

SUMMARY
The T cell receptor (TCR) and CD8 bind peptide-major histocompatibility complex (pMHC) glycoproteins to initiate adaptive immune responses, yet the trimolecular binding kinetics at the T cell membrane is unknown. Using a micropipette adhesion frequency assay, we show that this kinetic has two stages. The first consists of TCR-dominant binding to agonist pMHC. This triggers a second stage consisting of a step increase in adhesion following a one second delay. The second-stage binding requires Src family kinase activity to initiate CD8 binding to the same pMHC engaged by the TCR. This induced-trimeric-cooperative interaction enhances adhesion synergistically to favor potent ligands, which further amplifies discrimination. Our data reveal a TCR-CD8 positive feedback loop involved in initial signaling steps that is sensitive to a single pMHC, is rapid, reversible, synergistic, and peptide-discriminative.
INTRODUCTION
Antigen recognition by the T cell is central to its development and activation. Together with its coreceptor, the T cell receptor (TCR) discriminates various peptide-Major Histocompatibility Complex (pMHC) ligands, e.g., peptides that differ by even a single amino acid, to differentially trigger intracellular signals, leading to a wide range of T cell responses (Davis et al., 2007; Evavold and Allen, 1991; Evavold et al., 1993). Related to this specificity is an exquisite sensitivity; the T cells are capable of detecting a single agonist pMHC to initiate transient calcium fluxes (Irvine et al., 2002; Purbhoo et al., 2004). Furthermore, these responses are remarkably fast, as TCR microclusters (Campi et al., 2005; Yokosuka et al., 2005), intracellular calcium fluxes and phophorylation of linker for activation of T cells (LAT) are observed within a few seconds upon TCR contact with pMHC (Huse et al., 2007).
The coreceptor on cytotoxic T cells is CD8, which binds the α3 conservative domain of the MHC without peptide contact. This binding property is distinct from the TCR which binds the α1 and α2 domains as well as the peptide (Gao et al., 1997; Kern et al., 1998). CD8 has much lower affinity than TCR for agonist pMHC (Garcia et al., 1996; Wyer et al., 1999), suggesting that CD8 binding mostly helps weak ligands that have low TCR affinities (Laugel et al., 2007). CD8 associates with the Src family kinase p56lck (Lck) (Davis et al., 2003; Palacios and Weiss, 2004) and its absence impairs T-cell responses (Delon et al., 1998; Xu et al., 2001). However, it remains unclear whether and, if so, how CD8 facilitates peptide discrimination.
It is also unclear how pMHC binds the TCR and CD8, e.g., concurrently, sequentially, independently, or cooperatively. It has been proposed that binding of one receptor (CD8 or TCR) holds the ligand (MHC) to an optimal configuration, thereby accelerating the association of the other receptor (TCR or CD8) to MHC (Gakamsky et al., 2005; Gao et al., 2002; Pecht and Gakamsky, 2005). Alternatively, co-engagement of both TCR and CD8 with pMHC may stabilize the trimolecular bond, thereby decelerating the dissociation of either or both receptors (Campanelli et al., 2002; Garcia et al., 1996; Norment et al., 1988). A surface plasmon resonance study found that CD8 enhances the TCR-pMHC interaction by reducing the off-rate (Garcia et al., 1996). However, another study found that CD8 and TCR binds pMHC independently and the TCR-pMHC interaction is unaffected by the presence of CD8 (Wyer et al., 1999). Results from studies using pMHC tetramers indicate that CD8 plays a direct or indirect role in pMHC binding (Daniels and Jameson, 2000; Wooldridge et al., 2003). However, a major limitation of these studies is that they measure binding of soluble molecules in a fluid phase, i.e., three-dimensional (3D) binding. In reality, pMHC on an antigen presenting cell (APC) binds the TCR and/or CD8 on a T cell at the cross-junctional interface, i.e., two-dimensional (2D) binding, which may be impacted by the cellular environment (Huang et al., 2007; Huang et al., 2010; Huppa et al., 2010).
We recently used mechanically-based 2D assays with single-bond sensitivity and subsecond temporal resolution to measure the bimolecular interactions of a panel of pMHC ligands with the CD8 (Huang et al., 2007) or TCR (Huang et al., 2010) on the T cell membrane. Here we extend these studies to situations that allow TCR-pMHC-CD8 trimolecular interactions and show them to be cooperative upon induction. This induced TCR-CD8 cooperation enhanced adhesion synergistically to favor potent ligands, which amplified discrimination.
RESULTS
To measure 2D interactions, a CD8+ T cell (Fig. 1A, left) expressing either the OT1 or F5 TCR (Fig. 1B, left) was aspirated by a micropipette and moved in and out of contact with a red blood cell (RBC, Fig. 1A, right) bearing pMHC (Fig. 1B, right) held stationary by an apposing micropipette with a controlled duration and area. The RBC served as not only a surrogate APC but also an adhesion sensor as its membrane would be stretched by (a) molecular bond(s) on T cell retraction (Movie S1). Each contact resulted in a binary adhesion score (0 or 1) and its average over 50 contacts of the same duration t gives an adhesion frequency Pa, which is specific (Fig. 1C).
Figure 1. Micropipette adhesion frequency assay.
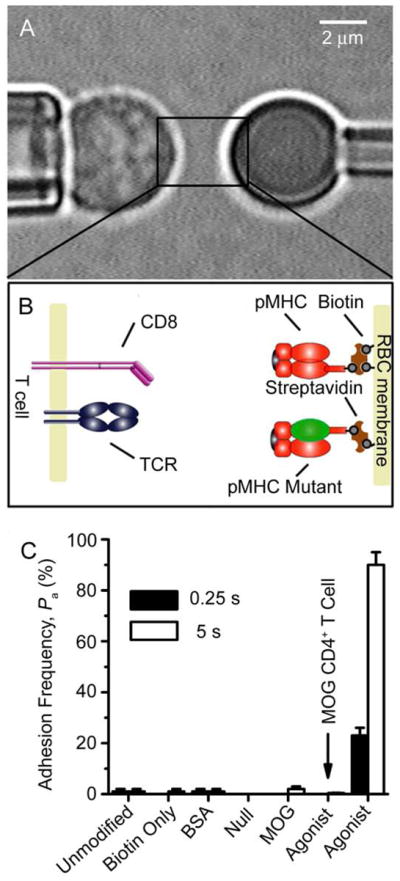
A, Micrograph of the micropipette assay. A T cell (left) was aspirated by a pipette and aligned with a pMHC-coated RBC held stationary by another pipette (right) (see Movie S1). B, Schematics of TCR and CD8 expressed on a T cell (left) and of pMHC coated on a RBC via biotin-streptavidin coupling (right). C, Specificity controls at contact duration of 0.25 s (solid bars) or 5 s (open bars) of adhesion frequencies between OT1 T cells and unmodified RBCs, biotinylated RBCs without coating, biotinylated RBCs coated with BSA, null pMHC-I (VSV:H-2Kb), pMHC-II (MOG:I-Ab) or agonist pMHC-I (OVA:H-2Kb), or between MOG CD4+ T cells and biotinylated RBCs coated with OVA:H-2Kb. Each T cell-RBC pair was tested repeatedly for 50 contact-retract cycles at a given contact duration to estimate an adhesion frequency, and 3-5 cell pairs were tested for each t to calculate a mean Pa ± s.e.m. See also Movie S1.
TCR-pMHC-CD8 trimolecular interaction proceeds in two stages
Plots of Pa vs. t exhibited two stages, starting with a low plateau and jumping to a high plateau after ~1 s with a <0.1 s transient phase before each equilibrium (Fig. 2A, B, ■), indicating rapid kinetics for both stages (Chesla et al., 1998). This two-stage kinetic, observed for both OT1 (Fig. 2A, ■) and F5 (Fig. 2B, ■) T cells interacting with RBCs bearing the appropriate agonistat both 25°C and 37°C (Fig. 2C, D), was distinct from the single-stage kinetic previously observed for the TCR-pMHC (Huang et al., 2010) and pMHC-CD8 (Huang et al., 2007) bimolecular interactions. We used selective inhibition to dissect the respective contributions of the TCR and CD8 to the two-stage curve. Anti-TCR completely blocked adhesion (Fig. 2A, B, △). This TCR requirement was consistent with the low CD8 effective 2D affinities (~10−6 and <10−8 μm4, for H-2Kb and H-2Db, respectively) (Huang et al., 2007). Because the low MHC densities (30 H-2Kb/μm2 and 14 H-2Db/μm2) used predicted 2% and <0.0074% adhesion frequencies contributed by the CD8 that was expressed at densities of at 250 and 426 μm−2 on OT1 and F5 T cells, respectively (Methods, Eqs. 1 and 2). By comparison, the TCR effective 2D affinities were 0.5 and 2.8 × 10−4 μm4 for ovalbumin (OVA):H-2Kb and influenza virus nucleoprotein (NP68):H-2Db, respectively, as calculated from the first plateau Pa and TCR densities (210 and 62 μm−2 on OT1 and F5 T cells). These results suggest that the first-stage curve is dominated by TCR-pMHC binding with negligible pMHC-CD8 contribution.
Figure 2. Two-stage kinetics of TCR-pMHC-CD8 trimolecular interaction.

A, B, Pa vs. t T1 (A) or F5 (B) T cells interacting with RBCs bearing 25 OVA:H-2Kb/μm2 (A) or 14 NP68:H-2Db/μm2 (B) in the absence (■) or presence of anti-CD8 (○) or anti-TCR (△) reagents. Also included in A are data for OT1 T cells interacting with RBCs bearing 25 OVA:H-2Kbα3A2/μm2 (◇). Curves are trend lines. C, D, Pa vs. t data of OT1 (C) or F5 (D) T cells interacting with RBCs bearing 12 OVA:H-2Kb/μm2 or 14 NP68:H-2Db/μm2, respectively, at 25 (□) or 37 (○) °C. Representative data (measured by the same method as that in Fig. 1) of three repeated experiments are shown. See also Figure S1.
By comparison, the CD8 blocking mAb CT-CD8a eliminated the adhesion increment in the second stage but did not affect the first stage (Fig. 2A, B, ○). This resulted in single-stage curves similar to that obtained using an MHC mutant. This mutant substituted the α3 domain in the wild-type mouse H-2Kb with the α3 domain of human HLA-A2 (H-2Kbα3A2) to abrogate the binding of mouse CD8 (Fig. 2A, ⋄) (Huang et al., 2010). Using CT-CD8a Fab produced the same effect as whole antibody (Fig. S1), excluding crosslinking as the cause of inhibition. Although these data demonstrate the requirement of CD8 for the second-stage adhesion increment, the complete abrogation of adhesion by the anti-TCR excludes the independent concurrent binding model (Zhu and Williams, 2000) of generating the second stage by simply adding pMHC-CD8 bonds to the same TCR-pMHC bonds as in the first stage (Methods, Eq. 3). Furthermore, to attribute the second-stage adhesion increment to pMHC-CD8 binding alone would require ~40- and >10,000-fold higher CD8 affinities for H-2Kb and H-2Db, respectively, than those measured in the absence of TCR binding (Huang et al., 2007). Nevertheless, the increased contribution to the second stage identified a CD8-dependent upregulated functional state. Since TCR binding was still required for the second stage, the upregulated adhesion must be induced by TCR engagement and may involve cooperation between the TCR and CD8 for pMHC binding.
The upregulated adhesion requires signaling
The distinctive shape, i.e., a rapid transition from one equilibrium to another after a 1-s delay, suggests that the two-stage curve may not be governed solely by the reaction kinetics of the TCR, CD8, and pMHC molecular triad. It may involve other molecules, e.g., elements of the signaling cascade whose (de)phosphorylation may take up some time, giving rise to a delay from the first to the second stage. We therefore employed targeted inhibition of key T cell signaling molecules to test whether they were required for the upregulated adhesion. Treating T cells with a Src tyrosine kinase inhibitor PP2 (Hanke et al., 1996) resulted in a single-stage pattern (Fig. 3A), suggesting a role for Lck, a Src family protein tyrosine that associates with CD8 (Davis et al., 2003; Palacios and Weiss, 2004). As a further confirmation, we inhibited the protein tyrosine phosphatase CD45, which activates Lck by dephosphorylating its inhibitory pY505 (Weiss and Littman, 1994; Zamoyska, 2007). Inhibition of CD45 also abolished the upregulated adhesion (Fig. 3B). In contrast, two-stage kinetics were unaffected by treatment of T cells with a Ca2+ chelator BAPTA-AM (Fig. 3C), an inhibitor of MAP kinase kinase-1 (Fig. 3D), and two inhibitors of PI3 kinase (Fig. S2), which represent signaling events downstream of Lck activity (Dong et al., 2002; Kane and Weiss, 2003; Wulfing et al., 1997).
Figure 3. TCR-induced CD8-dependent increased adhesion can be differentially inhibited.
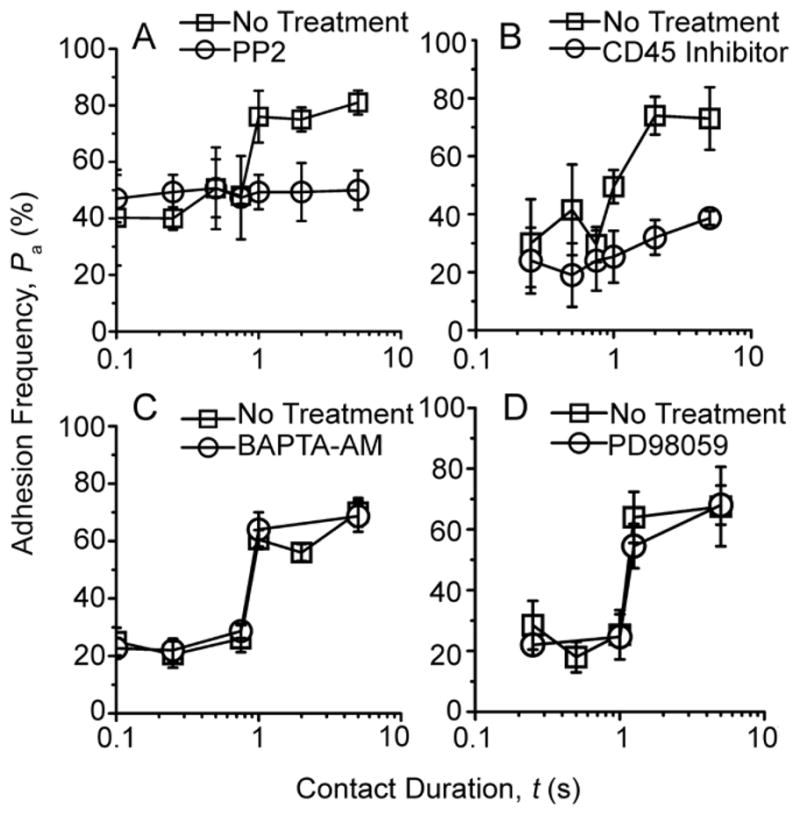
Pa vs. t data (measured by the same method as that in Fig. 2) of OT1 (A, B) or F5 (C, D) T cells interacting with pMHC-coated RBCs in the absence (□) and presence (○) of PP2 (A, 3.5 OVA:H-2Kb/μm2), inhibitor for protein tyrosine phosphatase CD45 (B, 14 OVA:H-2Kb/μm2), Ca2+ chelator BAPTA-AM (C, 32 NP68:H-2Db/μm2), or MAP kinase kinase-1 inhibitor PD98059 (D, 32 NP68:H-2Db/μm2). DMSO treatment alone did not inhibit the second-stage adhesion increment. See also Figure S2.
Thus, the TCR first binds agonist pMHC to trigger proximal signaling, which induces a CD8-dependent adhesion upregulation after a 1-s delay. This signaling process is downstream of Lck but upstream of Ca2+ fluxes. Since Ca2+ fluxes can be triggered by the TCR-pMHC interaction itself and are further enhanced by the CD8-dependent binding (Delon et al., 1998). Thus our data reveal a TCR-CD8 positive feedback loop involved in the initial steps of the signaling network.
Upregulation of adhesion by an anti-CD8
Some anti-CD8 reagents have been reported to enhance T cell binding to pMHC (Daniels and Jameson, 2000; Devine et al., 2004; Luescher et al., 1995). Incubating OT1 T cells with one such monoclonal antibody (mAb), 53-6.7 increased their adhesion to RBCs bearing OVA:H-2Kb to the higher second-stage level at the shortest contact time tested (Fig. 4A, compare □ and ○). Using OVA:H-2Kbα3A2 to prevent CD8 binding eliminated the 53-6.7 mAb-enhanced adhesion, yielding a lower first-stage level (Fig. 4B, △) identical to that obtained without 53-6.7 (Fig. 4B, ○). A Fab fragment of 53-6.7 had similar effect as the whole antibody (compare Fig. 4A and D, ○), thus excluding crosslinking as the cause of enhancement. TCR binding was also important as blocking with an anti-TCR (Fig. 4A, △), replacing H-2Kb by H-2Db (Fig. 4C), or using a CD8+-TCR− hybridoma (Yachi et al., 2006) (Fig. S3A) substantially reduced the adhesion enhanced by 53-6.7. Interestingly, inhibition of Lck with PP2 abrogated the increased adhesion by 53-6.7 (Fig. 4D). The role for Lck was further supported by the finding that the Fab fragment of 53-6.7, but not of CT-CD8a, increased Lck activation (Fig. S3B), consistent with previous reports that some anti-CD8 reagents generate intracellular activation signals resulting in cytotoxic T cell effector function (Tomonari and Spencer, 1990; Wooldridge et al., 2003). Together, these data suggest that the antibody, 53-6.7, also induced upregulated pMHC binding by the CD8 and TCR, which may share some mechanistic elements with the second-stage increased adhesion shown in the preceding section.
Figure 4. Effect of anti-CD8 clone 53-6.7 on CD8 adhesion and its inhibition.

Pa vs. t data (measured by the same method as that in Fig. 2) of OT1 T cells interacting with RBCs bearing indicated densities of OVA:H-2Kb (A, D), OVA:H-2Kbα3A2 (B), or GP33:H-2Db (C) in the absence (□) and presence (○, △) of the indicated mAb(s) or inhibitor. Note that the one-order-of-magnitude higher pMHC density used in panel C than in other panels translates to an order of magnitude lower binding (average number of bonds formed per pMHC density) between OT1 T cells and RBCs bearing H-2Db than H-2Kb (Methods, Eq. 3). See also Figure S3.
TCR engagement with a single pMHC is sufficient to induce cooperation with CD8
The requirement of direct pMHC binding by both TCR and CD8 suggests a mechanism for the upregulated second-stage adhesion, which might involve signaling-induced cooperation between the TCR and CD8. To test this hypothesis and to assess the sensitivity of signaling, we took advantage of the single-bond sensitivity of the micropipette assay (Chesla et al., 1998; Zhu et al., 2002) by limiting the ligand density to prevent TCR and CD8 from binding different pMHC molecules. The two plateau levels changed with the changing pMHC density as expected from mass action (Fig. S4). But the qualitative patterns remained even when the density was reduced to 3 pMHC/μm2 (Fig. 5A, B), which predicts a 0.58-μm average distance between neighboring pMHCs. To achieve this low density, D-biotin was used to block excess biotin binding-sites on streptavidin bound to RBCs, which minimized the chance of forming dimeric pMHC by capturing two pMHC monomers onto the same streptavidin. The resulting first-stage adhesion frequencies were only 6% (Fig. 5A, △) or 2% (Fig. 5B, △), so that 97% (for OT1) or 99% (for F5) of them were single-bond adhesions as predicted by Poisson distribution (Chesla et al., 1998). Yet, the second-stage adhesion was clearly evident, reaching 31% (for OT1) or 16% (for F5) (Fig. 5A, B, △). Therefore, a single TCR-pMHC bond is sufficient to induce the upregulated functional state.
Figure 5. TCR engagement with a single pMHC is sufficient to induce cooperation with CD8.

A, B, Pa vs. t data of OT1 (A) or F5 (B) T cells interacting with RBCs bearing indicated densities of OVA:H-2Kb or NP68:H-2Db. Curves are trend lines. C, Pa vs. t data of OT1 T cells interacting with RBCs bearing a mixture of OVA:H-2Kbα3A2 and VSV:H-2Kb at indicated ratios but the same total density (70 sites/μm2). Adhesion frequencies were measured by the same method as that in Fig. 2. See also Figure S4.
The limited availability of pMHC favors CD8 binding to the pMHC molecules that also interacted with TCR, suggesting trimeric cooperative interaction as a possible mechanism for the upregulated functional state. To further test this hypothesis, we prevented the TCR and CD8 from binding the same pMHC by coating RBCs with a mixture of OVA:H-2Kbα3A2 and vesicular stomatitis virus (VSV):H-2Kb at various ratios. The former allowed TCR recognition but not CD8 binding whereas the latter allowed CD8 binding but not TCR recognition. The control with 100% of OVA:H-2Kbα3A2 showed a single-stage curve (Fig. 5C, □) similar to that shown previously (Fig. 2A, ⋄). 100% of VSV:H-2Kb abolished binding (Fig. 5C, △), confirming the requirement for TCR. Importantly, the 5:95 ratio also produced only a single-stage curve (Fig. 5C, ○). The lack of the second stage was not due to restricted TCR binding, as the first-stage binding was 20% in adhesion frequency. Nor was it due to the lack of CD8 binding-sites, as there were 67 VSV:H-2Kb/μm2 on the RBCs. Thus, our results indicate it is necessary for the TCR and CD8 to bind the same pMHC to initiate the upregulated second-stage adhesion.
The TCR-CD8 positive signaling feedback loop is rapidly reversible
It has recently been reported that TCR signaling could be triggered by applied forces (Kim et al., 2009; Li et al., 2010). To exclude the possibility that pulling on the TCR in repeated contacts caused the two-stage binding, a “first-contact” experiment was performed in which each cell pair was contacted only once. The adhesion frequency was calculated by dividing the number of adherent cell pairs by 50 cell pairs tested at each contact duration. Values at five contact durations were measured using a total of 250 cell pairs. The result was a two-stage binding curve indistinguishable from that obtained using 50 repeated contacts per cell pair and 3–4 cell pairs at each contact duration (Fig. 6A), ruling out the possibility that repeated-contacts caused two-stage kinetics by mechanical stimulation. The data also indicated that the TCR signal required for the second-stage increased binding was turned off quickly, as it did not accumulate in the repeated contacts to yield higher adhesion frequencies than those obtained by single contacts.
Figure 6. Second-stage binding does not need accumulation of TCR-CD8-pMHC trimolecular interaction and is reversible.

A, For each contact duration, 50 pairs of F5 T cells and NP68:H-2Db-coated RBCs (32 μm−2) were each contacted once to estimate Pa from percent adherent pairs (□) to compare with mean Pa ± s.e.m. measured using 3-4 pairs of cells from the same batch each repeatedly contacted 50 times (○). B, F5 T cells and NP68:H-2Db-coated RBCs (32 μm−2) were repeatedly contacted 50 times first to measure Pa at a long duration (≥1 s, indicated by different symbols) and then another 50 times to measure anther Pa at a short duration (≤0.75 s, indicated by matched symbols) (□) to compare with Pa measured using cells from the same batch but repeatedly contacted 50 times at only one duration per pair (○). Data (mean ± s.e.m. of 3-4 cell pairs) at the first plateau overlap. C, Three pairs of F5 T cells and NP68:H-2Db-coated RBCs (23 μm−2) were each contacted 100 times with durations alternating between 0.25 and 5 s to measure two mean Pa ± s.e.m. (one at each contact duration) by dividing the number of adhesions resulting from contacts of the same duration by 50 (solid bars), which were significantly different (p = 0.0056, Student t-test). However, no statistical differences (p > 0.25) were found for both 0.25 and 5 s groups between these Pa values and those measured using 10 pairs of cells from the same batch each consecutively contacted 50 times (open bars). See also Movie S2.
We performed two independent experiments to examine how rapid this activation signal is regulated and how long it sustains. In the “long-then-short” experiment, a cell pair was first repeatedly contacted for a long (>1 s) duration 50 times to estimate one adhesion frequency and then repeatedly contacted for a short (<1 s) duration another 50 times to estimate a second adhesion frequency. Three pairs of long and short durations were chosen to cover the entire range, resulting in the same two-stage curve indistinguishable from that obtained using different cell pairs for different contact durations (Fig. 6B). This suggests the signal triggered by TCR is rapidly reversible and short-lived.
In the “alternating” experiment, we programmed the adhesion assay by alternating the contact duration between 0.25 and 5 s in 100 repeated contacts (Movie S2). Each test included a 1.5-s cycling time between the instant when the T cell was retracted away from the RBC to the instant when it was brought back to contact the RBC again. Adhesions generated by contacts of the same duration (0.25 or 5 s) were divided by 50 to calculate an adhesion frequency for that contact duration. The two adhesion frequencies so measured (Fig. 6C, solid bars) were significantly different. However, they were indistinguishable from the respective adhesion frequencies obtained from 50 consecutive contacts of 0.25 and 5 s (Fig. 6C, open bars) for both the 0.25-s and 5-s groups. This result indicates that the activation signal generated in the 5-s contact was rapidly turned off within the 1.5-s cycling time. The activation signal was not turned on in the next 0.25-s contact because this duration was too short, but it was turned on again when the contact duration was switched back to 5 s. This rapid reversibility also suggests that the second-stage binding is not caused by upregulated expression of CD8 as surface expression is unlikely to be turned over so fast. The 1-s delay of outset of the second-stage adhesion enhancement and its elimination by a 1.5-s contact time gap reveal the speed, duration, and timing of the signal generation and regulation.
Trimolecular binding is synergistic
Since synergy is a hallmark of cooperativity, we examined whether TCR-pMHC-CD8 trimolecular interactions generate synergy over the simple sum of the TCR-pMHC and pMHC-CD8 bimolecular interactions. Because a T cell may adhere to a pMHC-coated RBC via TCR-pMHC, pMHC-CD8, and/or TCR-pMHC-CD8 bonds or their combinations, we calculated the average number of total bonds <n> formed between an OT1 T cell and a pMHC-coated RBC from the adhesion frequency and divided it by the ligand density ml to obtain normalized adhesion bonds <n>/ml (Methods, Eq. 4). In our previous experiments where T-cell-RBC adhesion was mediated by TCR-pMHC or CD8-pMHC bimolecular interactions only (Huang et al., 2007; Huang et al., 2010), normalized adhesion bonds equaled the effective 2D affinity multiplied by the receptor (TCR or CD8) density (Methods, Eq. 2). However, a single affinity is insufficient in the present case because there are potentially three different types of bonds. Nevertheless, using normalized adhesion bonds allowed us to compare the propensities of the TCR-/pMHC, pMHC-CD8, and TCR-pMHC-CD8 interactions for a panel of pMHCs.
For pMHC-CD8 biomolecular interactions (obtained using TCR blockade), similar curves of normalized adhesion bonds were observed in spite of the variable peptide potencies (Fig. 7A–F, △), consistent with our previous report (Huang et al., 2007). For TCR-pMHC biomolecular interactions (obtained using H-2Kbα3A2 to abrogate CD8 binding), widely ranged curves of normalized adhesion bonds were observed that matched the peptide potencies (Fig. 7A–F, ○), also consistent with our previous report (Huang et al., 2010). Adding these two data sets for each peptide predicts the normalized adhesion bonds that would have been formed between OT1 T cells and RBCs bearing wild-type pMHC without TCR blocking. This assumes that the two TCR-pMHC and pMHC-CD8 bimolecular interactions would have occurred concurrently and independently, i.e., without cooperation (Method, Eq. 3). However, much higher normalized adhesion bonds were formed between the OT1 T cells and pMHC-coated RBCs than this simple sum (Fig. 7B–F and Fig. S5, comparing □ and dashed curves), demonstrating synergy between the TCR and CD8 for pMHC binding. Although synergy was observed for all peptides tested, the resulting normalized adhesion bonds were much higher for strong ligands than weak ligands. To compensate for the low TCR-pMHC-CD8 binding propensities of the weak ligands, substantially higher ligand densities were used to engage the TCR, which likely increased the chance for the CD8 to bind pMHC cooperatively with the TCR. Therefore, TCR induced CD8 cooperation generated synergy over the simple sum of the TCR-pMHC and pMHC-CD8 bimolecular interactions.
Figure 7. Synergy between TCR and CD8 amplifies T cell discrimination.

A–F, Pa vs. t data (measured by the same methods as that in Fig. 2) for OT1 T cells interacting with RBCs bearing H-2Kb (□) or H-2Kbα3A2 (○) complexed with OVA (A), A2 (B), G4 (C), E1 (D), V-OVA (E), and R4 (F). Also presented are data measured using RBCs bearing H-2Kb in the presence of the anti-TCR reagent B20.1 (△). Pa was converted to normalized adhesion bonds using Eq. 4 (Methods) and presented as mean ± s.e.m. Dashed curves represent trendlines to the sum of TCR-pMHC (○) and pMHC-CD8 (△) data. G, Steady-state (plateau values at 5 s) of the differential normalized adhesion bonds, △(<n>/mpMHC), formed between OT1 T cells and pMHC-coated RBCs minus the sum of those formed by the two bimolecular interactions for a panel of pMHC ligands with increasing potencies. H, The differential reciprocal concentrations required to reach half-maximal T cell proliferation △(1/EC50), measured in the absence of blocking antibody minus that measured in the presence of Fab of anti-CD8 mAb CT-CD8a, is plotted vs. △(<n>/mpMHC). See also Figure S5.
TRC-CD8 cooperation amplifies peptide discrimination
To determine the biological relevance of the TCR-CD8 positive feedback loop, we examined whether the increased propensities of TCR-pMHC-CD8 trimolecular interactions for a panel of peptides correspond to the peptide potencies of triggering T cell responses. The degree of synergy was quantified by the differential normalized adhesion bonds actually formed between the T cells and RBCs over the sum of those formed by the TCR-pMHC and pMHC-CD8 bimolecular interactions, Δ<n>αmpMHCα (Methods, Eqs. 3 and 4). Importantly, the level of synergy increased with the peptide potency (Fig. 7G). To further define the physiological significance of the TCR-CD8 cooperation, we quantified the differential functional response due to this cooperation. To do this we measured the reciprocal peptide concentration required to induce half maximal T cell proliferation, 1/EC50, without and with CD8 blocking (by CT-CD8a Fab) and calculated the difference, Δ(1/EC50), due to signaling-induced cooperation between TCR and CD8 for pMHC binding. Remarkably, a strong correlation was observed in the Δ (1/EC50) vs. Δ(<n>/mpMHC) plot (Fig. 7H), suggesting that the induced TCR-CD8 cooperation amplifies peptide discrimination.
DISCUSSION
Effective cytotoxic T cell activation requires engagement of the TCR and contribution of the CD8 coreceptor. Using the micropipette 2D assay to directly measure T cell adhesion to pMHC, we uncovered a two-stage kinetics of a signaling-induced cooperation between the TCR and CD8 to enhance binding of agonist pMHC, which is sensitive to a single ligand. This interaction was found to be very fast, short-lived, reversible, synergistic, and peptide-discriminative.
Without cooperation, TCR and CD8 should have interacted with pMHC concurrently but independently, generating a single-stage adhesion curve from the addition of two types of bimolecular bonds (Methods, Eq. 3) (Zhu and Williams, 2000). This appeared to be the case for the first stage of the two-stage curve where the TCR interaction with agonist pMHC (Huang et al., 2010) dominated due to its much higher affinity than the pMHC-CD8 interaction (Huang et al., 2007). However, this was not the case for the second-stage adhesion because many more bonds were formed than the simple sum of TCR-pMHC plus pMHC-CD8 bonds, thus revealing cooperative binding that required both the TCR and CD8.
Cooperative binding could result in either enhanced biomolecular interactions of TCR-pMHC and/or pMHC-CD8 or trimolecular complex with co-engagement of TCR and CD8 for the same pMHC. To distinguish these scenarios we prevented TCR and CD8 from binding different pMHCs by limiting their density. Two-stage binding was still observed, demonstrating that binding of TCR and CD8 to the same pMHC was sufficient to generate cooperation. Using mixed ratios of agonist pMHC encoding a mutated CD8 binding-site and null pMHC unrecognized by TCR to force TCR and CD8 to bind distinct pMHCs eliminated cooperation. This result suggests a 1:1:1 stoichiometry of functional binding among TCR, CD8 and MHC, and indicates a spatial relationship between TCR and CD8 required for binding to the same pMHC (Gao et al., 1997). The cooperation also raises the possibility that the co-receptor may directly associate with the TCR although structural modeling does not seem to support this hypothesis (Gao et al., 2002; Rudolph et al., 2006). Another possibility is that the TCR and CD8 may be indirectly associated via Lck and other signaling molecules.
Unlike classical cooperative binding, the cooperation between TCR and CD8 for binding of agonist pMHC is induced. Initial evidence for induction came from the two-stage curve, where transition between the first and second stage did not occur until after a 1-s delay. This was not due to slow pMHC binding kinetics for TCR and/or CD8 because both the TCR-dominant first stage and the CD8-dependent second stage rapidly reached equilibrium. Rather, the 1-s period may be required for intracellular signals triggered by TCR engagement to enable TCR and CD8 cooperation. The finding that a single TCR-pMHC bond at the first stage is sufficient to induce the increased binding in the second stage highlights the importance of the induced cooperation since T-cell responses need to function optimally at low antigen density.
As more definitive evidence, Lck activity was necessary for the second-stage CD8-dependent adhesion increase, yet its inhibition did not affect the first-stage TCR-pMHC binding. To further support the role of Lck, functional CD45, necessary for dephosphorylating pY505 to activate Lck, was also required for the signaling-induced cooperation between TCR and CD8 for pMHC binding. Lck may act via its interactions with immunoreceptor tyrosine-based activation motifs (ITAMs) of CD3. The initial TCR-pMHC binding may expose some of the CD3 ITAMs buried in the membrane (Xu et al., 2008) to allow access by Lck. The action of Lck on CD3 could in turn modify orientation, conformation, and/or clustering of TCR and/or CD8 at the membrane to enable their cooperative binding to pMHC. The enhanced binding further amplifies the Ca2+ and MAPK downstream signaling events, although these are not needed for the induced TCR-CD8 cooperation. Thus, the induced cooperation represents a positive feedback loop between the TCR and CD8 in the initial signaling events. This is consistent with the proposal that Lck serves as an adaptor to regulate the interactions of coreceptors with MHC and TCR (Xu and Littman, 1993). Additionally, it supports the model that Lck mediates the recruitment of CD4 or CD8 to the TCR-CD3ζ complex upon pMHC recognition as a means to increase binding (Thome et al., 1995; Thome et al., 1996).
Of interest, the anti-CD8 clone 53-6.7 that improved T-cell binding by pMHC tetramers also induced TCR and CD8 cooperation. While future studies are required to elucidate how 53-6.7 works, evidence suggests that the 53-6.7 effects may share some mechanistic elements with the TCR-induced CD8-dependent second-stage increased adhesion. Indeed, H-2Db binding to CD8 on H-2Kb-restricted OT1 T cells was increased moderately by 53-6.7, consistent with a change in CD8 orientation, conformation, and/or clustering. Replacing H-2Db by H-2Kb to allow OT1 TCR binding resulted in a substantial increase in normalized adhesion bonds to the second-stage level without the 1-s delay. This would be consistent with TCR-pMHC-CD8 cooperative binding induced by pre-bound 53-6.7. Importantly, the Fab fragment of 53-6.7, but not of the control CT-CD8a, increased Lck activation. Thus, our data have provided insights to the discrepant effects of some anti-CD8 antibodies on the binding of monomeric (Luescher et al., 1995) or tetrameric (Holman et al., 2005; Wooldridge et al., 2003) pMHC by the TCR and/or CD8 on T cells.
Our recent analysis of TCR-pMHC interactions on the T cell surface has revealed a strong correspondence between the 2D binding parameters and T cell responsiveness (Huang et al., 2010), suggesting that the TCR-pMHC interaction initiates peptide discrimination. The much higher affinities for agonist pMHCs (OVA and A2) enables the TCR to bind these strong ligands at low densities and use induced cooperation with CD8 to further enhance binding. By comparison, 1–2 orders of magnitude higher densities are required for weak ligands (E1, V-OVA, and R4) to bind the TCR due to the low TCR-pMHC affinities, which resulted in even fewer normalized adhesion bonds than pMHC-CD8 interactions (Fig. 7D–F, compare Δ and ○). The substantially higher densities of pMHC may increase the chance for TCR-CD8 cooperation even in the classical sense without the 1-s delay to produce more relative increases in the normalized adhesion bonds (Fig. 7, compare panel A–B with D–F). However, the induced TCR-CD8 cooperation produced more absolute increases in the normalized adhesion bonds for strong ligands than weak ligands (Fig. 7G). In fact, the adhesion enhancements for a panel of pMHC ligands match the corresponding enhancements in T-cell responses, suggesting an additional level of peptide discrimination. It is reasonable to hypothesize that different initial TCR-pMHC binding characteristics have to be converted into differential biological signals, be amplified by the signaling cascade, and pass some fidelity checkpoints before committing the T cell to distinct responses. Our results thus reveal a mechanism for TCR-induced cooperation with CD8 to amplify the initial discriminative signals by positive feedback.
EXPERIMENTAL PROCEDURES
Reagents
Naïve CD8+ T cells of OT1 or F5 TCR transgenic mice (Huang et al., 2007) and MOG-reactive CD4+ T cells (Ford and Evavold, 2003) were generated using Emory University IACUC approved protocols. CD8+-TCR- OT1 hybridoma(Yachi et al., 2006) was from N. R. Gascoigne (Scripps Research Institute). The following peptides were synthesized: ovalbumin-derived peptides OVA, A2, G4, E1, V-OVA and R4, and a vesicular stomatitis virus-derived peptide VSV (Alam et al., 1996; Huang et al., 2010) as well as the influenza virus nucleoprotein-derived peptide NP68 (Smyth et al., 2002) and the lymphocytic choriomeningitis virus-derived peptide GP33 (Kerry et al., 2003). Monomeric mouse pMHC-I (OVA, A2, G4, E1, V-OVA, R4, or VSV on H-2Kb or H-2Kbα3A2 and NP68 or GP33 on H-2Db) and pMHC-II (MOG38-49:I-Ab) (Sabatino et al., 2008) with C-terminal biotin-tags were produced by the NIH Tetramer Core Facility.
PE-conjugated/-unconjugated anti-mouse TCR Vα2 (B20.1) and Vβ 11 (RR3-15) mAbs were from BD Biosciences (San Jose, CA), and CD8α (CT-CD8a) and CD8β (CT-CD8b) mAbs were from Invitrogen (Carlsbad, CA). Anti-mouse CD8 clone 53-6.7 was from eBioscience (San Diego, CA). Anti-mouse H-2Kb (3H2672, PE-conjugated) and H-2Db (BCDb, FITC-conjugated) were from US Biological (Swampscott, MA) and Biocarta (San Diego, CA), respectively. Anti-biotin (Bio3-18E7.2, PE-conjugated) was from Miltenyi Biotec (Auburn, CA). Anti-mouse Lck pY394 (9A6) was from Milliore (Billerica, MA). PE-conjugated goat anti-mouse was from Sigma-Aldrich (St. Louis, MO). Fab fragments of mAbs were prepared using a kit following the manufacturer’s instruction (Pierce, Rockford, IL).
PP2 (4-Amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine) was from Biomol (Plymouth Meeting, PA). Protein tyrosine phosphatase CD45 inhibitor (N-(9, 10-Dioxo-9, 10-dihydro-phenanthren-2-yl)-2, 2-dimethyl-propionamide), intracellular calcium chelator BAPTA-AM, MAP kinase kinase-1 inhibitor PD98059, and PI3 kinase inhibitor Ly294002 and wortmanin were from EMD (San Diego, CA).
Coating pMHC onto RBC surface
RBCs were isolated using a Georgia Institute of Technology IRB approved protocol as described (Chesla et al., 1998). To coat different pMHC densities, RBCs were biotinylated using Biotin-X-NHS (EMD) at different concentrations as described (Huang et al., 2007). Biotinylated RBCs were incubated with excess streptavidin (EMD or Pierce) for 30 min and with saturating amount of pMHC for another 30 min after removing unbound streptavidin. To achieve low pMHC densities and prevent dimeric pMHC formation, excessive biotin binding-sites were partially blocked by D-biotin (Sigma-Aldrich). To separate TCR and CD8 binding-sites on distinct pMHC monomers, RBCs were incubated with saturating amount of OVA:H-2Kbα3A2 and VSV:H-2Kb at a 5:95 ratio. Tetrameric streptavidin has four biotin binding-sites, two on each side. One side binds the biotinylated RBC and the other side binds pMHC or D-biotin. The 5:95 ratio predicts 0.2% of OVA:H-2Kbα3A2/OVA:H-2Kbα3A2 homodimers, 9.6% of OVA:H-2Kbα3A2/VSV:H-2Kb heterodimers, and 90.2% VSV:H-2Kb/VSV:H-2Kb homodimers on the same streptavidin.
Site density and Lck activation measurements
The densities of pMHC, TCR, and CD8 were determined as described (Huang et al., 2007). To evaluate Lck activation by 53-6.7, OT1 T cells were incubated in modified flow cytometry buffer (PBS+5% FCS+0.02% NaN3) with 10 μg/ml Fab of 53.6.7 or control mAb CT-CD8a and 50 μg/ml anti-TCR mAb B20.1 for 30 min at 4°C. Cells were fixed by 4% paraformaldehyde, incubated with 10 μg/ml anti-Lck pY394 in SAP buffer (HBSS+0.1% saponin+0.05% NaN3+5% FBS), stained with a PE-conjugated secondary antibody, resuspended in modified flow cytometry buffer, and analyzed by flow cytometry.
Adhesion frequency assay
This assay has been described (Chesla et al., 1998; Huang et al., 2007). Briefly, a T cell and a RBC were aspirated by respective pipettes (Fig. 1A) and driven to contact with controlled area and duration. Adhesion was observed from stretching of the RBC on T cell retraction. This contact-retraction cycle was either repeated 50 times for a given contact duration (Movie S1) or alternated between two contact durations for 100 times (Movie S2) on each cell pair and 3–5 cell pairs were used to estimate an adhesion frequency Pa (mean ± s.e.m.). Experiments were performed at 25°C (most cases) or 37°C (indicated). In some experiments, T cells were pretreated for 30 min at 25°C (for mAbs) or 37°C (for inhibitors) with 10 μg/ml anti-CD8 blocking (CT-CD8a) or activating (53-6.7) mAb or equivalent amount of their Fab, 50 μg/ml anti-TCR Vα2 (B20.1, for blocking the OT1 TCR) or 25 μg/ml Vβ 11 (RR3-15, for blocking the F5 TCR) mAb, 10 μM protein tyrosine phosphatase CD45 inhibitor, 50 μM PD98059, 10 μM Ly294002, or 100 nM wortmannin and the experiments were performed in the continuous presence of these agents. To inhibit Src family kinases, T cells were preincubated with 10 μM PP2 for 10 min at 25°C and tested in the next 10 min with continuous presence of PP2. For chelating Ca2+, T cells were preloaded with 50 μM of BAPTA-AM for 20 min at 37°C in the presence of 1 mM EGTA and with 1mM EGTA supplemented to chamber medium.
Calculating effective 2D affinity and synergy
The adhesion frequency Pa is related to the average bond number <n> by
| (1) |
When adhesion is mediated by a single receptor-ligand specie and Pa has reached a plateau,
| (2) |
where mr and ml are receptor and ligand densities (Chesla et al., 1998). The product of the contact area Ac (a few percents of 3 μm2, kept constant in experiments) and binding affinity Ka is called effective 2D affinity (in μm4). For adhesion mediated by dual receptor-ligand species, e.g., TCR and CD8, <n> includes contributions from both species and is expressed at a per pMHC density basis as the sum of two bimolecular interactions (Zhu and Williams, 2000):
| (3) |
provided that TCR and CD8 bind pMHC concurrently and independently. However, Eq. 3 no longer applies if TCR and CD8 bind pMHC cooperatively. Nevertheless, Pa can be converted to <n> and divided by ligand density to obtain normalized adhesion bonds,
| (4) |
The level of synergy can be calculated from the difference between <n>/mpMHC determined using the TCR-pMHC-CD8 trimolecular Pa and the sum of <n>/mpMHC values determined using the TCR-pMHC and pMHC-CD8 bimolecular Pa’s.
Quantifying peptide potency
Naïve OT1 splenocytes (3×105/well) were cultured in 96-well plates with the desired peptide at 37°C in the absence or presence of 10 μg/ml anti-CD8 - (CT-CD8a) Fab. After 48 h, 0.4 μCi/well of [3H] thymidine was added. After another 18 h, cells were harvested on a FilterMate harvester (PerkinElmer) and analyzed on a Matrix 96 Direct Beta Counter (PerkinElmer). EC50 values were calculated using GraphPad Prism.
Supplementary Material
Acknowledgments
We thank S. Sambhara for providing F5 T cells, N. R. Gascoigne for providing CD8+/TCR- OT1 hybridoma, J. Altman for providing the H-2Kbα3A2 construct, H. He for experimental advice, V. Zarnitsyna for helping with the PP2 inhibition, J. Lou for structural analysis, J. Plowden and F. Zhang for technical assistance, and the NIH Tetramer Core Facility at Emory University for providing MHC monomers. This work was supported by NIH grants AI38282 and AI060799 (to C.Z.) and AI056017 and National Multiple Sclerosis Society Grant RG4047-A-3 (to B.D.E.).
Footnotes
We have no conflict of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alam SM, Travers PJ, Wung JL, Nasholds W, Redpath S, Jameson SC, Gascoigne NR. T-cell-receptor affinity and thymocyte positive selection. Nature. 1996;381:616–620. doi: 10.1038/381616a0. [DOI] [PubMed] [Google Scholar]
- Campanelli R, Palermo B, Garbelli S, Mantovani S, Lucchi P, Necker A, Lantelme E, Giachino C. Human CD8 co-receptor is strictly involved in MHC-peptide tetramer-TCR binding and T cell activation. Int Immunol. 2002;14:39–44. doi: 10.1093/intimm/14.1.39. [DOI] [PubMed] [Google Scholar]
- Campi G, Varma R, Dustin ML. Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J Exp Med. 2005;202:1031–1036. doi: 10.1084/jem.20051182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesla SE, Selvaraj P, Zhu C. Measuring two-dimensional receptor-ligand binding kinetics by micropipette. Biophys J. 1998;75:1553–1572. doi: 10.1016/S0006-3495(98)74074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels MA, Jameson SC. Critical role for CD8 in T cell receptor binding and activation by peptide/major histocompatibility complex multimers. J Exp Med. 2000;191:335–346. doi: 10.1084/jem.191.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MM, Krogsgaard M, Huppa JB, Sumen C, Purbhoo MA, Irvine DJ, Wu LC, Ehrlich L. Dynamics of cell surface molecules during T cell recognition. Annu Rev Biochem. 2003;72:717–742. doi: 10.1146/annurev.biochem.72.121801.161625. [DOI] [PubMed] [Google Scholar]
- Davis MM, Krogsgaard M, Huse M, Huppa J, Lillemeier BF, Li QJ. T cells as a self-referential, sensory organ. Annu Rev Immunol. 2007;25:681–695. doi: 10.1146/annurev.immunol.24.021605.090600. [DOI] [PubMed] [Google Scholar]
- Delon J, Gregoire C, Malissen B, Darche S, Lemaitre F, Kourilsky P, Abastado JP, Trautmann A. CD8 expression allows T cell signaling by monomeric peptide-MHC complexes. Immunity. 1998;9:467–473. doi: 10.1016/s1074-7613(00)80630-5. [DOI] [PubMed] [Google Scholar]
- Devine L, Hodsdon ME, Daniels MA, Jameson SC, Kavathas PB. Location of the epitope for an anti-CD8alpha antibody 53.6.7 which enhances CD8alpha-MHC class I interaction indicates antibody stabilization of a higher affinity CD8 conformation. Immunol Lett. 2004;93:123–130. doi: 10.1016/j.imlet.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu Rev Immunol. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- Evavold BD, Allen PM. Separation of IL-4 production from Th cell proliferation by an altered T cell receptor ligand. Science. 1991;252:1308–1310. doi: 10.1126/science.1833816. [DOI] [PubMed] [Google Scholar]
- Evavold BD, Sloan-Lancaster J, Allen PM. Tickling the TCR: selective T-cell functions stimulated by altered peptide ligands. Immunol Today. 1993;14:602–609. doi: 10.1016/0167-5699(93)90200-5. [DOI] [PubMed] [Google Scholar]
- Ford ML, Evavold BD. Regulation of polyclonal T cell responses by an MHC anchor-substituted variant of myelin oligodendrocyte glycoprotein 35–55. J Immunol. 2003;171:1247–1254. doi: 10.4049/jimmunol.171.3.1247. [DOI] [PubMed] [Google Scholar]
- Gakamsky DM, Luescher IF, Pramanik A, Kopito RB, Lemonnier F, Vogel H, Rigler R, Pecht I. CD8 kinetically promotes ligand binding to the T-cell antigen receptor. Biophys J. 2005;89:2121–2133. doi: 10.1529/biophysj.105.061671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao GF, Rao Z, Bell JI. Molecular coordination of alphabeta T-cell receptors and coreceptors CD8 and CD4 in their recognition of peptide-MHC ligands. Trends Immunol. 2002;23:408–413. doi: 10.1016/s1471-4906(02)02282-2. [DOI] [PubMed] [Google Scholar]
- Gao GF, Tormo J, Gerth UC, Wyer JR, McMichael AJ, Stuart DI, Bell JI, Jones EY, Jakobsen BK. Crystal structure of the complex between human CD8alpha(alpha) and HLA-A2. Nature. 1997;387:630–634. doi: 10.1038/42523. [DOI] [PubMed] [Google Scholar]
- Garcia KC, Scott CA, Brunmark A, Carbone FR, Peterson PA, Wilson IA, Teyton L. CD8 enhances formation of stable T-cell receptor/MHC class I molecule complexes. Nature. 1996;384:577–581. doi: 10.1038/384577a0. [DOI] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- Holman PO, Walsh ER, Jameson SC. Characterizing the impact of CD8 antibodies on class I MHC multimer binding. J Immunol. 2005;174:3986–3991. doi: 10.4049/jimmunol.174.7.3986. [DOI] [PubMed] [Google Scholar]
- Huang J, Edwards LJ, Evavold BD, Zhu C. Kinetics of MHC-CD8 interaction at the T cell membrane. J Immunol. 2007;179:7653–7662. doi: 10.4049/jimmunol.179.11.7653. [DOI] [PubMed] [Google Scholar]
- Huang J, Zarnitsyna VI, Liu B, Edwards LJ, Jiang N, Evavold BD, Zhu C. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature. 2010;464:932–936. doi: 10.1038/nature08944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppa JB, Axmann M, Mortelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, Klein LO, Schutz GJ, Davis MM. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963–967. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse M, Klein LO, Girvin AT, Faraj JM, Li QJ, Kuhns MS, Davis MM. Spatial and temporal dynamics of T cell receptor signaling with a photoactivatable agonist. Immunity. 2007;27:76–88. doi: 10.1016/j.immuni.2007.05.017. [DOI] [PubMed] [Google Scholar]
- Irvine DJ, Purbhoo MA, Krogsgaard M, Davis MM. Direct observation of ligand recognition by T cells. Nature. 2002;419:845–849. doi: 10.1038/nature01076. [DOI] [PubMed] [Google Scholar]
- Kane LP, Weiss A. The PI-3 kinase/Akt pathway and T cell activation: pleiotropic pathways downstream of PIP3. Immunol Rev. 2003;192:7–20. doi: 10.1034/j.1600-065x.2003.00008.x. [DOI] [PubMed] [Google Scholar]
- Kern PS, Teng MK, Smolyar A, Liu JH, Liu J, Hussey RE, Spoerl R, Chang HC, Reinherz EL, Wang JH. Structural basis of CD8 coreceptor function revealed by crystallographic analysis of a murine CD8alphaalpha ectodomain fragment in complex with H-2Kb. Immunity. 1998;9:519–530. doi: 10.1016/s1074-7613(00)80635-4. [DOI] [PubMed] [Google Scholar]
- Kerry SE, Buslepp J, Cramer LA, Maile R, Hensley LL, Nielsen AI, Kavathas P, Vilen BJ, Collins EJ, Frelinger JA. Interplay between TCR affinity and necessity of coreceptor ligation: high-affinity peptide-MHC/TCR interaction overcomes lack of CD8 engagement. J Immunol. 2003;171:4493–4503. doi: 10.4049/jimmunol.171.9.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ST, Takeuchi K, Sun ZY, Touma M, Castro CE, Fahmy A, Lang MJ, Wagner G, Reinherz EL. The alphabeta T cell receptor is an anisotropic mechanosensor. J Biol Chem. 2009;284:31028–31037. doi: 10.1074/jbc.M109.052712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laugel B, van den Berg HA, Gostick E, Cole DK, Wooldridge L, Boulter J, Milicic A, Price DA, Sewell AK. Different T cell receptor affinity thresholds and CD8 coreceptor dependence govern cytotoxic T lymphocyte activation and tetramer binding properties. J Biol Chem. 2007;282:23799–23810. doi: 10.1074/jbc.M700976200. [DOI] [PubMed] [Google Scholar]
- Li YC, Chen BM, Wu PC, Cheng TL, Kao LS, Tao MH, Lieber A, Roffler SR. Cutting Edge: mechanical forces acting on T cells immobilized via the TCR complex can trigger TCR signaling. J Immunol. 2010;184:5959–5963. doi: 10.4049/jimmunol.0900775. [DOI] [PubMed] [Google Scholar]
- Luescher IF, Vivier E, Layer A, Mahiou J, Godeau F, Malissen B, Romero P. CD8 modulation of T-cell antigen receptor-ligand interactions on living cytotoxic T lymphocytes. Nature. 1995;373:353–356. doi: 10.1038/373353a0. [DOI] [PubMed] [Google Scholar]
- Norment AM, Salter RD, Parham P, Engelhard VH, Littman DR. Cell-cell adhesion mediated by CD8 and MHC class I molecules. Nature. 1988;336:79–81. doi: 10.1038/336079a0. [DOI] [PubMed] [Google Scholar]
- Palacios EH, Weiss A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 2004;23:7990–8000. doi: 10.1038/sj.onc.1208074. [DOI] [PubMed] [Google Scholar]
- Pecht I, Gakamsky DM. Spatial coordination of CD8 and TCR molecules controls antigen recognition by CD8+ T-cells. FEBS Lett. 2005;579:3336–3341. doi: 10.1016/j.febslet.2005.04.025. [DOI] [PubMed] [Google Scholar]
- Purbhoo MA, Irvine DJ, Huppa JB, Davis MM. T cell killing does not require the formation of a stable mature immunological synapse. Nat Immunol. 2004;5:524–530. doi: 10.1038/ni1058. [DOI] [PubMed] [Google Scholar]
- Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- Sabatino JJ, Jr, Shires J, Altman JD, Ford ML, Evavold BD. Loss of IFN-gamma enables the expansion of autoreactive CD4+ T cells to induce experimental autoimmune encephalomyelitis by a nonencephalitogenic myelin variant antigen. J Immunol. 2008;180:4451–4457. doi: 10.4049/jimmunol.180.7.4451. [DOI] [PubMed] [Google Scholar]
- Smyth LA, Ardouin L, Williams O, Norton T, Tybulewicz V, Kioussis D. Inefficient clustering of tyrosine-phosphorylated proteins at the immunological synapse in response to an antagonist peptide. Eur J Immunol. 2002;32:3386–3394. doi: 10.1002/1521-4141(200212)32:12<3386::AID-IMMU3386>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Thome M, Duplay P, Guttinger M, Acuto O. Syk and ZAP-70 mediate recruitment of p56lck/CD4 to the activated T cell receptor/CD3/zeta complex. J Exp Med. 1995;181:1997–2006. doi: 10.1084/jem.181.6.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome M, Germain V, DiSanto JP, Acuto O. The p56lck SH2 domain mediates recruitment of CD8/p56lck to the activated T cell receptor/CD3/zeta complex. Eur J Immunol. 1996;26:2093–2100. doi: 10.1002/eji.1830260920. [DOI] [PubMed] [Google Scholar]
- Tomonari K, Spencer S. Epitope-specific binding of CD8 regulates activation of T cells and induction of cytotoxicity. Int Immunol. 1990;2:1189–1194. doi: 10.1093/intimm/2.12.1189. [DOI] [PubMed] [Google Scholar]
- Weiss A, Littman DR. Signal transduction by lymphocyte antigen receptors. Cell. 1994;76:263–274. doi: 10.1016/0092-8674(94)90334-4. [DOI] [PubMed] [Google Scholar]
- Wooldridge L, Hutchinson SL, Choi EM, Lissina A, Jones E, Mirza F, Dunbar PR, Price DA, Cerundolo V, Sewell AK. Anti-CD8 antibodies can inhibit or enhance peptide-MHC class I (pMHCI) multimer binding: this is paralleled by their effects on CTL activation and occurs in the absence of an interaction between pMHCI and CD8 on the cell surface. J Immunol. 2003;171:6650–6660. doi: 10.4049/jimmunol.171.12.6650. [DOI] [PubMed] [Google Scholar]
- Wulfing C, Rabinowitz JD, Beeson C, Sjaastad MD, McConnell HM, Davis MM. Kinetics and extent of T cell activation as measured with the calcium signal. J Exp Med. 1997;185:1815–1825. doi: 10.1084/jem.185.10.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyer JR, Willcox BE, Gao GF, Gerth UC, Davis SJ, Bell JI, van der Merwe PA, Jakobsen BK. T cell receptor and coreceptor CD8 alphaalpha bind peptide-MHC independently and with distinct kinetics. Immunity. 1999;10:219–225. doi: 10.1016/s1074-7613(00)80022-9. [DOI] [PubMed] [Google Scholar]
- Xu C, Gagnon E, Call ME, Schnell JR, Schwieters CD, Carman CV, Chou JJ, Wucherpfennig KW. Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell. 2008;135:702–713. doi: 10.1016/j.cell.2008.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Littman DR. A kinase-independent function of Lck in potentiating antigen-specific T cell activation. Cell. 1993;74:633–643. doi: 10.1016/0092-8674(93)90511-n. [DOI] [PubMed] [Google Scholar]
- Xu XN, Purbhoo MA, Chen N, Mongkolsapaya J, Cox JH, Meier UC, Tafuro S, Dunbar PR, Sewell AK, Hourigan CS, et al. A novel approach to antigen-specific deletion of CTL with minimal cellular activation using alpha3 domain mutants of MHC class I/peptide complex. Immunity. 2001;14:591–602. doi: 10.1016/s1074-7613(01)00133-9. [DOI] [PubMed] [Google Scholar]
- Yachi PP, Ampudia J, Zal T, Gascoigne NR. Altered peptide ligands induce delayed CD8-T cell receptor interaction--a role for CD8 in distinguishing antigen quality. Immunity. 2006;25:203–211. doi: 10.1016/j.immuni.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Yokosuka T, Sakata-Sogawa K, Kobayashi W, Hiroshima M, Hashimoto-Tane A, Tokunaga M, Dustin ML, Saito T. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat Immunol. 2005;6:1253–1262. doi: 10.1038/ni1272. [DOI] [PubMed] [Google Scholar]
- Zamoyska R. Why is there so much CD45 on T cells? Immunity. 2007;27:421–423. doi: 10.1016/j.immuni.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Zhu C, Long M, Chesla SE, Bongrand P. Measuring receptor/ligand interaction at the single-bond level: experimental and interpretative issues. Ann Biomed Eng. 2002;30:305–314. doi: 10.1114/1.1467923. [DOI] [PubMed] [Google Scholar]
- Zhu C, Williams TE. Modeling concurrent binding of multiple molecular species in cell adhesion. Biophys J. 2000;79:1850–1857. doi: 10.1016/S0006-3495(00)76434-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.