J. Sequence motifs and antimotifs in beta-barrel membrane proteins from a genome-wide analysis: The Ala-Tyr dichotomy and chaperone binding motifs. J. Mol. Biol. 2006;363:611–623. doi: 10.1016/j.jmb.2006.07.095. [DOI] [PubMed] [Google Scholar]
- [37].Jackups R, Liang J. Interstrand pairing patterns in beta-barrel membrane proteins: The positive-outside rule, aromatic rescue, and strand registration prediction. J. Mol. Biol. 2005;354:979–993. doi: 10.1016/j.jmb.2005.09.094. [DOI] [PubMed] [Google Scholar]
- [38].Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta(2)-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EYT, Velasquez J, Kuhn P, Stevens RC. A specific cholesterol binding site is established by the 2.8 angstrom structure of the human beta(2)-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 1994;238:777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- [41].Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, Ijzerman AP, Stevens RC. The 2.6 Angstrom crystal structure of a human A(2A) adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shinoda T, Ogawa H, Cornelius F, Toyoshima C. Crystal structure of the sodium-potassium pump at 2.4A resolution. Nature. 2009;459:446–450. doi: 10.1038/nature07939. [DOI] [PubMed] [Google Scholar]
- [43].Chen YL, Cai T, Wang HJ, Li ZC, Loreaux E, Lingrel JB, Xie ZJ. Regulation of intracellular cholesterol distribution by Na/K-ATPase. J. Biol. Chem. 2009;284:14881–14890. doi: 10.1074/jbc.M109.003574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ferguson AD, Hofmann E, Coulton JW, Diederichs K, Welte W. Siderophore-mediated iron transport: Crystal structure of FhuA with bound lipopolysaccharide. Science. 1998;282:2215–2220. doi: 10.1126/science.282.5397.2215. [DOI] [PubMed] [Google Scholar]
- [45].Mamat U, Seydel U, Grimmecke D, Holst O, Rietschel ET. In: Comprehensive Natural Products Chemistry. Pinto M, editor. Elsevier Science; Amsterdam: 1999. pp. 179–239. [Google Scholar]
- [46].Holst O. In: Endotoxin in Health and Disease. Brade H, Morrison DC, Opal S, Vogel S, editors. Marcel Dekker Inc.; New York: 1999. pp. 115–154. [Google Scholar]
- [47].Ferguson AD, Welte W, Hofmann E, Lindner B, Holst O, Coulton JW, Diederichs K. A conserved structural motif for lipopolysaccharide recognition by procaryotic and eucaryotic proteins. Structure. 2000;8:585–592. doi: 10.1016/s0969-2126(00)00143-x. [DOI] [PubMed] [Google Scholar]
- [48].Jackups R, Jr, Cheng S, Liang J. Sequence motifs and antimotifs in β-barrel membrane proteins from a genome-wide analysis: the Ala-Tyr dichotomy and chaperone binding motifs. J. Mol. Biol. 2006;363:611–623. doi: 10.1016/j.jmb.2006.07.095. [DOI] [PubMed] [Google Scholar]
- [49].Mileykovskaya E, Dowhan W. Cardiolipin membrane domains in prokaryotes and eukaryotes. Biochim. Biophys. Acta. 2009;1788:2084–2091. doi: 10.1016/j.bbamem.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trezeguet V, Lauquin GJM, Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426:39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- [51].Hoffmann B, Stockl A, Schlame M, Beyer K, Klingenberg M. The reconstituted ADP/ATP carrier activity has an absolute requirement for cardiolipin as shown in cysteine mutants. J. Biol. Chem. 1994;269:1940–1944. [PubMed] [Google Scholar]
- [52].Nury H, Dahout-Gonzalez C, Trézéguet V, Lauquin G, Brandolin G, Pebay-Peyroula E. Structural basis for lipid-mediated interactions between mitochondrial ADP/ATP carrier monomers. FEBS Letters. 2005;579:6031–6036. doi: 10.1016/j.febslet.2005.09.061. [DOI] [PubMed] [Google Scholar]
- [53].Gennis RB. Biomembranes: molecular structure and function. Springer-Verlag; New York: 1989. [Google Scholar]
- [54].Wang Y, Zhang Y, Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature. 2006;444:179–180. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- [55].Phillips R, Ursell T, Wiggins P, Sens P. Emerging roles for lipids in shaping membrane-protein function. Nature. 2009;459:379–385. doi: 10.1038/nature08147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Shi T, Huang HW, Barker MS. Ancient genome duplications during the evolution of kiwifruit (Actinidia) and related Ericales. Ann. Bot. 2010;106:497–504. doi: 10.1093/aob/mcq129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhou T, Gu WJ, Wilke CO. Detecting positive and purifying selection at synonymous sites in yeast and worm. Mol. Biol. Evol. 2010;27:1912–1922. doi: 10.1093/molbev/msq077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Toft C, Andersson SG. Evolutionary microbial genomics: insights into bacterial host adaptation. Nat. Rev. Genet. 2010;11:465–475. doi: 10.1038/nrg2798. [DOI] [PubMed] [Google Scholar]
- [59].Hou ZC, Xu GY, Su Z, Yang N. Purifying selection and positive selection on the myxovirus resistance gene in mammals and chickens. Gene. 2007;396:188–195. doi: 10.1016/j.gene.2007.03.017. [DOI] [PubMed] [Google Scholar]
- [60].Schirmer A, Link D, Cognat V, Moury B, Beuve M, Meunier A, Bragard C, Gilmer D, Lemaire O. Phylogenotic analysis of isolates of Beet necrotic yellow vein virus collected worldwide. J. Gen. Virol. 2005;86:2897–2911. doi: 10.1099/vir.0.81167-0. [DOI] [PubMed] [Google Scholar]
- [61].Doring Y, Zechner U, Roos C, Rosenkranz D, Zischler H, Herlyn H. Accelerated evolution of Fetuin-A (FETUA, also AHSG) is driven by positive Darwinian selection, not GC-biased gene conversion. Gene. 2010;463:49–55. doi: 10.1016/j.gene.2010.04.018. [DOI] [PubMed] [Google Scholar]
- [62].Liang GM, Jiang XP. Positive selection drives lactoferrin evolution in mammals. Genetica. 2010;138:757–762. doi: 10.1007/s10709-010-9456-x. [DOI] [PubMed] [Google Scholar]
- [63].Foltz DW, Mah CL. Recent relaxation of purifying selection on the tandem-repetitive early-stage histone H3 gene in brooding sea stars. Mar. Genom. 2009;2:113–118. doi: 10.1016/j.margen.2009.06.002. [DOI] [PubMed] [Google Scholar]
- [64].Anbazhagan P, Purushottam M, Kumar HBK, Mukherjee O, Jain S, Sowdhamini R. Phylogenetic analysis and selection pressures of 5-HT receptors in human and non-human primates: receptor of an ancient neurotransmitter. J. Biomol. Struct. Dyn. 2010;27:581–598. doi: 10.1080/07391102.2010.10508573. [DOI] [PubMed] [Google Scholar]
- [65].Bardet C, Delgado S, Sire JY. MEPE evolution in mammals reveals regions and residues of prime functional importance. Cell. Mol. Life Sci. 2010;67:305–320. doi: 10.1007/s00018-009-0185-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Scharer MA, Grutter MG, Capitani G. CRK: an evolutionary approach for distinguishing biologically relevant interfaces from crystal contacts. Proteins. 2010;78:2707–2713. doi: 10.1002/prot.22787. [DOI] [PubMed] [Google Scholar]
- [67].Durand PM, Naidoo K, Coetzer TL. Evolutionary Patterning: a novel approach to the identification of potential drug target sites in Plasmodium falciparum. PLoS One. 2008;3:12. doi: 10.1371/journal.pone.0003685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Burger K, Gimpl G, Fahrenholz F. Regulation of receptor function by cholesterol. Cell. Mol. Life Sci. 2000;57:1577–1592. doi: 10.1007/PL00000643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Paila YD, Chattopadhyay A. The function of G-protein coupled receptors and membrane cholesterol: specific or general interaction? Glycoconj. J. 2009;26:711–720. doi: 10.1007/s10719-008-9218-5. [DOI] [PubMed] [Google Scholar]
- [70].Simmonds AC, East JM, Jones OT, Rooney EK, McWhirter J, Lee AG. Annular and non-annular binding sites on the (Ca2+- Mg2+)-ATPase. Biochim. Biophys. Acta. 1982;693:398–406. doi: 10.1016/0005-2736(82)90447-3. [DOI] [PubMed] [Google Scholar]
- [71].Singh DK, Rosenhouse-Dantsker A, Nichols CG, Enkvetchakul D, Levitan I. Direct regulation of prokaryotic Kir channel by cholesterol. J. Biol. Chem. 2009;284:30727–30736. doi: 10.1074/jbc.M109.011221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Prasad R, Paila YD, Chattopadhyay A. Membrane cholesterol depletion enhances ligand binding function of human serotonin(1A) receptors in neuronal cells. Biochem. Biophys. Res. Commun. 2009;390:93–96. doi: 10.1016/j.bbrc.2009.09.072. [DOI] [PubMed] [Google Scholar]
- [73].Lyman E, Higgs C, Kim B, Lupyan D, Shelley JC, Farid R, Voth GA. A Role for a specific cholesterol interaction in stabilizing the apo configuration of the human A2A adenosine receptor. Structure. 2009;17:1660–1668. doi: 10.1016/j.str.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Paila YD, Tiwari S, Chattopadhyay A. Are specific nonannular cholesterol binding sites present in G-protein coupled receptors? Biochim. Biophys. Acta. 2009;1788:295–302. doi: 10.1016/j.bbamem.2008.11.020. [DOI] [PubMed] [Google Scholar]
- [75].Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AGW, Tate CG, Schertler GFX. Structure of a β1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].O’Malley MA, Lazarova T, Britton ZT, Robinson AS. High-level expression in Saccharomyces cerevisiae enables isolation and spectroscopic characterization of functional human adenosine A(2)a receptor. J. Struct. Biol. 2007;159:166–178. doi: 10.1016/j.jsb.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Aoyama H, Muramoto K, Shinzawa-Itoh K, Hirata K, Yamashita E, Tsukihara T, Ogura T, Yoshikawa S. A peroxide bridge between Fe and Cu ions in the O2 reduction site of fully oxidized cytochrome c oxidase could suppress the proton pump. Proc. Natl. Acad. Sci. U.S.A. 2009;106:2165–2169. doi: 10.1073/pnas.0806391106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Ramakrishnan M, Pocanschi CL, Kleinschmidt JH, Marsh D. Association of spin-labeled lipids with β-barrel proteins from the outer membrane of Escherichia coli. Biochemistry. 2004;43:11630–11636. doi: 10.1021/bi048858e. [DOI] [PubMed] [Google Scholar]
- [79].Locher KP, Rosenbusch JP. Oligomeric states and siderophore binding of the ligand-gated FhuA protein that forms channels across Escherichia coli outer membranes. Eur. J. Biochem. 1997;247:770–775. doi: 10.1111/j.1432-1033.1997.t01-1-00770.x. [DOI] [PubMed] [Google Scholar]
- [80].Marius P, Zagnoni M, Sandison ME, East JM, Morgan H, Lee AG. Binding of anionic lipids to at least three nonannular sites on the potassium channel KcsA is required for channel opening. Biophys. J. 2008;94:1689–1698. doi: 10.1529/biophysj.107.117507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.