

Abstract
Six new analogues of 1α,25-dihydroxy-19-norvitamin D3 (3a-4b, 5 and 6) were prepared by a convergent synthesis applying the Wittig-Horner reaction as a key step. The influence of methyl groups at C-22 on their biological activity was examined. It was established that both in vitro and in vivo activity is strongly dependent on the configuration of the stereogenic centers at C-20 and C-22. Introduction of the second methyl group at C-22 (analogues 5 and 6) generates the compounds that are slightly more potent than 1α,25-(OH)2D3 in the in vitro tests but much less potent in vivo. The greatest in vitro and in vivo biological activity was achieved when the C-20 is in the S-configuration and the C-22 is in the R configuration. The building blocks for the synthesis, the respective (20R,22R)-, (20R,22S)-, (20S,22R)- and (20S,22S)-diols were obtained by fractional crystallization of mixtures of the corresponding diastereomers. Structures and absolute configurations of the diols 21a, 21b and 22a as well as analogues 3a, 5 and 6 were confirmed by the X-ray crystallography.
Keywords: Vitamin D analogues, 19-Norvitamin D, Vitamin D receptor, Cellular HL-60 differentiation, calcemic activity
Introduction
Vitamin D3 must be metabolized to its active form, 1α,25-dihydroxyvitamin D3 [calcitriol, 1α,25-(OH)2D3 (1); Figure 1] before it is functionally active.1 This hormonal form of vitamin D3 is responsible for calcium and phosphorus homeostasis but it also plays a role in other biological systems that are still under investigation.2 A number of analogues of 1α,25-dihydroxyvitamin D3 have been successfully developed as pharmaceuticals used in the treatment of renal osteodystrophy, osteoporosis and vitamin D-resistant rickets.3 Although these compounds are an improvement over previous therapies, further improvements are clearly possible.
Figure 1.
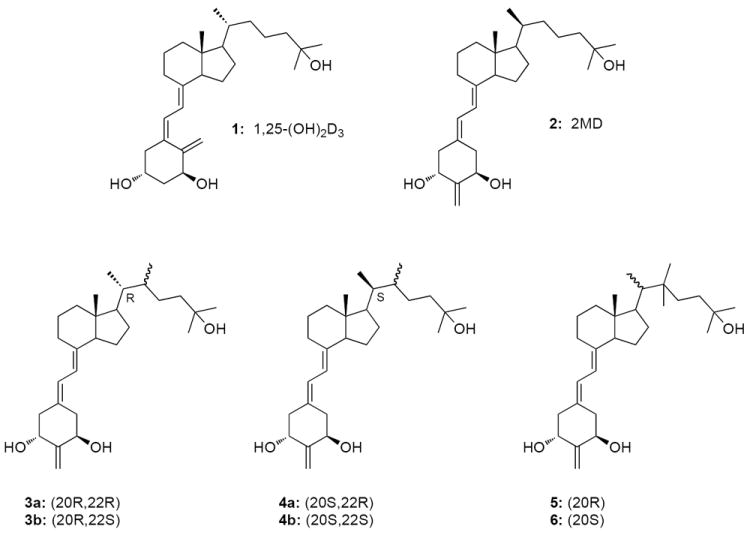
Structures of the 1α,25-(OH)2D3 (1), 2MD (2) and vitamin D analogues (3a-6).
Modification of the configuration of vitamin D compounds definitely impacts biological activity. Inverting the configuration at C-20 usually results in increased activity both in vivo and in vitro.4 Yamada et al. designed conformationally restricted vitamin D analogues postulating that introduction of a methyl group at C-22 hinders rotation around the C(20)-C(22) bond.5 Moreover, restricting the side chain rotation by the introduction of a C(17)-C(20) double bond has a marked impact on biological activity, with the Z-configuration strongly favoring calcemic potency.6
In 1998 we discovered A-ring modification of vitamin D compounds consisting in “shift” of the exomethylene unit at C-10 to carbon 2.7 One of the most potent analogues of this series proved to be (20S)-1α,25-dihydroxy-2-methylene-19-norvitamin D3 (2, 2MD) showing a very strong activity in bone and, moreover, inducing bone formation in vitro and in vivo.7,8 Recently, Yamamoto et al. described 2-methylene-19-norvitamin D compounds bearing a 22S-butyl and 22S-ethyl group and discovered that they can act as agonists or antagonists for the VDR.9
In the present study we examined all possible conformers 3a-4b resulting from C-20 epimerization and introduction of a methyl group at the two available positions on C-22 in the 1α,25-dihydroxy-2-methylene-19-norvitamin D3. We also used the attachment of two methyl groups on C-22 (analogues 5 and 6) to help understand the biological impact of the double substitution of this side-chain carbon. Our results show that superior biological activity both in vitro and in vivo is obtained in the case of 20S,22R-configuration. How this occurs is discussed in relation to the ligand interaction with the receptor.
Results
Synthesis
The first approach to prepare 22-methyl-1α,25-(OH)2D3 analogues was based on the stereoselective conjugate addition of organocuprate to the steroidal (E)- and (Z)-22-ene-24-ketones.10 We describe herein a convergent synthesis of all the four diastereomers of 22-methyl-1α,25-(OH)2D3 where the key precursors are nitriles 9 and 10, readily obtainable from the Inhoffen-Lythgoe diol.11 In contrast to similar synthetic approaches, independently proposed by Fujishima12 and Mouriño,13 we used crystallization, avoiding the expensive separation of the C,D-ring isomers by flash chromatography or HPLC. Since only a few 22-alkyl-19-nor-1α,25-dihydroxyvitamin D3 analogues have been synthesized to the date,9 we have also decided to prepare and test the analogues double-substituted at C-22 (Scheme 1).
Scheme 1.
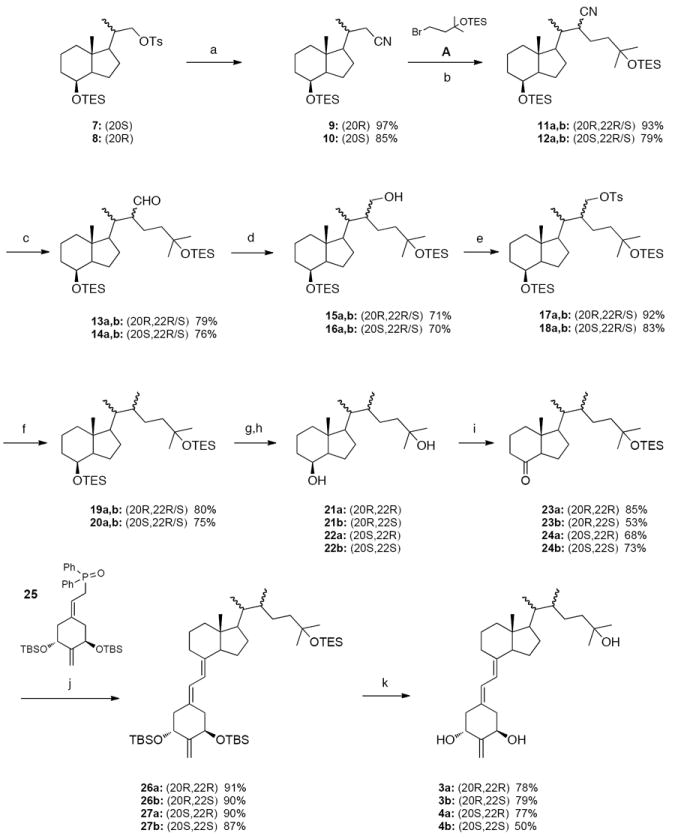
(a) NaCN, DMSO; (b) LDA, THF; (c) DIBAl-H, CH2Cl2; (d) NaBH4, MeOH; (e) p-TsCl, pyridine; (f) LiAlH4, Et2O; (g) TBAF, THF; (h) crystallization; (i) (1) PDC, CH2Cl2 or NMO, TPAP, 4Å mol. sieves, CH2Cl2; (2) TESOTf, 2,6-lutidine, CH2Cl2; (j) PhLi, THF; (k) 48% aq. HF, MeCN, THF.
As shown in Scheme 1, the vitamin D analogues 3a and 3b were prepared from the 20S-tosylate 7.11 Its substitution with cyanide provided the nitrile 9 (in 97% yield) that was alkylated at the α-position with bromide A using LDA as a base to give the compounds 11a,b (93% yield) being a mixture of epimers at C-22. The two consecutive reductions, first with DIBALH, then with NaBH4, afforded the epimeric mixture of the alcohols 15a,b in 56% yield (over 2 steps). The hydroxy group in these compounds was removed in a two-step reaction sequence: conversion into the tosylates 17a,b followed by reduction with LiAlH4 (80% overall yield). Removal of both silyl protecting groups in the formed products 19a,b afforded a mixture of the epimeric diols 21a and 21b in a ratio of 1:2, respectively (as established by 1H NMR). The stereoisomers were separated by crystallization from ethyl acetate, and their absolute configurations were determined by X-ray crystallography (Figure 2). This is one of rare examples of the process known as fractional crystallization used in chemical engineering for purification or analysis. In this method, based on differences in solubility, two or more substances dissolved in a solvent crystallize from the solution at different rates.14 In our case, the prevailing (20R,22S)-diol 21b crystallized first. Then, pure crystals of the (20R,22R)-diol 21a were obtained from the mother liquors. The diol 21a was subsequently oxidized with tetrapropylammonium perruthenate in the presence of 4-methylmorpholine N-oxide and, in the formed product the 25-hydroxy group was protected as a TES ether to give the Grundmann ketone 23a in 85% overall yield. The epimeric diol 21b was oxidized with PDC and, after hydroxy protection, the hydrindanone 23b was obtained in 53% yield (over 2 steps).
Figure 2.
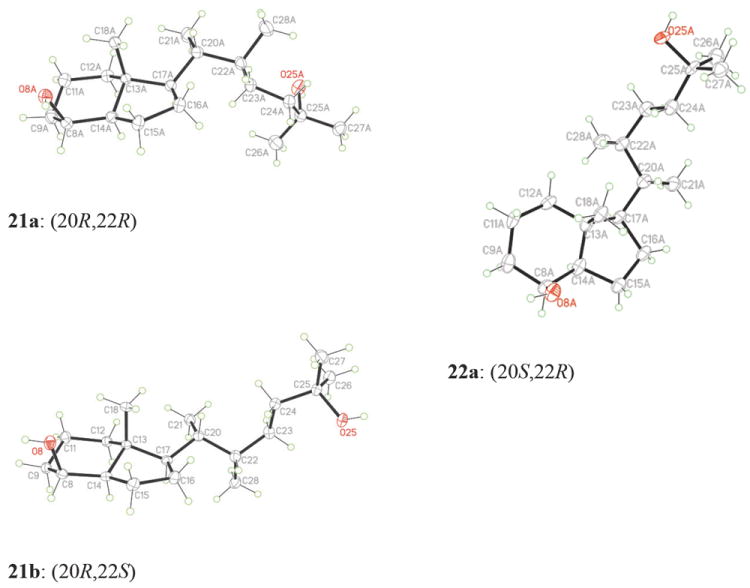
ORTEP drawings derived from the single-crystal X-ray analysis of the (20R,22R)-diol 21a, (20R,22S)-diol 21b and (20S,22R)-diol 22a.
Coupling of a phosphine oxide A-ring with a C,D-ketone is one of the most useful methods for the synthesis of vitamin D analogues.15 In this approach, first developed by Lythgoe,16 an anion of an allylic phosphine oxide reacts with the Grundmann ketone via the Wittig-Horner reaction. In our case, this method proved to be very useful. The anion, generated from the known phosphine oxide 257 with phenyllithium, was coupled with the ketones 23a and 23b to give the corresponding protected 19-norvitamin D analogues 26a and 26b in 91% and 90% yield, respectively. The silyl protecting groups were removed with hydrofluoric acid to give the final compounds 3a and 3b in 78% and 79% yield, respectively. The structure and absolute configuration of the vitamin 3a was confirmed by X-ray crystallography (Figure 3).
Figure 3.
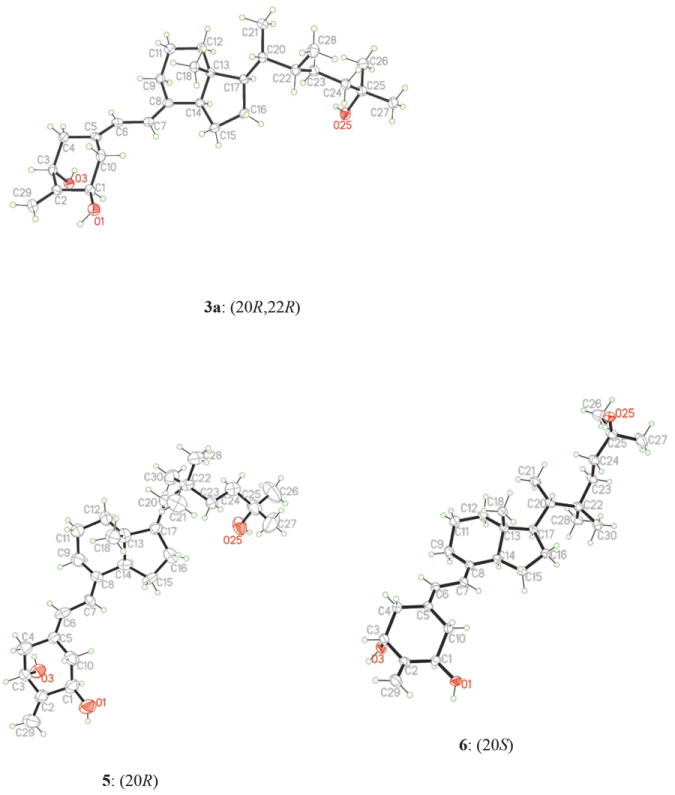
ORTEP drawings derived from the single-crystal X-ray analysis of the vitamins 3a, 5 and 6.
The synthesis of the diastereomeric vitamins 4a and 4b started from the 20R-tosylate 811 and followed the analogous path. Also in this case, fractional crystallization from ethyl acetate of the epimeric pair of 20,22-diols 22a and 22b, obtained in a ratio of 2:1, played a crucial role. The absolute (20S,22R)-configuration of the prevailing diol 22a, which crystallized first, was established by X-ray analysis (Figure 2). The pure (20S,22S)-diol 22b was obtained in the crystalline form from the mother liquors. Both diols were then separately converted into the respective protected 25-hydroxy Grundmann ketones 24a,b by the same consecutive processes as described above. Finally, they were subjected to the Wittig-Horner coupling with the anion generated from 25 and, after removal of the silyl protecting groups in the products 27a,b, the corresponding vitamin D3 analogues 4a and 4b were obtained in 77% and 50% yield, respectively.
The synthesis of 22,22-dimethyl vitamin D analogues 5 and 6, performed for the compounds in the 20R- and 20S-series separately, is shown in Scheme 2. Thus, α-alkylation of the 20R-nitrile 9 with methyl iodide was achieved using LDA as a base, and the product 28 double-substituted at C-20 was obtained in 56% yield. The conversion of the 20S-nitrile 10 into 29 was analogous, but significantly more efficient. The reduction of the obtained nitriles with DIBALH afforded the respective aldehydes 30 and 31 in 75% and 95% yield, respectively. These were next subjected to the Wittig-Horner-Emmons reaction with the triethylphosphonoacetate anion to give the α,β-unsaturated esters 32 and 33 (55% and 91% yield, respectively), which were subsequently hydrogenated using a palladium catalyst. The esters 34 and 35, obtained in 68% and 95% yield, were treated with methylmagnesium bromide to give the diols 36 and 37 in 100% and 84% yield, respectively. These diols were in turn oxidized to the ketones with tetrapropylammonium perruthenate in the presence of 4-methylmorpholine N-oxide. The terminal 25-hydroxy group in the formed products was protected as a triethylsilyl ether. The obtained Grundmann ketones 38 and 39 (86% and 78% overall yield) were coupled with a lithium phosphinoxy carbanion derived from the phosphine oxide 25 to give the protected 19-norvitamins 40 and 41 in 78% and 91% yield, respectively. Acidic cleavage of the silyl groups furnished the final 20R- and 20S-vitamins 5 and 6 in 79% and 75% yield, respectively. The structures and absolute configurations of these compounds were confirmed by X-ray crystallography (Figure 3).
Scheme 2.
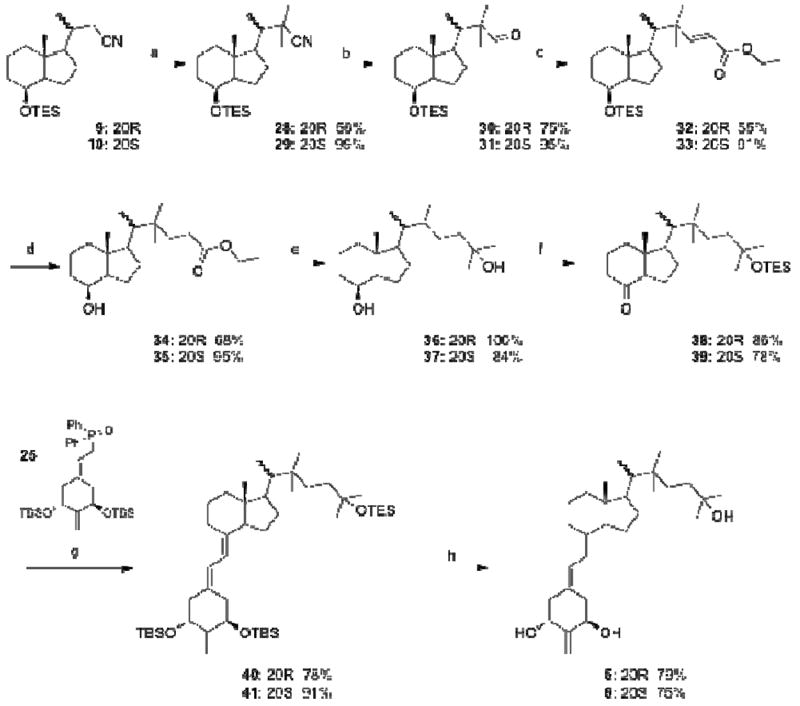
(a) LDA, THF, CH3I; (b) DIBAl-H, CH2Cl2; (c) LDA, THF, (EtO)2P(O)CH2COOEt; (d) 10% Pd/C, H2, MeOH; (e) CH3MgBr, Et2O; (f) (1) NMO, TPAP, 4Å mol. sieves, CH2Cl2; (2) TESOTf, 2,6-lutidine, CH2Cl2; (g) PhLi, THF; (h) 48% aq. HF, MeCN, THF.
Biological Evaluation
Biological activities in vitro are summarized in Table 1. Single 22-methylation of the (20R)-25-hydroxylated side chain did not significantly change the affinity of the analogues 3a and 3b to the nuclear receptor, as compared to their parent compound, 2-methylene-19-nor-1α,25-(OH)2D3.7 However, addition of a 22-methyl group to the analogue 2 (2MD), characterized by an “unnatural” 20S-side chain, caused a much stronger effect - the 22R-compound 4a bound 2.5 times stronger than 2 and it proved to be 250-fold more potent than its 22-epimer 4b. Also, clear distinctions in the biological activity appeared in the cell differentiation assay, where the analogue 3b with an additional 22S-methyl group in the “natural” side chain was tenfold more active than its 22-epimer 3a, whereas, in the case of 20-epi compounds, the 22R-isomer 4a was found fourfold more potent than 2MD and, compared to its 22S-counterpart 4b, showed the potency higher by three orders of magnitude. A similar pattern of relative activities of the four diastereomeric at C-20 and C-22, 22-methyl vitamins 3a,b-4a,b was found in the transcriptional assay. The transactivation potencies of the analogues 3b and 4a were significantly higher compared to the compounds 3a and 4b being their 22-epimers.
Table 1.
VDR Binding Properties,a HL-60 Differentiating Activities,b and Transcriptional Activitiesc of the Vitamin D Hormone (1), 2MD (2) and the vitamin D Analogues 3a-6.
| Compd No. | Side-chain structure | Spatial regiond | VDR binding | HL-60 differentiation | 24OHase transcription | |||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Ki (nM) | ratio | ED50 (nM) | ratio | ED50 (nM) | ratio | |||
| 1 |
|
A & G | 0.1 | 1 | 2 | 1 | 0.2 | 1 |
| 2 |
|
EA & EG | 0.1 | 1 | 0.08 | 25 | 0.007 | 29 |
| 3a |
|
G | 0.2 | 0.5 | 10 | 0.2 | 5 | 0.04 |
| 3b |
|
A | 0.1 | 1 | 0.9 | 2.2 | 0.2 | 1 |
| 4a |
|
EA | 0.04 | 2.5 | 0.02 | 100 | 0.007 | 29 |
| 4b |
|
EG | 10 | 0.01 | 20 | 0.1 | 4 | 0.05 |
| 5 |
|
EG & G | 0.06 | 1.7 | 1 | 2 | 0.6 | 0.3 |
| 6 |
|
A & G | 0.02 | 5 | 0.9 | 2.2 | 0.03 | 7 |
Competitive binding of 1α,25-(OH)2D3 (1) and the synthesized vitamin D analogues to the full-length recombinant rat vitamin D receptor. The experiments were carried out in duplicate on two different occasions. The Ki values are derived from the dose-response curves and represent the inhibition constant when radiolabeled 1α,25-(OH)2D3 is present at 1 nM and a Kd of 0.2 nM is used. The binding ratio is the average ratio of the 1α,25-(OH)2D3 Ki to the Ki for the analogue.
Induction of differentiation of HL-60 promyelocytes to monocytes by 1α,25-(OH)2D3 (1) and the synthesized vitamin D analogues. Differentiation state was determined by measuring the percentage of cells reducing nitro blue tetrazolium (NBT). The experiment was repeated in duplicate two times. The ED50 values are derived from the dose-response curves and represent the analogue concentration capable of inducing 50% maturation. The differentiation activity ratio is the average ratio of the 1α,25-(OH)2D3 ED50 to the ED50 for the analogue.
Transcriptional assay in rat osteosarcoma cells stably transfected with a 24-hydroxylase gene reporter plasmid. The ED50 values are derived from dose-response curves and represent the analogue concentration capable of increasing the luciferase activity by 50%. The luciferase activity ratio is the average ratio of the 1α,25-(OH)2D3 ED50 to the ED50 for the analogue.
Active side-chain spatial regions occupied by 25-hydroxy group, as defined by Yamada.5
Noteworthy, the introduction of two methyl groups at the 22 position, regardless of the configuration at the 20-carbon, created the vitamins 5 and 6 having higher binding affinities to VDR compared to their parent vitamins. This was especially notable in the case of the latter compound.7 However, in the two remaining in vitro tests, double-substitution at C-22 of the “natural” side chain resulted in a marked increase of potency of the analogue (6)7 but, for the corresponding 20-epi compound (2 versus 5), this modification caused a reversed effect. Thus, the 22,22-dimethyl analogue 5 showed cell differentiating potency and transcriptional activity lower by one and two orders of magnitude, respectively, than the structurally related analogue, 2MD.
In vivo biological activities are shown in Figures 5 and 6. The activity profiles in bone calcium mobilization partially paralleled the results observed in the in vitro assays, where 3a and 4b exhibited lower potency than the native hormone, and 4a was substantially more potent than 1α,25-(OH)2D3. Despite the fact that 3b, 5 and 6 appear to have the VDR binding affinity and the cell differentiation activity similar to or slightly higher than 1α,25-(OH)2D3, their ability to cause the release of bone calcium in vitamin D-deficient rats was at least 50 times lower. In the intestine, the order of potency of the analogues was the same to that observed for the bone.
Figure 5.
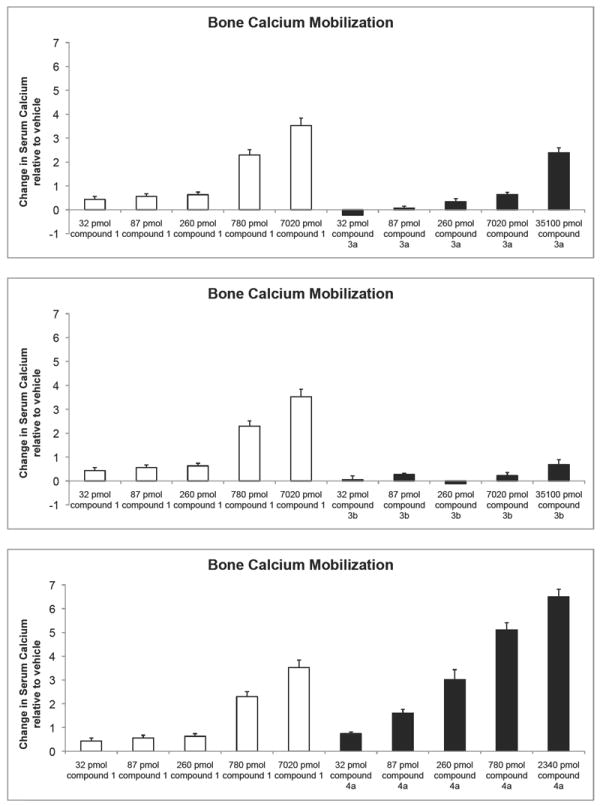
Total serum calcium levels reflecting the ability of each analog to release bone calcium stores. Note: the values shown are the difference from the vehicle controls. Each value represents the mean ± SEM of 6 values.
Figure 6.
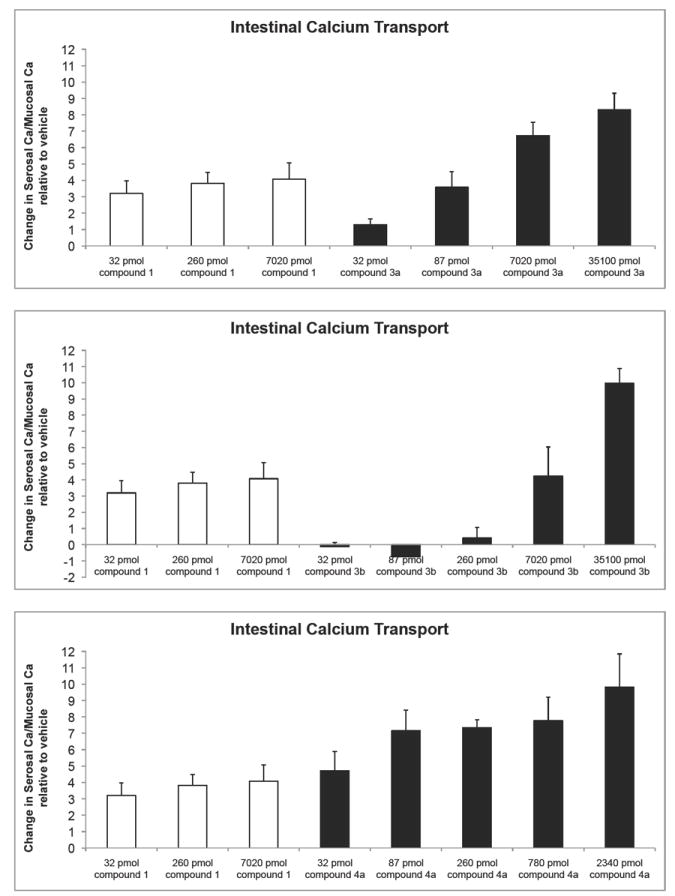
In vivo intestinal calcium transport compared to the native hormone. Note: the values shown are the difference from the vehicle controls and represent the average ± SEM of 6 measurements.
Discussion
The above-presented results of biological tests, performed for the synthesized vitamins mono-substituted at C-22, indicated that the order of their in vitro activity was 4a≫3b>3a>4b. Such an activity pattern could be quite successfully predicted taking into account the published data on the closely related diastereomers of 22-methyl-1α,25-(OH)2D3 differing in configurations at C-20 and C-22. On the basis of conformational analyses of 25-hydroxylated side chains present in these compounds and other side-chain restricted vitamin D analogues, Yamada and collaborators developed an “active space region” concept.5,17 They created three-dimensional “dot maps” showing the spatial regions which can be potentially occupied by the 25-oxygen present in the energetically preferred conformations of the vitamin D compounds possessing different side chains. It was established that the 25-hydroxy group occupied only one spatial region (A, G, EA, and EG) in each of the four C-20 and C-22 diastereomers of the 22-methylated hormone. Interestingly, further conformational studies allowed the Japanese scientists to correlate these defined regions with the biological activities of vitamin D compounds such as the receptor (VDR) affinity, cell differentiation, and transactivation potency.5,17 Thus, in terms of the space region, the orders of activity were established as follows: EA>A>G>EG (affinity for the VDR), and EA>A>EG≥G (cell differentiation and gene transactivation). Inspired by the above described findings, we performed a conformational analysis of the model 8-methylene des-A,B-compounds 42a,b,c possessing 25-hydroxy-22-methyl-substituted side chains of the vitamins, whose synthesis is presented in this study. Compound 42a was chosen to prove that our calculations provide the same spatial region that was ascribed by Yamada for the side chain of analogous (20S,22R)-isomer of 22-methyl-1α,25-(OH)2D3. We carried out the MM+ force field calculations of the model 8-methylene compounds by using the HyperChem molecular modeling program. The conformational searches were performed according to protocol described by us previously in detail.18 For the lowest energy conformers of (20S,22R)-compound 42a, only the gauche(+) conformation in relation to the dihedral angle C(16-17-20-22) was found. On the contrary, in the case of 22,22-dimethylated compounds 42b and 42c, only the anticlinal and antiperiplanar, respectively, conformation was present in respect to this torsion angle. Figure 4 shows the result of superimposition of the most stable side-chain conformers of 42a,b,c, falling into 1 kcal/mol energy window. As a consequence of the different torsion angles adopted by low-energy side chain conformers of the examined compounds, the 25-oxygen tends to reside in the following regions: EA (42a), EG and G (42b), A and G (42c). The first result confirms the correctness of our calculations, because the most active front region EA was also found by Yamada for the 22-methylated vitamin having analogous (20S,22R)-configurations of the side-chain substituents.5 A comparison of the in vitro activities of the 22-monomethylated vitamins 3a,b-4a,b with the spatial regions occupied by their side chain hydroxy group (Table 1) indicates that their order of biological potencies is in agreement with the correlations found by Yamada, despite the fact that different A-ring fragments are present in these two series of compounds. When considering the biological in vitro activity of the analogues 5 and 6, conformational analysis also explains the effect of two additional alkyl substituents at C-22. The 25-oxygen in 5 is distributed over the inactive rear regions of EG-G, whereas in 6 it also tends to locate in the active front region A.
Figure 4.
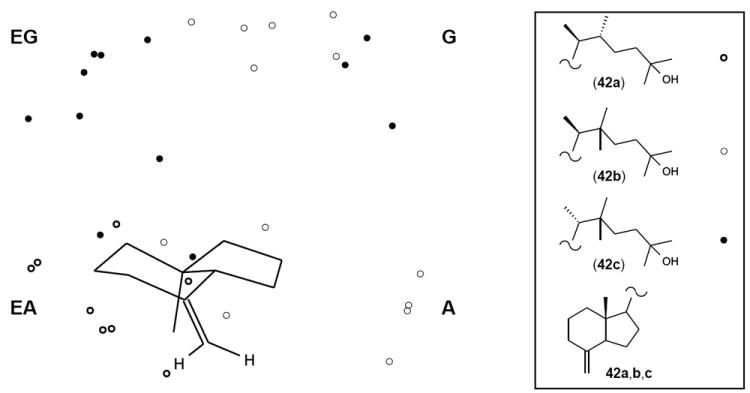
Stereoview illustrating side-chain conformational analysis of the model 8-ethylene compounds 42a,b,c possessing side chains identical as the vitamin D analogues 4a, 5 and 6, respectively. Energy-minimized conformations of these compounds (energy window 1 kcal/mol) were overlaid; the circles show the location of 25-oxygen atoms in the corresponding conformers. Side-chain carbons and hydrogen atoms are omitted for clarity. There are indicated spatial regions EG, G, EA and A as defined by Yamada.5
Summarizing, the vitamin D analogues presented in this work are interesting candidates in the structure-function studies. Moreover, they have shown a promising therapeutic value that is under investigation now.
Experimental Section
Chemistry
Melting points (uncorrected) were determined on a Thomas-Hoover capillary melting point apparatus. Optical rotations were measured in chloroform using a Perkin-Elmer model 343 polarimeter at 22 °C. Ultraviolet (UV) absorption spectra were recorded with a Perkin-Elmer Lambda 3B UV-vis spectrophotometer in ethanol or hexane. 1H nuclear magnetic resonance (NMR) spectra were recorded in deuteriochloroform at 400 and 500 MHz with Bruker Instruments DMX-400 and DMX-500 Avance console spectrometers. In the case of diastereomeric mixtures of compounds, proton signals belonging to the major isomer are listed; selected signals of the minor isomer are marked in italic. 13C NMR spectra were recorded in deuteriochloroform at 100 and 125 MHz with the same Bruker Instruments. Chemical shifts (δ) are reported in parts per million relative to CH3Si (δ 0.00) as an internal standard. Abbreviations used are singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m). Numbers in parentheses following the chemical shifts in the 13C NMR spectra refer to the number of attached hydrogens as revealed by DEPT experiments. Electron impact (EI) mass spectra were obtained with a Micromass AutoSpec (Beverly, MA) instrument. HPLC was performed on a Waters Associates liquid chromatograph equipped with a model 6000A solvent delivery system, model U6K Universal injector, and model 486 tunable absorbance detector. Solvents were dried and distilled following standard procedures.
The purity of final compounds was determined by HPLC, and they were judged at least 99% pure. Two HPLC columns (9.4 mm × 25 cm Zorbax-Sil and Zorbax RX-C18) were used as indicated in Table 1 (Supporting Information). The purity and identity of the synthesized vitamins were additionally confirmed by inspection of their 1H NMR and high-resolution mass spectra.
(8S,20R)-Des-A,B-20-(cyanomethyl)-8β-[(triethylsilyl)oxy]-pregnane (9)
Sodium cyanide (2 g, 41 mmol) was added to a solution of the tosylate 7 (0.84 g, 1.75 mmol) in dry DMSO (8 mL). The resulting mixture was stirred at 90 °C for 3 h, then it was cooled, diluted with water, and extracted with ethyl acetate. The combined organic phases were dried (Na2SO4) and concentrated. The residue was purified by column chromatography on silica gel using 10% ethyl acetate/hexane to give the nitrile 9 (0.57 g, 97%).
(8S,20S)-Des-A,B-20-(cyanomethyl)-8β-[(triethylsilyl)oxy]-pregnane (10)
The substitution reaction of the tosylate 8 with cyanide, performed as described for 9, gave the nitrile 10 (85%).
(8S,20R,22R)- and (8S,20R,22S)-Des-A,B-22-cyano-8β,25-bis[(triethylsilyl)oxy]-cholestane (11a,b)
n-Butyllithium (1.6 M in hexane, 2.7 mL, 4.32 mmol) was added to a solution of diisopropylamine (0.6 mL, 0.43 g, 4.25 mmol) in THF (4 mL) at 0 °C. The resulting mixture was stirred at 0 °C for 30 min, then it was cooled to -78 °C, and a solution of the nitrile 9 (0.57 g, 1.70 mmol) in THF (5 mL) was added. The mixture was stirred at -78 °C for 30 min and a solution of the bromide A (0.96 g, 3.42 mmol) was added. The reaction mixture was stirred at -78 °C for 1 h and at 0 °C for 1 h, then it was quenched with a saturated aqueous NH4Cl solution and extracted with ethyl acetate. The combined organic phases were washed with brine, dried (Na2SO4) and concentrated. The residue was purified by column chromatography on silica gel (elution with 1.5%, 3% and 10% ethyl acetate/hexane) to give a mixture of nitriles 11a,b (0.85 g, 93%).
(8S,20S,22R)- and (8S,20S,22S)-Des-A,B-22-cyano-8b,25-bis[(triethylsilyl)oxy]-cholestane (12a,b)
Reaction of the nitrile 10 anion with the bromide A, carried out as described for 9, gave a mixture of nitriles 12a,b (79%).
(8S,20R,22R)- and (8S,20R,22S)-Des-A,B-22-formyl-8b,25-bis[(triethylsilyl)oxy]-cholestane (13a,b)
Diisobutylaluminum hydride (1.5 M in toluene, 1.4 mL, 2.1 mmol) was added to a solution of the nitriles 11a,b (0.81 g, 1.51 mmol) in dichloromethane (10 mL) at -10 °C. The reaction mixture was stirred at -10 °C for 1 h and it was quenched with a saturated aqueous potassium sodium tartrate solution (5 mL). The water phase was extracted with dichloromethane, and the combined organic layers were washed with brine, dried (Na2SO4) and concentrated. The residue was purified by column chromatography on silica gel (elution with 3% ethyl acetate/hexane) to give a mixture of the aldehydes 13a,b (0.64 g, 79%).
(8S,20S,22R)- and (8S,20S,22S)-Des-A,B-22-formyl-8β,25-bis[(triethylsilyl)oxy]-cholestane (14a,b)
DIBALH reduction of the nitrile 12a,b was performed as described for 11a,b. A mixture of the aldehydes 14a,b was obtained in 76% yield.
(8S,20R,22R)- and (8S,20R,22S)-Des-A,B-22-(hydroxymethyl)-8β,25-[(triethylsilyl)oxy]-cholestane (15a,b)
Sodium borohydride (0.44 g, 11.63 mmol) was added to a solution of the aldehydes 13a,b (0.64 g, 1.19 mmol) in methanol (10 mL) at 0 °C. The reaction mixture was warmed and stirred at room temperature for 2 h, then quenched with water and extracted with ethyl acetate. The combined organic layers were washed with brine, dried (Na2SO4) and concentrated. The residue was purified by column chromatography on silica gel (elution with 3% and 10% ethyl acetate/hexane) to give a mixture of the alcohols 15a,b (0.46 g, 71%).
(8S,20S,22R)- and (8S,20S,22S)-Des-A,B-22-(hydroxymethyl)-8β,25-[(triethylsilyl)-oxy]-cholestane (16a,b)
Sodium borohydride reduction of the aldehydes 14a,b was performed as described for 13a,b. A mixture of the alcohols 16a,b was obtained in 70% yield.
(8S,20R,22R)- and (8S,20R,22S)-Des-A,B-22-methyl-8β,25-[(triethylsilyl)oxy]-cholestane (19a,b)
A solution of tosyl chloride (0.66 g, 3.46 mmol) in pyridine (2 mL) was added to a solution of the alcohols 15a,b (0.46 g, 0.85 mmol) in dry pyridine (4 mL) at -20 °C. The reaction mixture was stirred at -20 °C for 1 h and at +4 °C for 18 h. Then it was poured into a saturated aqueous CuSO4 solution and extracted with dichloromethane. The combined organic phases were dried (Na2SO4) and concentrated. The residue was purified by column chromatography on silica gel (elution with 3% ethyl acetate/hexane) to give a mixture of the tosylates 17a,b (0.54 g, 92%).
LiAlH4 (0.4 g, 10.53 mmol) was added to a solution of the tosylates 17a,b (0.53 g, 0.76 mmol) in dry diethyl ether (10 mL) at 0 °C. The reaction mixture was stirred at +4 °C for 20 h. The excess of LiAlH4 was decomposed with water. The reaction mixture was diluted with diethyl ether and, subsequently, filtered through Celite. The filtrate was extracted with ethyl acetate, dried (Na2SO4) and concentrated. The residue was purified by column chromatography on silica gel (3% ethyl acetate/hexane) to give a mixture of the products 19a,b (0.32 g, 80%).
(8S,20S,22R)- and (8S,20S,22S)-Des-A,B-22-methyl-8β,25-[(triethylsilyl)oxy]-cholestane (20a,b)
Tosylation of the alcohols 16a,b and reduction of the formed tosylates with LiAlH4, performed as described for 15a,b, gave a mixture of the products 20a,b (96 mg, 75%).
(8S,20R,22R)- and (8S,20R,22S)-Des-A,B-22-methyl-cholestane-8β,25-diol (21a and 21b)
Tetrabutylammonium fluoride (1.0 M in THF, 3.4 mL, 3.4 mmol) was added to a solution of the compounds 19a,b (0.31 g, 0.59 mmol) in THF (3 mL) at 0 °C. The reaction mixture was stirred at +4 °C for 20 h, then it was diluted with water and extracted with ethyl acetate. The combined organic extracts were dried (Na2SO4) and concentrated. The residue was purified by column chromatography on silica gel (elution with 10% and 50% ethyl acetate/hexane) to give a mixture of diols 21a and 21b (0.17 g, 99%) in a 1:2 ratio, respectively (based on 1H NMR). The isomers were separated by crystallization from ethyl acetate, and the absolute configuration was established by X-ray analysis. Pure crystals (96 mg) of the 22S-isomer 21b were obtained after first crystallization. Then, pure crystals (44.6 mg) of the other 22R-isomer 21a were obtained from the concentrated mother liquors. Additionally, a second crop of pure crystals (16 mg) of the diol 21b was obtained from the filtrate after second crystallization.
(8S,20S,22R)- and (8S,20S,22S)-Des-A,B-22-methyl-cholestane-8β,25-diol (22a and 22b)
Deprotection of the silyl groups in the compounds 20a,b with tetrabutylammonium fluoride was performed analogously as described for 19a,b. The products were purified by column chromatography on silica gel (30% ethyl acetate/hexane) to give a mixture of the diols 22a and 22b (99%) in a 2:1 ratio, respectively (as established by 1H NMR). The isomers were separated by crystallization from ethyl acetate, and the absolute configuration was established by X-ray analysis. Pure crystals of the 22R-isomer 22a were obtained after two crystallizations, and the isomeric 22S-diol 22b was obtained from the mother liquors.
(20R,22R)-Des-A,B-22-methyl-25-[(triethylsilyl)oxy]-cholestan-8-one (23a)
Molecular sieves 4Å (60 mg) were added to a solution of 4-methylmorpholine oxide (33 mg, 0.282 mmol) in dichloromethane (0.25 mL). The mixture was stirred at room temperature for 15 min and tetrapropylammonium perruthenate (2 mg, 5.7 μmol) was added, followed by a solution of the diol 21a (16 mg, 54 μmol) in dichloromethane (300 + 250 μL). The resulting suspension was stirred at room temperature for 1 h. The reaction mixture was filtered through a Waters silica Sep-Pak cartridge (2 g) that was further washed with ethyl acetate. After removal of the solvent, the 25-hydroxy-8-ketone (14.4 mg, 89%) was obtained as a colorless oil.
Triethylsilyl trifluoromethanesulfonate (20 μL, 23 mg, 88 μmol) was added dropwise to a solution of the obtained 25-hydroxy-8-ketone (14.4 mg, 49 μmol) and 2,6-lutidine (20 μL, 18 mg, 0.17 mmol) in dichloromethane (2 mL) at -40 °C. The reaction mixture was stirred at -40 °C for 15 min, then it was diluted with dichloromethane and washed with water. The organic layer was dried (Na2SO4) and concentrated. The residue was applied on a Waters silica Sep-Pak cartridge (5 g). Elution with ethyl acetate/hexane (1:99, then 2:98) gave the protected ketone 23a (19 mg, 95%).
(20R,22S)-Des-A,B-22-methyl-25-[(triethylsilyl)oxy]-cholestan-8-one (23b)
Pyridinium dichromate (0.18 g, 0.48 mmol) and pyridinium p-toluenesulfonate (24 mg, 95 μmol) were added in one portion to a solution of diol 21b (24.9 mg, 84 μmol) in dry dichloromethane (5 mL). The reaction mixture was stirred at room temperature for 75 min, then it was quenched with water and extracted with dichloromethane. The combined organic layers were dried (Na2SO4) and concentrated. The residue was applied on a Waters silica Sep-Pak cartridge (2 g). Elution with dichloromethane gave the pure 25-hydroxy-8-ketone (23.6 mg).
Silylation of the 8-ketone with triethylsilyl trifluoromethanesulfonate was performed according to procedure described above. The crude product was applied to a Waters silica Sep-Pak cartridge (10 g). Elution with ethyl acetate/hexane (2:98, then 5:95) gave the protected ketone 23b (18.2 mg, 53% yield in 2 steps).
(8S,20S,22R)-Des-A,B-22-methyl-25-[(triethylsilyl)oxy]-cholestan-8-one (24a)
Oxidation of the diol 22a with tetrapropylammonium perruthenate and 4-methylmorpholine oxide, and the subsequent silylation of the resulted 25-hydroxy-8-ketone was performed as described for conversion of 21a into 23a. The protected ketone 24a was obtained in 68% yield.
(8S,20S,22S)-Des-A,B-22-methyl-25-[(triethylsilyl)oxy]-cholestan-8-one (24b)
Oxidation of the diol 22b and subsequent silylation of the 25-hydroxy-8-ketone was carried out according to the procedure described above for conversion of 21a into 23a, to give the protected ketone 24b in 73% yield.
(20R,22R)-1α,25-Dihydroxy-22-methyl-2-methylene-19-norvitamin D3 (3a)
Phenyllithium (1.8 M in di-n-butyl ether, 60 μL, 108 μmol) was added to a stirred solution of the phosphine oxide 25 (54 mg, 93 μmol) in anhydrous THF (500 μL) at -30 °C. After 30 min, the mixture was cooled to -78 °C and a precooled solution of the ketone 23a (19 mg, 47 μmol) in anhydrous THF (300 + 200 μL) was added. The reaction mixture was stirred under argon at -78 °C for 4 h and then at +4 °C for 19 h. Ethyl acetate was added and the organic phase was washed with brine, dried (Na2SO4) and concentrated. The residue was applied on a Waters silica Sep-Pak cartridge (5g). The cartridge was eluted with hexane and ethyl acetate/hexane (1:99) to give the protected vitamin 26a (32.64 mg, 91%).
The protected vitamin 26a (32.64 mg, 42 mmol) was dissolved in THF (4 mL) and acetonitrile (3 mL). A solution of aqueous 48% HF in acetonitrile (1:9 ratio, 4 mL) was added at 0 °C and the resulting mixture was stirred at room temperature for 2 h. Saturated aqueous NaHCO3 solution was added and the reaction mixture was extracted with dichloromethane. The combined organic phases were dried (Na2SO4) and concentrated under reduced pressure. The residue was diluted with 2 mL of hexane/ethyl acetate (7:3) and applied on a Waters silica Sep-Pak cartridge (5 g). An elution with hexane/ethyl acetate (7:3, then 1:1) gave the crude product 3a. The vitamin 3a was further purified by a normal-phase HPLC [9.4 × 250 mm Zorbax Silica column, 5 mL/min, hexane/2-propanol (85:15) solvent system, Rt= 6.5 min.] and a reversed-phase HPLC [9.4 × 250 mm Zorbax RX-C18 column, 3 mL/min, methanol/water (85:15) solvent system, Rt= 13.2 min.] to give the pure compound 3a (15.3 mg, 78%). Pure crystals of the analogue 3a were obtained after crystallization from hexane/2-propanol and they were characterized by an X-ray analysis.
(20R,22S)-1α,25-Dihydroxy-22-methyl-2-methylene-19-norvitamin D3 (3b)
The protected vitamin 26b was prepared in 90% yield by the Wittig-Horner reaction of the ketone 23b and the phosphine oxide 25, performed analogously to the process described above for the preparation of 26a. The protected vitamin 26b was hydrolyzed as described for 26a, and the product 3b was further purified by a normal-phase HPLC [9.4 mm × 25 cm Zorbax Silica column, 5 mL/min, hexane/2-propanol (85:15) solvent system, Rt = 8.5 min.] and a reversed-phase HPLC [9.4 × 25 cm Zorbax RX-C18 column, 3 mL/min, methanol/water (85:15) solvent system, Rt = 15.2 min] to give the pure compound 3b (13.5 mg, 79%).
(20S,22R)-1α,25-Dihydroxy-22-methyl-2-methylene-19-norvitamin D3 (4a)
The protected vitamin 27a was prepared from the ketone 24a in 90% yield analogously to the isomeric vitamin 26a. Hydrolysis of silyl protecting groups in 27a was performed as described for 26a and the obtained vitamin 4a was purified by a normal-phase HPLC [9.4 mm × 25 cm Zorbax-Sil column, 4 mL/min, hexane/2-propanol (85:15) solvent system, Rt = 7.9 min] and a reversed-phase HPLC [9.4 mm × 25 cm Zorbax RX-C18 column, 3 mL/min, methanol/water (85:15) solvent system, Rt = 14.7 min] to give the pure compound 4a (10.3 mg, 77%).
(20S,22S)-1α,25-Dihydroxy-22-methyl-2-methylene-19-norvitamin D3 (4b)
The protected vitamin 27b was prepared from the ketone 24b in 87% yield analogously to 26a. The silyl protecting groups in 27b were hydrolyzed as described for 26a and the obtained vitamin 4b was purified by a normal-phase HPLC [9.4 mm × 25 cm Zorbax-Sil column, 4 mL/min, hexane/2-propanol (85:15) solvent system, Rt = 7.3 min] and a reversed-phase HPLC [9.4 mm × 25 cm Zorbax RX-C18 column, 3 mL/min, methanol/water (85:15) solvent system, Rt = 11.7 min] to give the vitamin 4b (6.6 mg, 50%).
(8S,20R)-Des-A,B-20-(cyano-dimethyl-methyl)-8β-[(triethylsilyl)oxy]-pregnane (28)
n-Butyllithium (1.6 M in hexane, 2.4 mL, 3.8 mmol) was added to a solution of diisopropylamine (0.54 mL, 0.384 g, 3.8 mmol) in THF (2 mL) at 0 °C. The resulting mixture was stirred at 0 °C for 30 min, then it was cooled to -78 °C and a solution of the nitrile 9 (0.326 g, 0.973 mmol) in THF (2 mL) was added. The mixture was stirred at -78 °C for 30 min, then iodomethane (1.2 mL, 2.73 g, 19.2 mmol) was added. The reaction mixture was stirred at -78 °C for 1 h and at room temperature for 1 h, then quenched with saturated aqueous NH4Cl solution, and extracted with ethyl acetate. The combined organic phases were washed with brine, dried (Na2SO4) and concentrated. The residue was purified by column chromatography on silica gel (elution with 5%, then 10% ethyl acetate/hexane) to give the product 28 (0.197 g, 56%).
(8S,20S)-Des-A,B-20-(cyano-dimethyl-methyl)-8β-[(triethylsilyl)oxy]-pregnane (29)
Alkylation of the nitrile 10, performed as described for 9, gave the product that was purified by column chromatography on silica gel (elution with 10%, then 20% ethyl acetate/hexane) to furnish the compound 29 in 99% yield.
(8S,20R)-Des-A,B-20-(1’,1’-dimethyl-2’-oxo-ethyl)-8-[(triethylsilyl)oxy]-pregnane (30)
Diisobutylaluminum hydride (1.0 M in dichloromethane, 3.1 mL, 3.1 mmol) was added to a solution of the nitrile 28 (0.197 g, 0.543 mmol) in dichloromethane (4 mL) at -10 °C. The reaction mixture was stirred at -10 °C for 1 h, then it was quenched with a saturated aqueous potassium sodium tartrate solution (5 mL). The water phase was extracted with dichloromethane, the combined organic layers were washed with brine, dried (Na2SO4) and concentrated. The residue was purified by column chromatography on silica gel (elution with 10% ethyl acetate/hexane) to give the aldehyde 30 (0.15 g, 75%).
(8S,20S)-Des-A,B-20-(1’,1’-dimethyl-2’-oxo-ethyl)-8-[(triethylsilyl)oxy]-pregnane (31)
DIBALH reduction of the nitrile 29, performed as described for 28, gave the aldehyde 31 in 95% yield as colorless crystals.
(8S,20R)-Des-A,B-20-(1’,1’-dimethyl-3’-ethoxycarbonyl-allyl)-8β-[(triethylsilyl)oxy]-pregnane (32)
n-Butyllithium (1.6 M in hexane, 5.2 mL, 8.3 mmol) was added to a solution of diisopropylamine (1.2 mL, 0.84 g, 8.3 mmol) in dry THF (2 mL) at 0 °C. After 30 min the mixture was cooled to -10 °C and triethylphosphonoacetate (1.9 mL, 2.13 g, 9.5 mmol) was added. The reaction mixture was stirred at -10 °C for 30 min and then a solution of the aldehyde 30 (0.15 g, 0.41 mmol) in anhydrous THF (5 mL + 3 mL) was added via cannula. The mixture was stirred under argon at -10 °C for 1 h, then it was warmed at 37 °C for 2.5 h, and then stirred at room temperature overnight. Dichloromethane was added and the organic phase was washed with water, dried (Na2SO4), and concentrated. The product was purified on a Sep-Pak cartridge (5 g). Elution with ethyl acetate/hexane (2%) provided 32 (99 mg, 55%).
(8S,20S)-Des-A,B-20-(1’,1’-dimethyl-3’-ethoxycarbonyl-allyl)-8β-[(triethylsilyl)oxy]-pregnane (33)
The olefination reaction of the aldehyde 31 gave the product that was purified on a Sep-Pak cartridge (5 g). Elution with ethyl acetate/hexane (2%, then 3% and 5%) furnished 33 in 91% yield.
(8S,20R)-Des-A,B-20-(1’,1’-dimethyl-3’-ethoxycarbonyl-propyl)-pregnan-8β-ol (34)
A solution of the ester 32 (99 mg, 0.23 mmol) in methanol (5 mL) was hydrogenated in the presence of 10% palladium on powdered charcoal (10 mg) at room temperature for 20 h. Filtration of the reaction mixture through a Waters silica Sep-Pak cartridge (2 g) provided the ester 34 (50.4 mg, 68%).
(8S,20S)-Des-A,B-20-(1’,1’-dimethyl-3’-ethoxycarbonyl-propyl)-pregnan-8β-ol (35)
Hydrogenation of the ester 33, carried out as described above for 32, afforded the compound 35 in 95% yield as a colorless oil.
(8S,20R)-Des-A,B-22,22-dimethyl-cholestane-8β,25-diol (36)
Methyl-magnesium bromide (3.0 M solution in diethyl ether, 130 μL, 0.39 mmol) was added to a solution of the ester 34 50 mg, 0.154 mmol) in anhydrous diethyl ether (3 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 2 h and then at room temperature for 18 h. It was quenched with saturated aqueous NH4Cl solution, extracted with ethyl acetate, dried (Na2SO4) and concentrated. The residue was applied on a Waters silica Sep-Pak cartridge (5 g). Elution with ethyl acetate/hexane (1:1) gave the diol 36 (48 mg, 100%) as colorless crystals.
(8S,20S)-Des-A,B-22,22-dimethyl-cholestane-8β,25-diol (37)
Reaction of the ester 35 (24 mg, 0.074 mmol) with the Grignard reagent, performed analogously to that described above for 34, yielded the diol 37 (19.2 mg, 84%).
(20R)-Des-A,B-22,22-dimethyl-25-[(triethylsilyl)oxy]-cholestan-8-one (38)
Oxidation of the diol 36 with tetrapropylammonium perruthenate and 4-methylmorpholine oxide, and the subsequent silylation of the formed 25-hydroxy-8-ketone was carried out analogously as described for the conversion of 21a into 23a. The protected ketone 38 was obtained in 86% yield.
(20S)-Des-A,B-22,22-dimethyl-25-[(triethylsilyl)oxy]-cholestan-8-one (39)
Oxidation of the diol 37 with tetrapropylammonium perruthenate and 4-methylmorpholine oxide, and the subsequent silylation of the resulted 25-hydroxy-8-ketone was performed as described for the conversion of 21a into 23a. The protected ketone 39 was obtained in 78% yield.
(20R)-1α,25-Dihydroxy-22,22-dimethyl-2-methylene-19-norvitamin D3 (5)
The protected vitamin 40 was obtained by the Wittig-Horner reaction of the ketone 38 and the phosphine oxide 25, performed analogously to the process described above for preparation of 26a. The vitamin was purified by a normal-phase HPLC [9.4 mm × 25 cm Zorbax-Sil column, 4 mL/min, 2-propanol/hexane (0.1:99.9) solvent system, Rt = 3.2 min] to give the pure protected compound 40 (78%).
The protected vitamin 40 was hydrolyzed as described for 26a, and the product 5 was purified by a normal-phase HPLC [9.4 mm × 25 cm Zorbax-Sil column, 5 mL/min, hexane/2-propanol (85:15) solvent system, Rt = 7.4 min] and a reversed-phase HPLC [9.4 mm × 25 cm Zorbax RX-C18 column, 3 mL/min, methanol/water (85:15) solvent system, Rt = 13.3 min] to give the pure compound 5 (79%). m.p. 154 °C (from 2-propanol/hexane).
(20S)-1α,25-Dihydroxy-22,22-dimethyl-2-methylene-19-norvitamin D3 (6)
The protected vitamin 41 was prepared by the Wittig-Horner reaction of the ketone 39 and the phosphine oxide 25, performed analogously to the process described above for the preparation of 26a. The vitamin was purified by a normal-phase HPLC [9.4 mm × 25 cm Zorbax-Sil column, 4 mL/min, 2-propanol/hexane (0.1:99.9) solvent system, Rt = 3.4 min] to give the pure protected compound 41 (91%).
The protected vitamin 41 was hydrolyzed as described for 26a, and the product 6 was purified by a normal-phase HPLC [9.4 mm × 25 cm Zorbax-Sil column, 4 mL/min, hexane/2-propanol (85:15) solvent system, Rt = 7.8 min] and a reversed-phase HPLC [9.4 mm × 25 cm Zorbax RX-C18 column, 3 mL/min, methanol/water (85:15) solvent system, Rt = 15.7 min] to give the pure compound 6 (75%).
Biological Studies
1. In vitro Studies
VDR binding, HL-60 differentiation, and 24-hydroxylase transcription assays were performed as previously described and are shown in the footnote of Table 1. 19,20
2. In vivo Studies
Bone calcium mobilization and intestinal calcium transport
Male, weanling Sprague-Dawley rats were purchased from Harlan (Indianapolis, IN). The animals were group housed and placed on Diet 11 (0.47% Ca as CaCO3) + AEK oil for one week followed by Diet 11 (0.02% Ca) + AEK oil for 3 weeks. The rats were then switched to a diet containing 0.47% Ca21 for one week followed by two weeks on a diet containing 0.02% Ca. Dose administration began during the last week on 0.02% Ca diet. Four consecutive intraperitoneal doses were given approximately 24 hours apart. Twenty-four hours after the last dose, blood was collected from the severed neck and the concentration of serum calcium determined as a measure of bone calcium mobilization. Because there is essentially no calcium in the intestine (the diet contains 0.02% calcium), the rise in serum calcium derives from mobilization of calcium from bone. Further, no calcium is found in urine when a low-calcium diet is fed; thus the rise in serum calcium cannot be the result of increased renal reabsorption. The first 10 cm of the intestine was also collected for the intestinal calcium transport analysis using the everted gut sac method.20 Statistical significance was ascertained by application of the Student’s “t” test.
Molecular Modeling
The molecular mechanism studies were used to establish the energy-minimized structures of the model compounds 42a,b,c. The calculation of optimized geometries and steric energies was carried out using the algorithm from the MM+ HyperChem (release 8.0) software package (Autodesk, Inc.). MM+ is an all-atom force field based on the MM2 functional form. The procedure used for generation of the respective side-chain conformers and finding the global minimum structures was analogous to that described by us previously18 and involved the Conformational Search module.
Crystallographic studies
Crystal data (for the diol 21a)
C19H36O2, M = 296.48, T = 100 (1) K, monoclinic, P2 (1), a = 11.394 (2) Å, b = 16.535 (3) Å, c = 11.450 (2) Å, αγ = 90°, β = 119.26 (3)°, V = 1881.9 (7) Å3, Z = 4, Dx = 1.046 Mg/m3, μ = 0.497 mm-1, F (000) = 664, two molecules of the diol 21a were present in the asymmetric unit.
Crystal data (for the diol 21b)
C19H36O2, M = 296.48, T = 100 (1) K, monoclinic, C2, a = 26.391 (5) Å, b = 6.0830 (12) Å, c = 12.688 (3) Å, αγ = 90°, β = 118.38 (3)°, V = 1792.1 (6) Å3, Z = 4, Dx = 1.099 Mg/m3, μ = 0.522 mm-1, F (000) = 664.
Crystal data (for the diol 22a)
C19H36O2, M = 296.48, T = 100 (1) K, monoclinic, P2 (1), a = 9.6840 (19) Å, b = 19.156 (4) Å, c = 9.6870 (19) Å, αγ = 90°, β = 91.27 (3)°, V = 1796.6 (6) Å3, Z = 4, Dx = 1.096 Mg/m3, μ = 0.521 mm-1, F (000) = 664.
Crystal data (for the vitamin 3a)
C31H54O4, M = 490.74, T = 100 (1) K, monoclinic, C2 (1), a = 27.039 (5) Å, b = 6.4790 (13) Å, c = 17.412 (4) Å, αγ = 90°, β = 103.35 (3)°, V = 2967.9 (10) Å3, Z = 4, Dx = 1.098 Mg/m3, μ = 0.544 mm-1, F (000) = 1088, in addition to one molecule of the vitamin 3a, there was also one molecule of 2-propanol in the asymmetric unit.
Crystal data (for the vitamin 5)
C32H56O4, M = 504.77, T = 298 (1) K, monoclinic, C2, a = 27.3382 (16) Å, b = 6.6860 (13) Å, c = 19.221 (10) Å, αγ = 90°, β = 113.57 (3)°, V = 3320.3 (11) Å3, Z = 4, Dx = 1.041 Mg/m3, μ = 0.513 mm-1, F (000) = 1120, in addition to one molecule of the vitamin 5, there was also one molecule of 2-propanol in the asymmetric unit.
Crystal data (for the vitamin 6)
C33H58O4, M = 518.79, T = 100 (1) K, monoclinic, P2 (1), a = 7.5780 (15) Å, b = 14.792 (3) Å, c = 14.481 (3) Å, αγ = 90°, β = 102.22 (3)°, V = 1586.5 (6) Å3, Z = 2, Dx = 1.086 Mg/m3, μ = 0.532 mm-1, F (000) = 576, in addition to one molecule of the vitamin 6, there was also one molecule of diethyl ether in the asymmetric unit.
Structure determination
The data were collected using a Bruker AXS Platinum 135 CCD detector controlled with the PROTEUM software suite (Bruker AXS Inc., Madison, WI). The X-ray source was CuK radiation (1.54178 Å) from a Rigaku RU200 X–ray generator equipped with Montel optics, operated at 50 kV and 90 mA. The X-ray data were processed with SAINT version 7.06A (Bruker AXS Inc.) and internally scaled with SADABS version 2005/1 (Bruker AXS Inc.). The sample was mounted on a glass fiber using vacuum grease and cooled to 100 K. The intensity data were measured as series of phi and omega oscillation frames each of 1° for 5-20 sec/frame. The detector was operated in 512 × 512 mode and was positioned 4.5 cm from the sample. Cell parameters were determined from a non-linear least squares fit in the range of 4.0<theta<55°.
The space group was determined by systematic absences and statistical tests and verified by subsequent refinement. The structure was solved by direct methods22 and refined by the full-matrix least-squares methods on F2. The hydrogen atom positions were determined from difference peaks and ultimately refined by a riding model with idealized geometry. Non-hydrogen atoms were refined with anisotropic displacement parameters. The absolute structure was determined by refinement of the Flack parameter.23
Crystallographic data for the structures reported in this paper have been deposited at the Cambridge Crystallographic Data Center with the deposition numbers: CCDC 851675 (21a), CCDC 851674 (21b), CCDC 851677 (22a), CCDC 851676 (3a), CCDC 851679 (5) and CCDC 851678 (6).
Supplementary Material
Acknowledgments
We gratefully acknowledge Jennifer Vaughan, Jean Prahl and Erin Gudmundson for their excellent technical assistance. We also thank Dr. Mark Anderson for his assistance in recording NMR spectra. This study made use of the National Magnetic Resonance Facility at Madison, which was supported by the NIH grants P41RR02301 (BRTP/ NCRR) and P41GM66326 (NIGMS). Additional equipment was purchased with funds from the University of Wisconsin, the NIH (RR02781, RR08438), the NSF (DMB-8415048, OIA-9977486, BIR-9214394), and the USDA.
Abbreviations
- 1α,25-(OH)2D3
1α,25-dihydroxyvitamin D3
- VDR
vitamin D receptor
Footnotes
Supporting Information Available: Purity criteria of the vitamin D analogues 3a-4b, 5 and 6, their 1H and 13C NMR spectra, figures with dose-response curves derived from cellular differentiation assay, spectral data of the all synthesized compounds. Ordering information is given on any current masthead page.
References
- 1.Jones G, Strugnell SA, DeLuca HF. Current understanding of the molecular actions of vitamin D. Physiol Rev. 1998;78:1193–1231. doi: 10.1152/physrev.1998.78.4.1193. [DOI] [PubMed] [Google Scholar]
- 2.De Luca HF. Overview of general physiologic features and functions of vitamin D. Am J Clin Nutr. 2004;80(suppl):1689S–1696S. doi: 10.1093/ajcn/80.6.1689S. [DOI] [PubMed] [Google Scholar]
- 3.(a) Feldman D, Pike JW, Adams JS, editors. Vitamin D. 3. Elsevier Academic Press; San Diego, CA: 2011. [Google Scholar]; (b) Kubodera N. A new look at the most successful prodrugs for active vitamin D (D hormone): alfacalcidol and doxercalciferol. Molecules. 2009;14:3869–3880. doi: 10.3390/molecules14103869. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Peleg S, Posner G. Vitamin D analogs as modulators of vitamin D receptor action. Current Topics in Med Chem. 2003;3:1555–1572. doi: 10.2174/1568026033451691. [DOI] [PubMed] [Google Scholar]; (d) Bouillon R, Okamura WH, Norman AW. Structure-function relationships in the vitamin D endocrine system. Endocrine Rev. 1995;16:200–257. doi: 10.1210/edrv-16-2-200. [DOI] [PubMed] [Google Scholar]
- 4.Binderup L, Latini S, Binderup E, Bretting C, Calverley M, Hansen K. 20-Epi-vitamin D3 analogues: A novel class of potent regulators of cell growth and immune responses. Biochem Pharmacol. 1991;42:1569–1575. doi: 10.1016/0006-2952(91)90426-6. [DOI] [PubMed] [Google Scholar]
- 5.(a) Yamada S, G, Shimizu M, Yamamoto K. Structure-function relationships of vitamin D including ligand recognition by the vitamin D receptor. Med Res Rev. 2003;23:89–115. doi: 10.1002/med.10023. [DOI] [PubMed] [Google Scholar]; (b) Yamada S, Yamamoto K, Masuno H, Ohta M. Conformation-function relationship of vitamin D: conformational analysis predicts potential side-chain structure. J Med Chem. 1998;41:1467–1475. doi: 10.1021/jm970761l. [DOI] [PubMed] [Google Scholar]; (c) Yamamoto K, Yan Sun W, Ohta M, Hamada K, DeLuca HF, Yamada S. Conformationally restricted analogs of 1α,25-dihydroxyvitamin D3 and its 20-epimer: compounds for study of the three-dimensional structure of vitamin D responsible for binding to the receptor. J Med Chem. 1996;39:2727–2737. doi: 10.1021/jm9600048. [DOI] [PubMed] [Google Scholar]
- 6.(a) DeLuca HF, Tadi BP, Plum LA, Clagget-Dame M. 17,20(Z)-Dehydro vitamin D analogs and their uses. 7,241,748. 2007 Jul 10;; (b) Vanhooke JL, Tadi BP, Benning MM, Plum LA, DeLuca HF. New analogs of 2-methylene-19-nor-(20S)-1,25-dihydroxyvitamin D3 with conformationally restricted side chains: evaluation of biological activity and structural determination of VDR-bound conformations. Arch Biochem Biophys. 2007;460:161–165. doi: 10.1016/j.abb.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 7.Sicinski R, Prahl J, Smith C, DeLuca HF. New 1α,25-dihydroxy-19-norvitamin D3 compounds of high biological activity: synthesis and biological evaluation of 2-hydroxymethyl, 2-methyl, and 2-methylene analogues. J Med Chem. 1998;41:4662–4674. doi: 10.1021/jm9802618. [DOI] [PubMed] [Google Scholar]
- 8.Shevde NK, Plum LA, Clagett-Dame M, Yamamoto H, Pike JW, DeLuca HF. A potent analog of 1α,25-dihydroxyvitamin D3 selectively induces bone formation. Proc Natl Acad Sci U S A. 2002;99:13487–13491. doi: 10.1073/pnas.202471299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakamaki Y, Inaba Y, Yoshimoto N, Yamamoto K. Potent antagonist for the vitamin D receptor: vitamin D analogues with simple side chain structure. J Med Chem. 2010;53:5813–5826. doi: 10.1021/jm100649d. [DOI] [PubMed] [Google Scholar]
- 10.(a) Yamamoto K, Yamada S. Highly diastereoselective conjugate addition of organocuprate to acyclic E- and Z-enones: reversal stereoselectivity under kinetic and thermodynamic conditions. Tetrahedron Lett. 1992;33:7521–7524. [Google Scholar]; (b) Yamamoto K, Takahashi J, Hamano K, Yamada S. Stereoselective syntheses of (22R)- and (22S)-22-methyl-1α,25-dihydroxyvitamin D3: active vitamin D3 analogs with restricted side chain conformation. J Org Chem. 1993;58:2530–2537. [Google Scholar]
- 11.(a) Posner GH, Lee JK, White C, Hutchings RH, Dai H, Kachinski JL, Dolan P, Kensler TW. Antiproliferative hybrid analogs of the hormone 1α,25- dihydroxyvitamin D3: design, synthesis, and preliminary biological evaluation. J Org Chem. 1997;62:3299–3314. doi: 10.1021/jo970049w. [DOI] [PubMed] [Google Scholar]; (b) DeLuca HF, Flores A, Grzywacz P, Plum LA, Clagett-Dame M, Thoden JB, Holden HM. Diastereomers of 2-methylene-19-nor-22-methyl-1α,25-dihydroxyvitamin D3. 0,237,557. 2011 Sep 29;
- 12.Fujishima T, Zhaopeng L, Konno K, Nkagawa K, Okano T, Yamaguchi K, Takayama H. Highly potent cell differentiation-including analogues of 1α,25-dihydroxyvitamin D3: synthesis and biological activity of 2-methyl-1,25-dihydroxyvitamin D3 with side-chain modifications. Bioorg Med Chem. 2001;9:525–535. doi: 10.1016/s0968-0896(00)00267-4. [DOI] [PubMed] [Google Scholar]
- 13.Fall Y, Fernandez C, González V, Mouriño A. Stereoselective synthesis of (22R)- and (22S)-methyl-1α,25-dihydroxy-vitamin D3. Synlett. 2001:1567–1568. [Google Scholar]
- 14.Kelley SP, Fábián L, Pratt Brock C. Failures of fractional crystallization: ordered cocrystals of isomers and near isomers. Acta Cryst. 2011;B67:79–93. doi: 10.1107/S0108768110048135. [DOI] [PubMed] [Google Scholar]
- 15.(a) Zhu GD, Okamura WH. Synthesis of vitamin D (calciferol) Chem Rev. 1995;95:1877–1952. [Google Scholar]; (b) Dai H, Posner G. Synthetic approaches to vitamin D. Synthesis. 1994:1383–1398. [Google Scholar]
- 16.(a) Lythgoe B, Moran TA, Nambudiry MEN, Ruston S, Tideswell J, Wright PW. Allylic phosphine oxides as precursors of dienes of defined geometry: synthesis of 3-deoxyvitamin D2. Tetrahedron Lett. 1975;44:3863–3866. [Google Scholar]; (b) Lythgoe B, Nambudiry MEN, Tideswell J. Direct total synthesis of vitamins D2 and D3. Tetrahedron Lett. 1977;41:3685–3688. [Google Scholar]; (c) Lythgoe B, Moran TA, Nambudiry MEN, Tideswell J, Wright PW. Calciferol and its derivatives. Part 22. A direct synthesis of vitamin D2 and D3. J Chem Soc Perkin Trans. 1978;1:590–595. [Google Scholar]
- 17.(a) Yamamoto K, Inaba Y, Yoshimoto N, Choi M, DeLuca HF, Yamada S. 22-Alkyl-20-epi-1α,25-dihydroxyvitamin D3 compounds of superagonistic activity: syntheses, biological activities and interaction with the receptor. J Med Chem. 2007;50:932–939. doi: 10.1021/jm060889f. [DOI] [PubMed] [Google Scholar]; (b) Masuno H, Yamamoto K, Wang X, Choi M, Ooizumi H, Shinki T, Yamada S. Rational design, synthesis, and biological activity of novel conformationally restricted vitamin D analogues, (22R)- and (22S)-22-Ethyl-1α,25-dihydroxy-23,24-didehydro-24a,24b-dihomo-20-epivitamin D3. J Med Chem. 2002;45:1825–1834. doi: 10.1021/jm0105631. [DOI] [PubMed] [Google Scholar]; (c) Yamamoto K, Ooizumi H, Umesono K, Verstuyf A, Bouillon R, DeLuca HF, Yamada S. Three-dimensional structure-function relationship of vitamin D: side chain location and various activities. Bioorg Med Chem Lett. 1999;9:1041–1046. doi: 10.1016/s0960-894x(99)00129-8. [DOI] [PubMed] [Google Scholar]; (d) Yamamoto K, Ohta M, DeLuca HF, Yamada S. On the side chain conformation of 1α,25-dihydroxyvitamin D3 responsible for binding to the receptor. Bioorg Med Chem Lett. 1995;5:979–984. [Google Scholar]
- 18.Sicinski RR, DeLuca HF. Synthesis and biological activity of 22-iodo- and (E)-20(22)-dehydro analogues of 1α,25-dihydroxyvitamin D3. Bioorg Med Chem. 1999;7:2877–2889. doi: 10.1016/s0968-0896(99)00249-7. [DOI] [PubMed] [Google Scholar]
- 19.Chiellini G, Grzywacz P, Plum LA, Barycki R, Clagett-Dame M, DeLuca HF. Synthesis and biological properties of 2-methylene-19-nor-25-dehydro-1α-hydroxyvitamin D3-26,23-lactones – weak agonists. Bioorg Med Chem. 2008;16:8563–8573. doi: 10.1016/j.bmc.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glebocka A, Sicinski RR, Plum LA, Clagett-Dame M, DeLuca HF. New 2-alkylidene 1α,25-dihydroxy-19-norvitamin D3 analogues of high intestinal activity: synthesis and biological evaluation of 2-(3’-alkoxypropylidene)- and 2-(3’-hydroxypropylidene) derivatives. J Med Chem. 2006;49:2909–2920. doi: 10.1021/jm051082a. [DOI] [PubMed] [Google Scholar]
- 21.Suda T, DeLuca HF, Tanaka Y. Biological activity of 25-hydroxyergo-calciferol in rats. J Nutr. 1970;100:1049–1052. doi: 10.1093/jn/100.9.1049. [DOI] [PubMed] [Google Scholar]
- 22.(a) Sheldrick GM. SHELXTL Version 5 Reference Manual, Bruker AXS Inc 1994 [Google Scholar]; (b) International Tables for Crystallography. C. Kluwer; Boston: 1995. [Google Scholar]
- 23.Flack HD. On enantiomorph – polarity estimation. Acta Cryst A. 1983;39:876–881. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.