

Abstract
Purpose
Pulmonary Kv channels are thought to play a crucial role in the regulation of cell proliferation and apoptosis. Previous studies have shown that fluoxetine upregulated the expression of Kv1.5 and prevented pulmonary arterial hypertension in monocrotaline-induced or hypoxia-induced rats and mice. The current study was designed to test how fluoxetine regulates Kv1.5 channels, subsequently promoting apoptosis in human PASMCs cultured in vitro.
Materials and Methods
Human PASMCs were incubated with low-serum DMEM, ET-1, and fluoxetine with and without ET-1 separately for 72 h. Then the proliferation, apoptosis, and expression of TRPC1 and Kv1.5 were detected.
Results
In the ET-1 induced group, the upregulation of TRPC1 and down regulation of Kv1.5 enhanced proliferation and anti-apoptosis, which was reversed when treated with fluoxetine. The decreased expression of TRPC1 increased the expression of Kv1.5, subsequently inhibiting proliferation while promoting apoptosis.
Conclusion
The results from the present study suggested that fluoxetine protects against big endothelin-1 induced anti-apoptosis and rescues Kv1.5 channels in human pulmonary arterial smooth muscle cells, potentially by decreasing intracellular concentrations of Ca2+.
Keywords: Apoptosis, Kv1.5, human pulmonary arterial smooth muscle cells
INTRODUCTION
The development of pulmonary arterial hypertension (PAH) involves a complex constellation of multiple genes and molecules, which interact with each other and subsequently activate intracellular signaling pathways that eventually result in pulmonary remodeling. Vascular remodeling has been confirmed to be a hallmark pathological feature of PAH, and is characterized by changes in the pulmonary vascular structures associated with medial hypertrophy, which are mainly caused by an imbalance between the proliferation and apoptosis of pulmonary arterial smooth muscle cells (PASMCs).
Pulmonary Kv channels are thought to play a crucial role in the regulation of cell proliferation and apoptosis. K+ fluxes have been implicated in both the early and late stages of apoptosis, as the down regulation of Kv has been shown to induce increases in intracellular K+ concentrations ([K+]i) and tonically inhibit caspase, further suppressing apoptosis. As shown in human and animal models of PAH, resistance to apoptosis was further enhanced by the selective downregulation of Kv1.5 channels.1-3 However, increases in Kv channel activity and expression have been widely associated with apoptotic induction. And a previous study confirmed that the upregulation of Kv1.5 was correlated with an increase in apoptosis and inhibition of PAH.1,4
Endothelin-1 (ET-1) has been implicated in the pathogenesis of pulmonary hypertension. And, there was clear evidence of activation of the ET system in virtually all pre-clinical models of PAH, as well as in all categories of human PAH.5 Its levels have been shown to be closely correlated with the severity of pulmonary vascular remodeling.6 Interestingly, it was reported that extracellular application of ET-1 significantly reduced the amplitude of currents generated by K+ efflux through Kv1.5 channels. The inhibitory effect of ET-1 on Kv1.5 channels provided convincing evidence that the mitogenic effect of ET-1 may partially result from its inhibition of Kv1.5 channels in human PASMCs.7
Fluoxetine, the highly selective 5-hydroxytryptamine transporter (5-HTT) inhibitor, was reported to confer partial protection from PAH in chronically hypoxic mice.8 Intriguingly, fluoxetine was recently reported to prevent and reverse established PAH in monocrotaline (MCT)-induced hypertensive rats.9-11 In addition, it was also reported that the protective effect of fluoxetine against MCT-induced hypertension was potentially by upregulating Kv1.5 channels in rat. Therefore, the current study was designed to test the hypothesis that the antiproliferative and protective effects of fluoxetine are partially due to the upregulation of Kv1.5 channels and subsequent promotion of apoptosis in human PASMCs cultured in vitro.
MATERIALS AND METHODS
Cell preparation and culture
Human PASMCs from normal subjects were purchased from ATCC (Rockefeller, Mali organization, Manassas, VA, USA) and used at passages 6-8. PASMCs were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), and maintained at 37℃ in a humidified atmosphere of 5% CO2 in air. The growth of the cells was arrested by replacing 10% FBS DMEM with FBS-free DMEM for 24 hours. The cells were then incubated with low-serum DMEM (2% FBS), ET-1 (Enzo ALEXIS, ShangHai, China), and fluoxetine (F; Sigma-Aldrich, ShangHai, China) with and without ET-1 separately for 72 hours.
MTT assay
Cell proliferation was quantified by multiply-table tournament (MTT) assay. Briefly, human PASMCs were plated into 96-well microplates at the concentration of 2×103 cells/well and treated with the different drugs as described above. After incubation, 20 µL of the MTT reagent was added to each well and the multiwell plates were incubated in a humidified atmosphere for 4 hours. Then, the supernatant was removed from each well and 200 µL/well of dimethyl sulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO, USA) was added to solubilize the formed formazan salt crystals. The solubilized formazan product was spectrophotometrically quantified at 570 nm using an ELISA reader (Biorad, Hercules, CA, USA). Data were expressed as a % of the control.
Reverse transcription-polymerase chain reaction
Total RNA was isolated from human PASMCs using TRIzol reagent (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer's instructions. The specific primers were designed from the coding regions of human Kv1.5 (forward primer: 5'-TCCT CCG AGTCATCCG-3', reverse primer: 5'-ACAGCGAGCCCACGATC-3'). As a control for the integrity of RNA, the primers of glyceraldehydes phosphate dehydrogenase (GAPDH) were used (forward primer: 5'-A GGTGAAGG TCGGAG TCAAC-3' and reverse primer: 5'-CGCTCCTGGAAGATGG TGAT-3'). Amplified products were separated by 1.2% agarose gel electrophoresis and stained with ethidium bromide. PCR product bands were visualized by ultra violet light, and the intensity values were measured by densitometric analysis with the Quantity One program (Bio-Rad) and normalized to the intensity values of GAPDH for quantitative comparisons. The PCR product was sequenced and the amplified production of human Kv1.5 and GAPDH were 306 and 232 bp, respectively.
Western blotting
After 72 hours, cells were harvested in cell lysis solution (BioDev-Tech. Company, Beijing, China); then protein was extracted. The resultant protein concentrations were determined by BCA Protein Assay reagents (Beyotime Biotechnology, Jiangsu, China). The extracts were diluted in 5× loading buffer and heated at 95℃ for 5 minutes. Kv1.5 and TRPC1 proteins were detected using a standard Western blot protocol. Briefly, 30 µg proteins were separated by 8% SDS PAGE at 100 V for 0.5 hour and 80 V for 1.5 hours, and then transferred to a nitrocellulose membrane (Millipore, Billerica, MA, USA) at 4℃, 200 mA for 1 hour by a Western blot apparatus (Bio-Rad). The transferred membrane was blocked with 10% skimmed milk for 1 hour at room temperature, and then the blocked membrane was incubated with a primary antibody against Kv1.5 (dilution 1 : 700; Santa Cruz Biotechnology, Santa Cruz, CA, USA), TRPC1 (dilution 1 : 400; Santa Cruz Biotechnology) and GAPDH (dilution 1 : 700; Santa Cruz Biotechnology) overnight at 4℃, respectively. After incubation with the horseradish peroxidase-conjugated secondary antibody (dilutions of 1 : 5000; Beijing Zhong Shan-Golden Bridge Biological Technology Company, Beijing, China) for 1 hour at room temperature, the immunoblotting signals were visualized using a Western Luminescent Detection kit (Vigorous Biotechnology, Beijing, China). The results were quantified by densitometry and the density of immunoblotting was analyzed by scanning X-ray film with Quantitative One software. The values of the relative density of the Kv1.5 and TRPC1 bands were normalized to the density of GAPDH to represent the amount of Kv1.5 and TRPC1 protein. The ratio of the Blank group was regarded as 100%, and the results for the ET-1, F with or without ET-1 groups were expressed as a percentage of the value from the Blank group.
Flow cytometry
All samples were immunostained according to the protocol of the Annexin V/PI apoptosis kit. The apoptosis ratio was analyzed using flow cytometry.
Statistical analysis
All data are expressed as the mean±SEM. All experiments were performed at least with six independent Human PASMCs cultures. Statistical analyses were performed by one-way ANOVA. p-values <0.05 were considered significant.
RESULTS
Fluoxetine suppresses ET-1 induced human PASMCs proliferation
Plasma and lung ET-1 expression were increased in PAH, and correlated with disease severity, including the degree of PASMC proliferation.17 Fig. 1 shows the time course of human PASMC proliferation mediated by 2% FBS (Blank) (Fig. 1A) and ET-1 at concentrations of 0.01 to 1 µM (Fig. 1B). As shown in Fig. 1C, fluoxetine inhibited ET-1 (0.1 µM)-mediated increase of human PASMC numbers at the concentrations of 0.1 to 10 µM. Since the concentration of 1 µM fluoxetine suppressed proliferation approximately to that of seen in 2% FBS (Blank), shown in Fig. 1D, this concentration was used as the proper inhibitory dose. So, the final concentrations of ET-1 and fluoxetine were set at 0.1 µM and 1.0 µM, separately.
Fig. 1.

Fluoxetine mediated anti-proliferation of human PASMCs against ET-1. (A) Time course of the 2% FBS (Blank) and ET-1 (0.01 to 1 µM) induced human PASMCs proliferation (B). (C) The anti-proliferation of fluoxetine in a dose-dependent manner against ET-1 (0.1 µM). (D) The approximate proliferation between the 2% FBS and fluoxetine (1.0 µM) treated groups, induced by ET-1 (0.1 µM). PASMCs, pulmonary arterial smooth muscle cells; FBS, fetal bovine serum; ET-1, endothelin-1; MTT, multiply-table tournament.
Fluoxetine suppresses ET-1 induced upregulation of TRPC1 in human PASMCs
It has been reported that the activity and expression level of TRPC1 protein is directly correlated to [Ca2+]i. In the present study, fluoxetine suppressed the expression of the TRPC1 protein induced by ET-1. In Fig. 2, the expression of the TRPC1 markedly increased in the ET-1 induced group compared with the Blank (1.2448±0.2157 vs. 0.6572±0.1076, p<0.01). However, when treated with fluoxetine, it was down regulated in the ET-1 induced group (0.7904±0.1043 vs. 1.2448±0.2157, p<0.05).
Fig. 2.
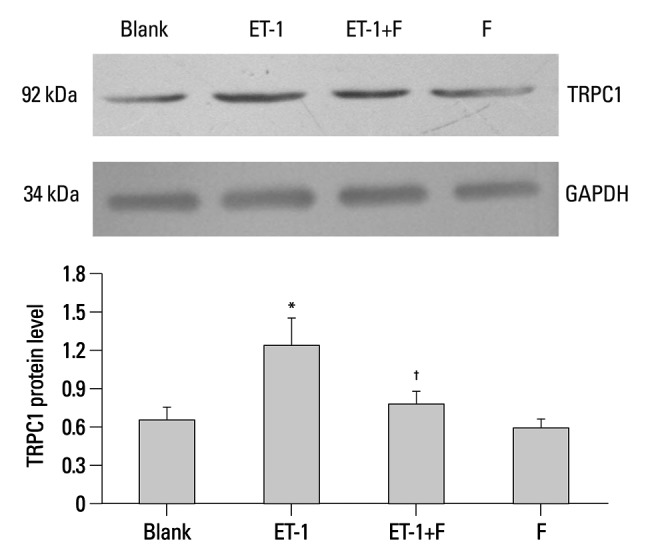
Fluoxetine down regulated the expression of TRPC1 protein in ET-1 induced human PASMCs. Western blot results are displayed for TRPC1 (92 kDa) and GAPDH (34 kDa) in the human PASMCs cultured with low-serum DMEM (2% FBS, Blank), ET-1, and fluoxetine with (ET-1+F) and without ET-1 (F) for 72 hours. Data, normalized to the amount of actin, are expressed as mean±SEM (n=7). *p<0.01 vs. Blank, †p<0.05 vs. ET-1. PASMCs, pulmonary arterial smooth muscle cells; FBS, fetal bovine serum; TRPC, transient receptor potential channels; ET-1, endothelin-1; GAPDH, glyceraldehydes phosphate dehydrogenase; SEM, standard error of mean.
Fluoxetine upregulates the expression level of Kv1.5 in ET-1 mediated human PASMCs
Pulmonary Kv1.5 channels are supposed to play a key role in the processes of proliferation and apoptosis. Results from semiquantitative RT-PCR analysis showed that the mRNA expression of Kv1.5 in ET-1 induced human PASMCs was decreased remarkably, similar to the protein expression seen with the Western blots. In Fig. 3, ET-1 significantly decreased the expression of Kv1.5 mRNA compared with the Blank (0.0303±0.0034 vs. 0.0661±0.0051, p<0.05), as well as the Kv1.5 protein (1.2198±0.1016 vs. 2.5717±0.1557, p<0.001), as seen in Fig. 4. Compared with the ET-1 induced group, the same mRNA and protein were increased remarkably in the fluoxetine treatment group (0.1648±0.0087 vs. 0.0303±0.0034, p<0.001; 2.1234±0.1766 vs. 1.2198±0.1016, separately, p<0.001). Although the Kv1.5 mRNA level was increased in the F group, compared with the Blank (0.1305±0.01478 vs. 0.0661±0.0051, p<0.01), the expression of Kv1.5 protein demonstrated no difference (2.5717±0.1557 vs. 2.2290±0.0337, p>0.05).
Fig. 3.
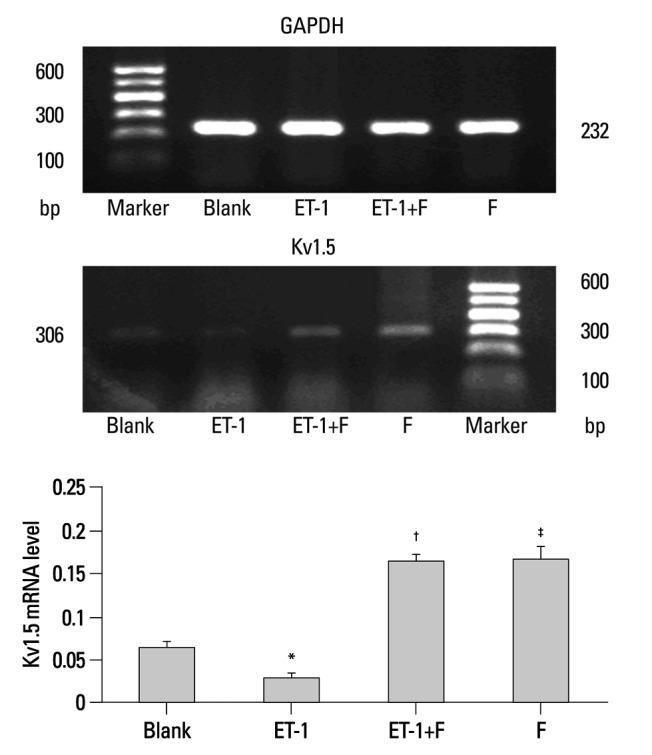
Fluoxetine rescued the expression of Kv1.5 mRNA that was down regulated by ET-1 in human PASMCs. PCR amplified products are displayed for Kv1.5 (306 bp) and GAPDH (232 bp) in the human PASMCs cultured with low-serum DMEM (2% FBS, Blank), ET-1, and fluoxetine with (ET-1+F) and without ET-1(F) for 72 hours. Data, normalized to the amount of GAPDH, are expressed as mean±SEM (n=7). *p<0.05 vs. Blank, †p<0.001 vs. ET-1, ‡p<0.001 vs. Blank. PASMCs, pulmonary arterial smooth muscle cells; ET-1, endothelin-1; GAPDH, glyceraldehydes phosphate dehydrogenase; DMEM, dulbecco's modified eagle medium; SEM, standard error of mean; PCR, polymerase chain reaction.
Fig. 4.
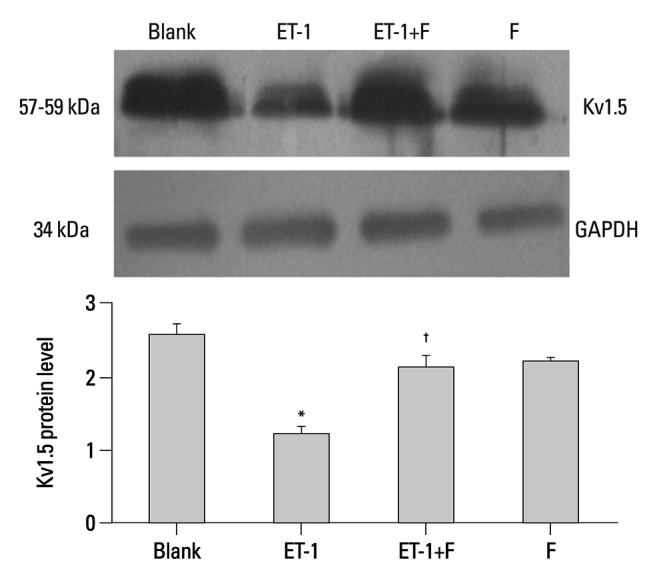
Fluoxetine reversed the level of Kv1.5 protein in ET-1 induced human PASMCs. Western blot results are displayed for Kv1.5 (57-59 kDa) and GAPDH (34 kDa) in the human PASMCs cultured with low-serum DMEM (2% FBS, Blank), ET-1, and fluoxetine with (ET-1+F) and without ET-1(F) for 72 h. Data, normalized to the amount of actin, are expressed as mean±SEM (n=9). *p<0.001 vs. Blank, †p<0.001 vs. ET-1. PASMCs, pulmonary arterial smooth muscle cells; FBS, fetal bovine serum; ET-1, endothelin-1; GAPDH, glyceraldehydes phosphate dehydrogenase; DMEM, dulbecco's modified eagle medium; SEM, standard error of mean.
Fluoxetine promotes the apoptosis ratio of ET-1 induced human PASMCs
The early stages of apoptosis in the human PASMCs treated above were detected by Flow Cytometry. Compared with the Blank, as shown in Fig. 5 and Fig. 6, ET-1 induced apoptosis inhibition was enhanced (1.1±0.1634 vs. 2.3834±0.0703, p<0.01). Apparently, the apoptosis ratio was increased in the fluoxetine treatment group in contrast with the ET-1 induced group (4.85±0.3852 vs. 1.1±0.1634, p<0.001). Also, there were no obvious changes between the Blank and fluoxetine only groups (2.2±0.1707 vs. 2.3834±0.0703, p>0.05).
Fig. 5.
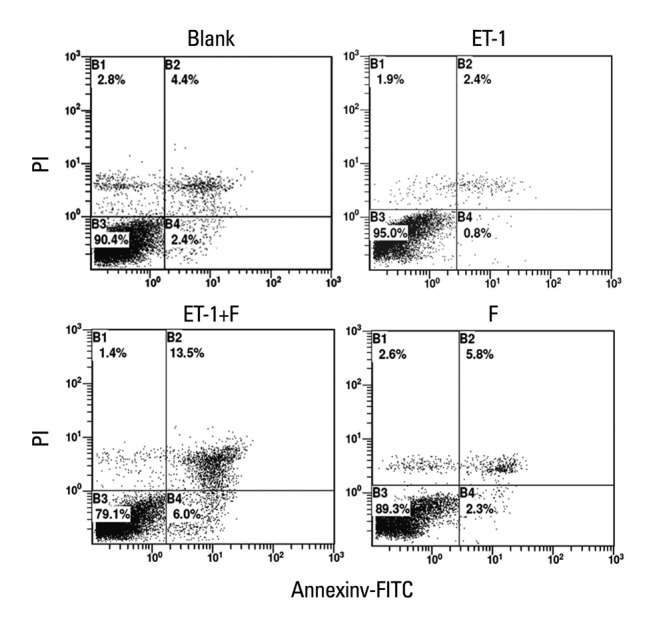
Fluoxetine enhanced the apoptosis ratio in ET-1induced human PASMCs. The result from Flow Cytometry are displayed for the ratio of apoptosis in the human PASMCs cultured with low-serum DMEM (2% FBS, Blank), ET-1, and fluoxetine with (ET-1+F) and without ET-1(F) for 72 h. PASMCs, pulmonary artery smooth muscle cells; FBS, fetal bovine serum; ET-1, endothelin-1; DMEM, dulbecco's modified eagle medium; PI, Propidium Iodide. FITC, fluoresceine isothiocyanate.
Fig. 6.

Fluoxetine enhanced the apoptosis ratio in ET-1induced human PASMCs. The result from Flow Cytometry are displayed for the ratio of apoptosis in the human PASMCs cultured with low-serum DMEM (2% FBS, Blank), ET-1, and fluoxetine with (ET-1+F) and without ET-1(F) for 72 h. Data are expressed as mean±SEM (n=6). *p<0.01 vs. Blank, †p<0.001 vs. ET-1. PASMCs, pulmonary artery smooth muscle cells; FBS, fetal bovine serum; ET-1, endothelin-1; DMEM, dulbecco's modified eagle medium; SEM, standard error of mean.
DISCUSSION
Pulmonary arterial hypertension is characterized by elevated pulmonary vascular resistance, smooth muscle remodeling and apoptosis, leading to right heart failure and death.4,12 Lumen narrowing and medial hypertrophy of small-sized pulmonary arteries are hallmarks of the pulmonary vascular remodeling process, which are mainly due to an increased number of pulmonary arterial smooth muscle cells.13,14 The imbalance between proliferation and apoptosis results in an augmentation on the number of PASMCs.4 Therefore, precise control of the balance between PASMC proliferation and apoptosis is important to maintaining the structural and functional integrity of the pulmonary vasculature. Guignabert, et al.9 confirmed the efficacy of fluoxetine in preventing and reversing pulmonary vascular remodeling in rats, which made fluoxetine a novel speculative therapeutic option for PAH. The protection of fluoxetine against PAH in MCT-induced rats was also previously demonstrated.10,11 In the present study, fluoxetine suppressed proliferation and enhanced apoptosis, reversing the imbalance between proliferation and apoptosis in human PASMCs induced by ET-1 in vitro.
Pulmonary Kv channels are thought to play a crucial role in the maintenance of resting membrane potentials, and subsequently the vascular tone of pulmonary arteries. Alterations in Kv channel function lead to several additional and interrelated consequences, including the regulation of cell proliferation and apoptosis, which ultimately leads to pulmonary vascular remodeling. It has been shown that dysfunction of Kv channels is closely linked to pulmonary vasoconstriction and pulmonary vascular remodeling in PAH.9,15 It was also becoming evident that proliferation of cultured human PASMCs was associated with membrane depolarization and down regulation of Kv currents.16 As shown in human and animal models of PAH, resistance to apoptosis was further enhanced by the selective down regulation of Kv1.5 channels.1-3 A similar phenomenon was also observed in persistent pulmonary hypertension of newborns.17 However, increases in Kv channel activity and expression has been widely associated with apoptotic induction. And a previous study confirmed that the up regulation of Kv1.5 was correlated with an increase in the apoptosis/proliferation ratio and inhibition of PAH.1,4 The available evidence presented to this point was quite strong regarding the role of Kv channels in vascular smooth muscle cell apoptosis. Conversely, there was also mounting evidence that Kv channel activation may also play a significant role in promoting proliferation.18
In the present study, the expression of the TRPC1 markedly increased in the ET-1 induced group compared with the Blank, and the expressions of Kv1.5 were decreased both in the levels of transcription and translation. Previously, reports demonstrated that [Ca2+]i inhibited K+ channels in canine pulmonary arteries,19 and it had also been reported that ET-1 induced-increases of [Ca2+]i were mainly caused by its upregulation of transient receptor potential channels (TRPC), especially TRPC1.20 And, when treated with fluoxetine, it was found that fluoxetine down regulated TRPC1 and rescued ET-1 induced Kv1.5 down-regulation in the levels of transcription and translation and promoted apoptosis in human PASMCs in vitro. The results from the present study demonstrated that the upregulation of TRPC1 down regulated the expression of Kv1.5 protein and mRNA in human PASMCs induced by ET-1, potentially by regulating the intracellular concentrations of Ca2+ in vitro. Furthermore, compared with the Blank, the apoptosis ratio of early stages was decreased, paralleling with the downregulation of Kv1.5 channels. Impressively, the human PASMCs treated with fluoxetine only changed in the mRNA expression of Kv1.5, but no changes on the expression of protein was observed, paralleling with the apoptosis ratio, compared with the Blank.
A detailed mechanism of the development of PAH is not yet known. The results from the present study showed that fluoxetine plays an important role in rescuing the expression of Kv1.5 channel in the ET-1 induced group. Potentially, the pharmacological blockade of 5-HTT may inhibit the activation of Ras/Rac system, down regulating the levels of TRPC1 and [Ca2+]i and rescue Kv1.5 channels.
In conclusion, fluoxetine plays an important role in improving pulmonary vascular remodeling, by suppressing proliferation, rescuing Kv1.5 channels and promoting apoptosis.
Footnotes
The authors have no financial conflicts of interest.
References
- 1.Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, et al. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci U S A. 2007;104:11418–11423. doi: 10.1073/pnas.0610467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Remillard CV, Tigno DD, Platoshyn O, Burg ED, Brevnova EE, Conger D, et al. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol. 2007;292:C1837–C1853. doi: 10.1152/ajpcell.00405.2006. [DOI] [PubMed] [Google Scholar]
- 3.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thébaud B, Bonnet S, et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation. 2006;113:2630–2641. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 4.McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, et al. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. 2004;95:830–840. doi: 10.1161/01.RES.0000145360.16770.9f. [DOI] [PubMed] [Google Scholar]
- 5.Michel RP, Langleben D, Dupuis J. The endothelin system in pulmonary hypertension. Can J Physiol Pharmacol. 2003;81:542–554. doi: 10.1139/y03-008. [DOI] [PubMed] [Google Scholar]
- 6.Rubens C, Ewert R, Halank M, Wensel R, Orzechowski HD, Schultheiss HP, et al. Big endothelin-1 and endothelin-1 plasma levels are correlated with the severity of primary pulmonary hypertension. Chest. 2001;120:1562–1569. doi: 10.1378/chest.120.5.1562. [DOI] [PubMed] [Google Scholar]
- 7.Shimoda LA, Sylvester JT, Booth GM, Shimoda TH, Meeker S, Undem BJ, et al. Inhibition of voltage-gated K(+) currents by endothelin-1 in human pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1115–L1122. doi: 10.1152/ajplung.2001.281.5.L1115. [DOI] [PubMed] [Google Scholar]
- 8.Marcos E, Adnot S, Pham MH, Nosjean A, Raffestin B, Hamon M, et al. Serotonin transporter inhibitors protect against hypoxic pulmonary hypertension. Am J Respir Crit Care Med. 2003;168:487–493. doi: 10.1164/rccm.200210-1212OC. [DOI] [PubMed] [Google Scholar]
- 9.Guignabert C, Raffestin B, Benferhat R, Raoul W, Zadigue P, Rideau D, et al. Serotonin transporter inhibition prevents and reverses monocrotaline-induced pulmonary hypertension in rats. Circulation. 2005;111:2812–2819. doi: 10.1161/CIRCULATIONAHA.104.524926. [DOI] [PubMed] [Google Scholar]
- 10.Zhu SP, Mao ZF, Huang J, Wang JY. Continuous fluoxetine administration prevents recurrence of pulmonary arterial hypertension and prolongs survival in rats. Clin Exp Pharmacol Physiol. 2009;36:e1–e5. doi: 10.1111/j.1440-1681.2009.05181.x. [DOI] [PubMed] [Google Scholar]
- 11.Zhai FG, Zhang XH, Wang HL. Fluoxetine protects against monocrotaline-induced pulmonary arterial hypertension: potential roles of induction of apoptosis and upregulation of Kv1.5 channels in rats. Clin Exp Pharmacol Physiol. 2009;36:850–856. doi: 10.1111/j.1440-1681.2009.05168.x. [DOI] [PubMed] [Google Scholar]
- 12.Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–1665. doi: 10.1056/NEJMra035488. [DOI] [PubMed] [Google Scholar]
- 13.Fantozzi I, Platoshyn O, Wong AH, Zhang S, Remillard CV, Furtado MR, et al. Bone morphogenetic protein-2 upregulates expression and function of voltage-gated K+ channels in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;291:L993–L1004. doi: 10.1152/ajplung.00191.2005. [DOI] [PubMed] [Google Scholar]
- 14.Krick S, Platoshyn O, McDaniel SS, Rubin LJ, Yuan JX. Augmented K(+) currents and mitochondrial membrane depolarization in pulmonary artery myocyte apoptosis. Am J Physiol Lung Cell Mol Physiol. 2001;281:L887–L894. doi: 10.1152/ajplung.2001.281.4.L887. [DOI] [PubMed] [Google Scholar]
- 15.Burg ED, Remillard CV, Yuan JX. Potassium channels in the regulation of pulmonary artery smooth muscle cell proliferation and apoptosis: pharmacotherapeutic implications. Br J Pharmacol. 2008;153(Suppl 1):S99–S111. doi: 10.1038/sj.bjp.0707635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Platoshyn O, Golovina VA, Bailey CL, Limsuwan A, Krick S, Juhaszova M, et al. Sustained membrane depolarization and pulmonary artery smooth muscle cell proliferation. Am J Physiol Cell Physiol. 2000;279:C1540–C1549. doi: 10.1152/ajpcell.2000.279.5.C1540. [DOI] [PubMed] [Google Scholar]
- 17.Konduri GG, Bakhutashvili I, Eis A, Gauthier KM. Impaired voltage gated potassium channel responses in a fetal lamb model of persistent pulmonary hypertension of the newborn. Pediatr Res. 2009;66:289–294. doi: 10.1203/PDR.0b013e3181b1bc89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neylon CB. Potassium channels and vascular proliferation. Vascul Pharmacol. 2002;38:35–41. doi: 10.1016/s1537-1891(02)00124-6. [DOI] [PubMed] [Google Scholar]
- 19.Post JM, Gelband CH, Hume JR. [Ca2+]i inhibition of K+ channels in canine pulmonary artery. Novel mechanism for hypoxia-induced membrane depolarization. Circ Res. 1995;77:131–139. doi: 10.1161/01.res.77.1.131. [DOI] [PubMed] [Google Scholar]
- 20.Wang C, Wang J, Zhao L, Wang Y, Liu J, Shi L, et al. Sildenafil inhibits human pulmonary artery smooth muscle cell proliferation by decreasing capacitative Ca2+ entry. J Pharmacol Sci. 2008;108:71–78. doi: 10.1254/jphs.08069fp. [DOI] [PubMed] [Google Scholar]