

Abstract
Mice deficient for the cellular prion protein (PrPC) do not develop prion disease; accordingly, gene-based strategies to diminish PrPC expression are of interest. We synthesized a series of chemically modified antisense oligonucleotides (ASOs) targeted against mouse Prnp messenger RNA (mRNA) and identified those that were most effective in decreasing PrPC expression. Those ASOs were also evaluated in scrapie-infected cultured cells (ScN2a) for their efficacy in diminishing the levels of the disease-causing prion protein (PrPSc). When the optimal ASO was infused intracerebrally into FVB mice over a 14-day period beginning 1 day after infection with the Rocky Mountain Laboratory (RML) strain of mouse prions, a prolongation of the incubation period of almost 2 months was observed. Whether ASOs can be used to develop an effective therapy for patients dying of Creutzfeldt–Jakob disease remains to be established.
Introduction
Over the past 3 decades, a distinct protein has been found to accumulate in the brains of patients suffering from each of the neurodegenerative diseases. Mutations have been found in the respective genes encoding the particular etiologic proteins responsible for the familial forms of the neurodegenerative diseases. Increasing evidence argues that each of the disease-causing proteins undergo posttranslational modification in a self-perpetuating process. This prion-like mechanism leads to the accumulation of the modified, etiologic proteins. In Creutzfeldt–Jakob disease of humans, scrapie of sheep, bovine spongiform encephalopathy, and chronic wasting disease, the cellular prion protein (PrPC) undergoes refolding into the disease-causing isoform (PrPSc). The progressive accumulation of PrPSc in the brain leads to central nervous system (CNS) dysfunction that is accompanied by neuronal vacuolation and astrocytic gliosis. In some cases, amyloid plaques composed of PrPSc are found within the CNS but such plaques are not an obligatory feature of these disorders.
Many lines of evidence converged to argue that PrPSc is the sole component of the infectious prion particle. Later, knockout of the PrP gene encoding PrPC was found to render mice resistant to experimental scrapie. This finding suggested that patients dying of Creutzfeldt–Jakob disease would benefit from therapeutics that lower the levels of PrPC and/or PrPSc. Both RNA interference (RNAi) and antisense oligonucleotides (ASOs) have been used to lower the levels of PrPC and PrPSc in scrapie-infected cultured cells (ScN2a). RNAi molecules have been found to lower PrP mRNA levels and hence PrPC.1,2,3,4 In ScN2a cells as well as in the brains of scrapie-infected mice, the levels of PrPSc were also lowered by exposure to sequence-specific RNAi molecules. In contrast to the RNAi results, ASOs are confounded by the ability of these polymers to lower PrPSc levels in ScN2a independent of their sequence.5,6
Treatment of prion diseases using transgenic (Tg) mice with inducible PrPC expression systems have been performed.7,8 Tg(NFH-Cre/MloxP) mice were inoculated with the Rocky Mountain Laboratory (RML) prions at 3–4 weeks of age.7 Cre-mediated recombination at 10–12 weeks of age was effectively used to suppress neuronal PrPC expression but not that in other CNS cells. Shutting off PrPC expression in neurons, even after mice developed signs of neurologic dysfunction, reversed spongiform degeneration and prevented clinical disease. Surprisingly, mice remained asymptomatic even though their brains were inundated with extraneuronal PrPSc deposits. In bigenic Tg(tTA:PrP+/0)3 mice, PrPC expression was regulated by oral doxycycline administration.8 PrPC expression was reduced by 95% in the brains of bigenic mice compared to wild-type mice, which extended survival times following prion inoculation from ~150 to ~430 days when the mice eventually developed clinical disease. In these mice, PrPC suppression prevented pyramidal nerve cell death and enhanced the clearance of PrPSc deposits. Recently, a single injection of small hairpin RNA targeting PrP into the hippocampus of Tg37 mice protected the thalamus and cortex, brain regions distal to the injection site, from prion-induced neurodegeneration and prolonged survival time from 85 to 105 days even though PrPC expression was reduced only at the border of the injection site.4
Several clinically desirable advantages over lentiviral injections of RNAi molecules are offered by the intracerebral ventricular (ICV) delivery of ASOs that are fully phosphorothioated (PS). The five base caps at each end of the 20mer ASO were methoxyethyl-modified to increase potency and stability as well as to decrease proinflammatory effects. The PS modification in the backbone of DNA, in which one non-bridging oxygen atom is replaced with a sulfur atom, increases nuclease resistance, resulting in enhanced stability both in vitro and in vivo. In addition, the PS modification improves tissue distribution and cell uptake in vivo. The methoxyethyl modification increases the affinity of the ASOs for RNA and protects the 20mer from exonuclease degradation, resulting in an extended duration of action in tissues due to a longer metabolic half-life.9 Once introduced into the cells, hybridization of the ASO to the target mRNA results in RNase H cleavage of the message and suppression of gene expression.10
Here, we report on ASOs targeting mouse Prnp mRNA that suppress PrPC expression and inhibit PrPSc formation in cell culture and scrapie-infected mice. Intraventricular infusion of an ASO, for 14 days beginning 1 day after inoculation with RML prions, extended incubation periods in prion-infected mice by almost 2 months.
Results
Strategy. We synthesized and screened 78 ASOs with methoxyethyl gapmer chemistry targeting the mouse (Mo) PrP message in a mouse endothelial cell line designated b.END. From this panel, we identified the 10 most active ASOs for PrP transcript knockdown by dose response. The three most-effective ASOs—742, 747, and 771—were then screened for efficacy in reducing PrPC and PrPSc in N2a and ScN2a cells, respectively. These three ASOs were injected intraperitoneally (ip) in uninfected FVB mice to evaluate tolerance as well as to verify in vivo transcript and protein knockdown in the liver. Next, the three ASOs were tested for RNA and protein knockdown in the brain by intracerebral administration into the left lateral ventricle through a cannula using an implanted Alzet osmotic pump. Of the three most effective ASOs, 771 was chosen for further study in scrapie-infected mice (Figure 1a).
Figure 1.

Identification of ASOs that diminish mouse Prnp mRNA expression measured by qRT-PCR. (a) Experimental design. (b) Prnp mRNA expression 24 hours post-transfection with 78 antisense oligonucleotides (120 nmol/l) targeting mouse Prnp mRNA in b.END cells. ASOs are displayed relative to their position on the 2,206-bp mouse Prnp mRNA (Accession NM_011170). (c) Dose-response of Prnp mRNA expression using the 10 most-effective ASOs in b.END cells. ASO concentrations ranged from 6.25–200 nmol/l, as indicated. Prnp mRNA is expressed as a percentage compared to untransfected control (UTC). ASO, antisense oligonucleotide; bp, base pair; ICV, intracerebral ventricular; mRNA, messenger RNA; qRT-PCR, quantitative real time-PCR.
In vitro screening. High-throughput screening of 78 ASOs by quantitative real-time (qRT) PCR identified the 10 with the greatest mRNA knockdown efficacy (Figure 1b). These 10 ASOs were then evaluated by measuring the dose-dependent responses in b.END cells; ASOs were added at increasing concentrations (6.5–200 nmol/l) in the presence of the lipofectamine transfection reagent (Figure 1c). ASOs 742, 747, and 771 had the greatest potency, with PrP mRNA levels reduced by 94, 97, and 97%, respectively, at the 200-nmol/l dose compared to cells treated with non–sequence-specific control ASO 923 (Table 1).
Table 1. Percentage inhibition of Prnp mRNA expression in mouse b.END cells by different doses of ASOs compared to control cells treated with ASO 923.
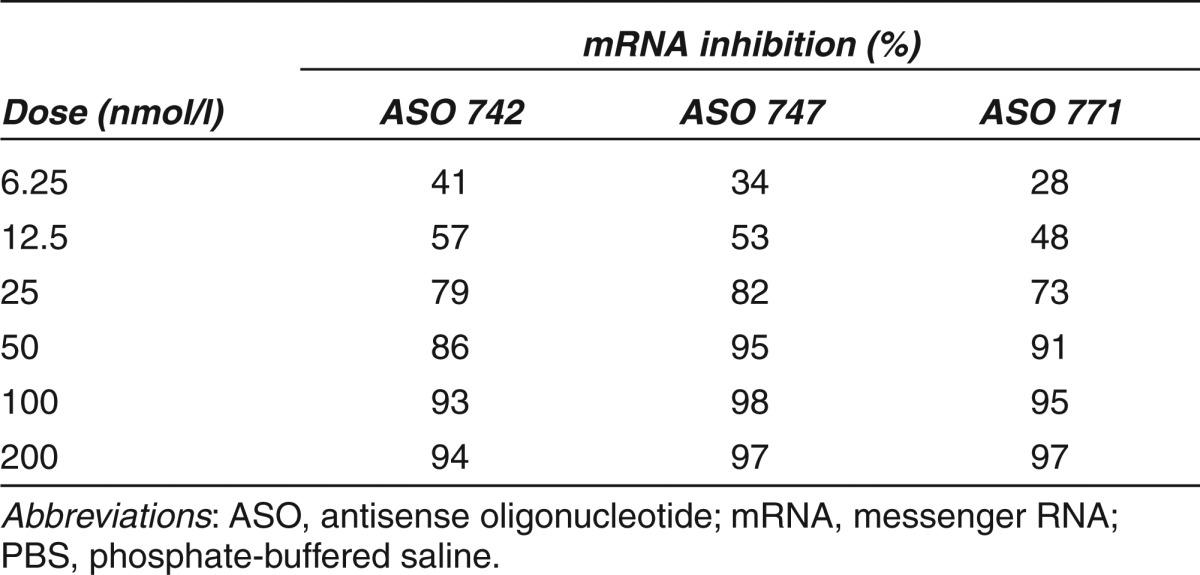
Based on reductions in PrP mRNA levels in b.END cells, ASOs 742, 747, and 771 were screened in N2a cells. These ASOs target three sites in the 3′ untranslated region (UTR) of MoPrP mRNA (NM_011170.1; Figure 2a). Furthermore, this experimental scenario better mimics in vivo delivery of the drug into the brain. Cells were cultured for 30 days in the continuous presence of 500 nmol/l of each ASO and lysates were collected at 2, 7, 14, and 28 days for analysis of PrPC expression by western immunoblots, which were quantified by densitometry (Figure 2b). ASO 771 reduced PrPC levels more rapidly than the other two ASOs, with a 60% reduction observed following 7 days of exposure and 84% reduction by 14 days. Control ASO 923 elicited low levels of non–sequence-specific reduction of PrPC expression, by ~20% at 7, 14, and 28 days.
Figure 2.

ASOs targeting mouse Prnp mRNA reduced PrPC and PrPSc expression in N2a and ScN2a cells. (a) The three lead ASOs 742, 747, and 771 identified in b.END cells are displayed relative to their positions on the 2,206-bp mouse Prnp mRNA (Accession NM_011170). All fall within exon 4 outside of the coding sequence in the 3′ UTR. (b,c) Kinetics of PrPC suppression in N2a cells is shown in b and protease-resistant PrPSc in ScN2a cells is shown in c exposed to 500 nmol/l of each ASO for 2, 7, 14, and 28 days. ASO 923 and PBS were used as controls. (d) Dose-response curves of PrPC in N2a and PrPSc in ScN2a cells incubated with 0–250 nmol/l of ASO 771. In b–d, PrP levels were quantified by densitometry of scanned western blot films using NIH Image J software and expressed as percentages relative to cells treated with PBS. Protein standards shown are 36, 30, and 22 kilodaltons. ASO, antisense oligonucleotide; mRNA, messenger RNA; ORF, open reading frame; PBS, phosphate-buffered saline; PrPC, cellular prion protein; PrPSc, disease-causing prion protein.
Antisense inhibition of PrP 27-30 in ScN2a cells. To determine whether ASO-mediated PrPC suppression affects PrPSc levels in prion-infected cells, ScN2a cells were cultured in the presence of 500 nmol/l of each lead ASO and lysates collected at 2, 7, 14, and 28 days. Following 2 days of treatment, levels of PrP 27-30, the protease-resistant core of PrPSc, were reduced by 81% with ASO 742, by 63% with ASO 747, and by 57% with ASO 771 (Figure 2c). After 7 days of exposure to ASOs 742, 747, or 771, PrP 27-30 was not detectable. Cells exposed to the control ASO 923 for 2 days showed a 41% reduction in PrP 27-30, and a 95% reduction in PrP 27-30 after 7 days. We and others had previously reported the reduction in PrPSc levels by sequence-scrambled PS oligonucleotides.5,6 Following 28 days of exposure to each of the four ASOs including the control ASO 923, PrPSc was undetectable. To determine whether these observations were specific to ScN2a cells, these experiments were also performed with prion-infected GT1 cells, ScGT1. PrP 27-30 in this cell line was completely eliminated after 7 days of exposure to each of ASOs including the control ASO 923 (data not shown).
To determine the ASO 771 concentration at which PrPC and PrPSc levels were suppressed by 50% (half maximal effective concentration (EC50)) in N2a and ScN2a cells, we treated cells with ASO 771 at concentrations ranging from 2 to 250 nmol/l, collected lysates after 7 days, then measured PrPC and PrPSc levels by western blot. Increasing ASO concentrations reduced the level of PrP 27-30 in ScN2a cells in an exponential manner (Figure 2d). The EC50 values for ASO 771 were 50 nmol/l for PrPC and 4 nmol/l for PrPSc. The control ASO 923 had an EC50 of 10 nmol/l for PrPSc due to non–sequence-specific effects (Figure 2c). It is noteworthy that ASO 771 was only able to reduce PrPC levels by 60% in cultured N2a cells at a concentration above 50 nnol/l (Figure 2d). This reduction was sequence-specific in contrast to the reduction of PrPSc, which exceeded 95% with less than 10 nmol/l of ASO 771.
ASOs reduced PrPC levels in vivo. Based on our findings in cell culture, we investigated whether ASOs 742, 747, and 771 would be well-tolerated and effective in diminishing mRNA and PrPC levels in vivo. We administered each ASO to eight groups of five FVB mice for 21 days via ip injections. Phosphate-buffered saline (PBS) and control ASO 847, which targets PTEN protein, were used as controls. The ASOs and PBS were administered ip twice weekly for 3 weeks, in doses of either 50 mg/kg/week or 100 mg/kg/week (Figure 3a). None of the mice showed negative effects through the course of ASO administration. All of the animals remained healthy until they were killed at 21 days. Routine clinical serum chemistry was performed and no abnormalities were observed (data not shown).
Figure 3.

Administration of ASOs targeting mouse Prnp mRNA reduced PrPC expression in the livers (a–c) and brains (d–h) of FVB mice. ASOs were administered by intraperitoneal inoculations for liver studies (n = 4 per dose) and by ICV infusion (n = 4) for brain studies. (a,d) Experimental design. ASOs were administered for 21 days at the specified doses, after which time, mice were killed, tissues taken, and mRNA and protein levels were analyzed. (b,f) Prnp mRNA levels were measured by qRT-PCR; (c,g,h) PrPC levels were analyzed by a capture ELISA using antibody D18 (c) or by densitometry (g) of western blots probed with antibody D18 (h). (e) Brain map showing specific brain regions that were cannulated (region 3), harvested for protein (region 1), and RNA (region 2). (g,h) ASO 771 more effectively reduced PrPC levels in brain compared to ASO 742. Different lanes represent four different animals. Bottom panel of western blot was probed with actin. Protein standards shown are 36, 30, and 22 kilodaltons. For all panels, PrP expression levels are shown as percentages relative to those measured in mice treated with PBS. ELISA, enzyme-linked immunosorbent assay; ICV, intracerebral ventricular; ip, intraperitoneal; mRNA, messenger RNA; PBS, phosphate-buffered saline; PrPC, cellular prion protein; qRT-PCR, quantitative real time-PCR.
Prnp mRNA expression in liver was determined by qRT-PCR; PrPC expression was measured by enzyme-linked immunosorbent assay (ELISA) using anti-PrP HuM-Fab D18 for detection. The greatest Prnp mRNA and PrPC diminutions in liver were found with the ASO 742: on 50 or 100 mg/kg/week, respective reductions of 75 and 85% in PrP mRNA (Figure 3b), and of 65 and 72% in PrPC, were found (Figure 3c). ASO 771 at 50 mg/kg/week reduced Prnp mRNA by 30% and PrPC by ~50% in liver, as measured by qRT-PCR and immunoblotting, respectively.
Intracerebral ventricular delivery of ASOs reduced brain PrPC. Encouraged by the safety profile and the effective reduction of PrPC by the ASOs in liver following systemic administration, we next characterized the tolerability and pharmacology of ASOs in the CNS via intracerebral ventricular (ICV) administration (Figure 3d). Two lead ASOs 771 and 742 were continuously infused into the right lateral ventricle (Figure 3e, region 3) of FVB mice using implanted Alzet osmotic pumps, at a dose of 75 µg/day for 21 days, after which mice were killed within 24 hours. A control group of mice was treated with PBS in the same manner for the same time period. The ASOs were well-tolerated; neither toxic side effects nor brain abscesses were observed. Histopathological analysis of a section of the midbrain, posterior to the cannulation site, showed no abnormalities (data not shown).
mRNA and PrPC levels were evaluated using different brain regions. RNA was isolated from a portion of brain adjacent to the cannulated lateral ventricle and ventral to the cannulation site (Figure 3e, region 2). For PrPC, a portion of the brain anterior to and on the ipsilateral side of cannulation (Figure 3e, region 1) was homogenized in PBS (10% w/v) and analyzed by densitometry of western blots. PrP mRNA measured by qRT-PCR was ~60% lower in mice receiving either ASO 742 or 771 compared to mice infused with PBS (Figure 3f). Administration of ASO 742 and 771 resulted in 30% and ~70% less PrPC, respectively, in the brain compared to mice receiving PBS control injections as shown by immunoblotting (Figure 3g,h).
Next, we conducted a dose-response study to determine the potency and tolerable dose compared to our previous, single-dose study for which 75 µg was administered per day. ASO 742 and 771, at doses of 25, 50, 75 and 100 µg/day, were continuously infused via ICV into the right lateral ventricle for 14 days (Figure 4a). A control group of mice received PBS via ICV delivery for the same time period. Mice were killed 24 hours after the end of the infusion. With the exception of 100 µg/day of ASO 742, all other doses were well-tolerated by the mice.
Figure 4.

Dose-dependent reduction of mouse Prnp mRNA and PrPC by ASOs administered by ICV infusion. (a) Experimental design. Doses of 25, 50, 75, or 100 µg/day were administered by ICV delivery for 14 days, and Prnp mRNA and PrPC levels analyzed at day 14. (b) PrP mRNA levels were quantified by qRT-PCR and expressed as the relative percentage measured in PBS-treated controls. (c) Regional expression of PrPC in coronal cryosections (10 µm) at the level of the septum, shown by histoblotting with antibody D18. (d) Immunohistochemistry of a hippocampal coronal section stained with anti-PS antibody that detects the PS modification in the ASO. Arrow indicates the cannulation side. ASO, antisense oligonucleotide; ICV, intracerebral ventricular; mRNA, messenger RNA; PBS, phosphate-buffered saline; PrPC, cellular prion protein; PS, phosphorothioate; qRT-PCR, quantitative real-time PCR.
PrP mRNA levels were analyzed in a brain section posterior to the cannulation site (Figure 3e, region 2) by qRT-PCR (Figure 4b). PrP mRNA expression levels decreased in an ASO dose-dependent manner. Delivery of ASO 742 at 50 µg/day reduced PrP mRNA levels by ~60% while ASO 771 at 100 µg/day decreased PrP mRNA by 70%.
Histoblots of coronal cryosections showed that PrPC suppression was most marked on the cannulated side of the brain, but was also found on the contralateral side (Figure 4c). Mice infused with ASO 771 at 75 µg/day showed the most pronounced decrease in PrPC expression compared to those receiving PBS. A midbrain section (Bregma −2.92 mm) taken from a mouse treated with 75 µg/day of ASO 771 was fixed and stained with the ASO antibody (Figure 4d, brown staining). The ASO dispersed from the injection site, travelling a distance of 2.92 mm Bregma to the midbrain and penetrated neurons bilaterally. Together, these results demonstrate that ASO 771 administered in vivo to the brain by ICV infusion is well-tolerated and reduces both Prnp mRNA and PrPC.
ASO 771 treatment of prion-inoculated mice. On day 0, mice were inoculated on the right side of the brain with RML prions. The next day, ICV infusion of ASO 771 (75 µg/d) or PBS was initiated on the left side of the brain and continued for 14 days. At 50 dpi (36 days after ICV treatment ended), six mice (three from the treated group and three from the control group) were killed in order to measure the levels of PrPC and PrPSc (Figure 5). Another group of mice (eight from the treatment group and six controls) were observed until they developed signs of neurological dysfunction (Figure 6).
Figure 5.

ASO 771 reduced PrPC expression and interfered with PrPSc deposition in prion-infected FVB mice. (a) Experimental design. On day 0, FVB mice were inoculated with RML prions on the right side of the brain. At day 1, ASO 771, ASO 923, or PBS was delivered ICV at 75 µg/day for 14 days on the left side of the brain. Mice were killed at day 50 dpi, brain sections taken and analyzed for PrPC and PrPSc levels. (b,c) Histoblot analysis was performed on 10-µm sections at the level of the septum, hippocampus, midbrain 1 and 2 regions, and stained with the D18 antibody. Sections were left undigested to visualize total PrP levels as shown in b or subjected to GdnHCl denaturation and PK digestion to visualize PrPSc levels as shown in c. (d) Western blots show total PrP (top) and PrPSc (bottom); each blot shows two different ASO-treated animals. (e) Densitometry of western blots using NIH Image J software, then expressed relative to PrP levels in the brains of mice treated with PBS. Protein standards shown are 36, 30, and 22 kDa. ASO, antisense oligonucleotide; ICV, intracerebral ventricular; PBS, phosphate-buffered saline; PrPC, cellular prion protein; PrPSc, disease-causing prion protein.
Figure 6.

Treatment with ASO 771 reduced PrPSc in the brain and extended incubation periods by 42%. (a) Experimental design. On day 0, FVB mice were inoculated with RML prions on the right side of the brain. At day 1, either ASO 771 or PBS was delivered ICV at 75 µg/day for 14 days on the left side of the brain. At day 126, some mice were killed, and their brains analyzed for PrPC and PrPSc. (b) Survival curves of FVB mice treated with PBS (black) or ASO 771 (gray). (c,d) Mice were killed at 126 dpi, their brains analyzed by histoblotting for (c) total PrP and (d) PrPSc. Sections were left undigested as shown in c or subjected to GdnHCl denaturation and PK digestion as shown in d. Membranes were stained with the D18 antibody. ASO, antisense oligonucleotide; PBS, phosphate-buffered saline; PrPSc, disease-causing prion protein.
In mice killed at 50 dpi, histoblots showed that ASO 771 treatment diminished PrPC expression (Figure 5b) and PrPSc levels (Figure 5c) compared to PBS-infused control mice. PrPC expression was primarily reduced on the left side where ASO 771 was released from the tip of the cannula. As shown, PrPC was reduced in the substantia nigra, striatum, cerebral cortex, hippocampus, caudate nucleus, amygdala, and the periaqueductal region (Figure 5b). A less marked reduction was observed in the pons and cerebellum (data not shown). It appears that there is a pharmacologically significant gradient from the ipsilateral to contralateral side (Figure 5b, bottom row).
To examine in situ PrPSc deposition, PrPC was degraded by limited digestion with proteinase K followed by GdnHCl denaturation of PrP 27-30 before immunostaining (Figure 5c). In the brains of PBS-treated mice killed at 50 dpi, PrPSc deposits were visible in the white matter tracts (corpus callosum, striatum), septum, hippocampus, and midbrain 1 and 2. The most intense immunostaining of PrPSc was seen in the thalamus at the site of inoculation. In the mice receiving ASO 771 and ASO 923, PrPSc deposits were minimal, indicating that the ASO inhibited prion replication and propagation bilaterally.
Western immunoblotting of brain homogenates revealed that only ASO 771–treated FVB mice had decreased PrPC expression (Figure 5d, top proteinase K (PK)−), whereas both ASO 771 and ASO 923 treatment resulted in diminished signals for PrPSc (Figure 5d, bottom PK+, two different gels with different animals). Densitometry of western blots quantified a ~50% reduction of total PrP expression throughout the brains of ASO 771–treated mice compared to mice infused with PBS and to mice treated with ASO 923 (Figure 5e, top). PrPSc levels were reduced by 96% in ASO 771–treated mice and by 92% in ASO 923–treated mice (Figure 5e, bottom). Although both sequence-specific ASO 771 and non–sequence-specific ASO 923 reduced PrPSc levels equally well in the brains of prion-inoculated mice at 50 dpi, only ASO 771 reduced the PrPC substrate.
The treated mice killed at 126 dpi showed a more advanced pathological deposition of PrPSc than those killed at 50 dpi described above. Histoblots of symptomatic control mice and asymptomatic ASO-treated mice at 126 dpi revealed abundant PrPSc in control mice but much less PrPSc in ASO-treated mice (Figure 6d). At the level of the septum (Bregma 0 mm), PrPSc was diminished in the frontal cerebral cortex and striatum/caudate nucleus on the side of the cannula. At the level of the hippocampus, the PrPSc signal was depressed bilaterally in the cerebral cortex, hippocampus, and amygdala. The ASO 771–treated mice killed at 126 dpi showed considerable PrPSc in the right thalamus where they had been inoculated with RML prions; they also harbored PrPSc deposits in the right corpus callosum. At the level of midbrain 1 (Bregma −2.92 mm), no PrPSc was detected in the cerebral cortex, substantia nigra, ventral hippocampus, or periaquel aquaduct. In the brain stem (midbrain/pons/medulla), PrPSc levels were reduced ~50% compared to PBS-treated mice (Figure 6d). Immunohistochemical staining for PrPSc showed more intense staining in the hippocampus on the contralateral side of cannulation (data not shown).
In a survival study, six control, prion-inoculated mice that received PBS developed neurological dysfunction with a mean incubation period of 136 ± 4 days. Eight mice treated with ASO 771 had a mean incubation period of 193 ± 10 days, representing a 40% prolongation in the incubation time (Figure 6b). One ASO-treated mouse exhibited signs of CNS dysfunction at 132 dpi while the other seven ASO-treated mice succumbed to disease between 183–228 dpi. ASO 771 administration for 14 days substantially reduced PrPSc levels in prion-infected mice, even 112 days after treatment ceased (126 dpi) at the time of killing (Figure 6d). ASO-treated mice exhibited PrPC levels that were slightly lower than those found in PBS-treated, control mice at 126 dpi (see Figure 6c). A persistent, ASO-induced reduction in PrPC expression was observed in periventricular regions as well as the hippocampus and cerebellum.
Delayed infusion of ASO 771. To assess the effectiveness of ASOs in mice with an established prion infection, we began infusing ASO 771 at 60 dpi. At this point in the incubation period, FVB mice remain asymptomatic but their brains show PrPSc accumulation as well as initial changes of astrocytic gliosis.11 Beginning 60 days after intracerebral inoculation with RML prions, the FVB mice received either 75 µg/day of ASO 771 (n = 24) or PBS (n = 8) by ICV. While 75 µg/day of ASO 771 was well-tolerated in uninfected FVB mice for 14 days, this dose was toxic in prion-infected mice, which had to be killed after 11 days. The toxicity of ASO 771 appears to be related to the stage of prion disease because similar ICV infusion started 1 dpi was well-tolerated (compare Figures 6 and 7). This illness was likely unrelated to prion disease. That the illness observed in prion-infected FVB mice at 11 days after ICV infusion was related to ASO 771 is supported by the well-tolerated injection of PBS in control, infected mice that survived for 2 months following 11–14 days of ICV infusion and developed signs of scrapie at 131 ± 1 days (n = 4).
Figure 7.

Treatment with ASO 771 at the midpoint of disease reversed PrPSc deposition but caused atypical illness. (a) Experimental design. On day 0, FVB mice were inoculated with RML prions. Beginning at 60 dpi, ASO 771 was delivered via ICV at 75 µg/day for 11 days, when they developed atypical illness. Mice were killed at 71 dpi and their brains analyzed for (c) PrPC and (b,d) PrPSc. (b) Immunohistochemistry for PrPSc was performed on formalin-fixed, paraffin-embedded tissue sections through the hippocampus by the hydrolytic autoclaving method and with recFab HuM-P. (c,d) Histoblots indicate that both PrPC and PrPSc were reduced following administration of ASO 771. Reduction of PrPC appeared more prominent on the cannulation side, whereas PrPSc depletion occurred bilaterally. Sections were left undigested to visualize total PrP levels as shown in c or subjected to GdnHCl denaturation and PK digestion to visualize PrPSc levels as shown in d, then stained with the D18 antibody. Bar in b represents 50 µm and applies to all the images in the panel. ASO, antisense oligonucleotide; cc, corpus collosum; hp, hippocampus; ICV, intracerebral ventricular; nc, neocortex; PBS, phosphate-buffered saline; PrPC, cellular prion protein; PrPSc, disease-causing prion protein.
The brains of ICV-infused mice were taken at 71 dpi and examined by immunohistochemistry to detect vacuolation and PrPSc plaque deposition (Figure 7b) as well as by histoblotting in order to measure PrPC and PrPSc (Figure 7c,d). In mice receiving PBS, PrPSc deposition was widely distributed on both sides of the brain while those treated with ASO 771 exhibited considerably less PrPSc. Notably, much of the PrPSc remaining after ASO infusion was found in plaque-like deposits along the corpus callosum (Figure 7b). Histoblots prepared from brains of ASO 771–treated mice showed bilateral reductions of PrPC in the septum, hippocampus, and midbrain 1 region, albeit with a greater suppression on the cannulated, left side (Figure 7c). PrPSc levels were also reduced bilaterally in the hippocampus, substantia nigra, ventral hippocampus, cerebral cortex, midbrain, and brain stem after treatment with ASO 771 (Figure 7d). PrPSc was cleared from the thalamus, amygdala, and caudate nucleus on the cannulated, left side, but persisted in the corpus callosum and the thalamus on the contralateral right side. The control FVB mice receiving the PBS infusion and killed at 71 dpi showed PrPC and PrPSc deposits throughout their brains.
Discussion
The dual action of ASO 771—(i) specific degradation of Prnp mRNA and (ii) nonspecific reduction of PrPSc—complicates any interpretation of the studies described here. The PrP-specific ASO 771 suppressed PrPC expression by binding to Prnp mRNA and rendering these transcripts susceptible to digestion by RNase H.10 Independent of the ASO sequence, these PS-DNA oligonucleotides reduced PrPSc levels, as previously reported for ScN2a cells.5,6 The mechanism by which ASOs diminish PrPSc remains to be determined; it is unclear whether ASOs exert their antiprion action by inhibiting nascent PrPSc formation or accelerating PrPSc clearance. In ScN2a cells, the EC50 value for ASO 771 in lowering PrPC levels was 50 nmol/l and in reducing PrPSc levels was 4 nmol/l. In earlier studies, the EC50 value for a CpG PS-DNA 22mer was 4,000 nmol/l for lowering PrPC and was 70 nmol/l for reducing PrPSc.6 This 22mer was not selected for its Prnp mRNA-lowering activity but rather for its ability to prolong the incubation times of mice inoculated ip with RML prions.12
While only sequence-specific ASOs targeting mouse Prnp reduced PrPC levels, both sequence-specific ASOs and scrambled ASO 923 cleared PrPSc in ScN2a cells (Figure 2b). Dose-response experiments in ScN2a cells revealed that slightly higher concentrations of ASO 923 were required to reduce PrPSc levels compared to those found with sequence-specific ASO 771 (Figure 2d). In addition, longer treatment periods with ASO 923 were required to achieve the same level of knockdown as ASO 771 (Figure 2c). A similar sequence-independent reduction in PrPSc levels was observed in vivo (Figure 5d,e). Together, the results suggest that PrP-targeted, PS-modified ASOs can reduce brain prion levels by two independent mechanisms: reduction of Prnp mRNA and diminution of PrPSc. The in vivo studies with ASOs reported here argue that the reduction in PrPSc was greater than the diminution in PrPC; however, we cannot deduce from our data the relative contribution of PrPC suppression to reducing the level of PrPSc.
The long tissue half-life of the ASOs (20–40 days in vivo) (ISIS unpublished data) and their ability to suppress PrPC and PrPSc expression for more than 100 days following cannula removal are noteworthy. When ASO 771 was administered immediately following prion inoculation for 14 days, PrPSc levels were reduced by 95% when measured 36 days after removal of the cannula (Figure 5e). Although ASO 771 administration significantly impeded PrPSc replication and extended survival times by almost 2 months, it did not eliminate PrPSc and the mice eventually succumbed to prion disease (Figure 6b). Interestingly, at 112 days after removal of the cannula, reduced levels of PrPC and PrPSc persisted, contending that ASO 771 was still present at sufficiently high concentrations to be biologically active (Figure 6c,d).
Intraventricular infusion of ASOs. ASOs dispensed directly into the ventricular system via the CSF of the lateral ventricle have been previously reported to be an effective way to achieve widespread delivery throughout the brain and penetrate the neural tissue of rodents, dogs, and non-human primates.13 Approximately half of the cannulated FVB mice died shortly after surgery—many of these deaths were due to hippocampal lesions resulting from accidental intraparenchymal catheter implantation. These mice were excluded from the study as well as the incubation time calculations. Those brain regions near the tip of the cannula were exposed to the highest levels of ASO 771, and reductions in PrPC and PrPSc occurred up to distances of 5.88 mm Bregma.
ICV injection of ASO 771 through a cannula on the left side of the brain produced greater reductions of PrPSc than PrPC on the contralateral, right side where the prions were inoculated into the thalamus (Figure 6c,d). The greater reduction in PrPSc levels compared to PrPC is readily explained by the sequence-independent, ASO-mediated diminution in PrPSc demonstrated in cultured cells (Figure 2d). Our findings are consistent with results from an earlier study reported by one of us (S.B.P.) where PS-DNA (22mers) containing a methylated CpG motif (non-immune reactive) and scrambled CpG motifs equally diminished PrPSc levels in ScN2a cells, indicating a sequence-independent reduction of PrPSc.6 The EC50 values for these 22mers were 5 µmol/l for PrPC and 70 nmol/l for PrPSc. Bioassays with treated cell extracts demonstrated that the prion titers were reduced in the PS-DNA–treated ScN2a cells.
In the work presented here, the EC50 value of the sequence-specific ASO 771 was 100-fold lower for PrPC (50 nmol/l) reduction than previously reported for the CpG motif 22mers (5 µmol/l). In addition, we did not observe a significant reduction of PrPC with the scrambled ASO 923, arguing that the PrPC reduction we observed with ASO 771 resulted from degradation of the Prnp transcripts.
In another study, a series of random-sequence, single-stranded PS-DNA 40mers was added to ScN2a cells, resulting in diminished levels of PrPSc.5 When Tg(SHaPrP)7 mice were given daily ip doses of one random-sequence PS-DNAs beginning 3 days before ip inoculation with SHa prions, the incubation times were prolonged from 88 to 330 days. After prion inoculation, the Tg7 mice received ip injections of the random-sequence PS-DNAs three times per week for 4 weeks. Since PS-DNAs administered ip did not traverse the blood-brain barrier, it is likely that the prolongation of incubation periods reported in these studies was due to systemic degradation of PrPSc outside of the CNS.
In the study described here, we intracerebrally inoculated RML prions into the right thalamus and 1 day later inserted a cannula on the left side of the brain, through which we infused ASO 771 for 14 days. This ASO treatment protocol increased the incubation time from 136 to 193 days (Figure 6b). In the ASO 771–treated mice that were killed at 126 dpi, the PrPSc levels were reduced throughout the brain compared to the untreated controls, except in the right thalamus where both control and treated mice had been inoculated.
It is instructive to compare the prolongation of the incubation time by ICV infusion of ASO 771–treated mice with that of Prnp+/0 mice where PrPC levels were reduced ~50%. In both cases, the mice had diminished PrPC from very early in the incubation period. In studies by one of us (S.B.P.), Prnp+/0/FVB mice inoculated with RML prions developed CNS dysfunction between 400 and 465 days after intracerebral inoculation compared to wt mice exhibiting neurologic signs at 146 days.14 In another study, Prnp+/0 mice developed CNS dysfunction between 260 and 350 days after receiving RML prions ic.15 Recently, a third study using the same gene knockout showed signs of neurological disease between 240 and 300 days after intracerebral inoculation of RML prions.16 In the third study, prion titers were measured in the brains of Prnp+/0 mice using endpoint titrations in cultured cells. At 100 days after intracerebral inoculation, the brain titers were found to reach a maximum but more than another 150 days passed before signs of brain dysfunction were observed. The authors argued that an isoform other than PrPSc is neurotoxic and that this alternative form induces clinical signs. Whether infusions of ASOs can help decipher the molecular pathogenesis of prion disease is unknown at present.
Concluding remarks. The sequence-independent inhibition of PrPSc formation complicated the interpretation of our findings using ASO 771. Delaying the infusion of ASOs until 60 days after inoculation resulted in many acute deaths (Figure 7), which prevented us from measuring the antiprion effect of these PS-modified oligonucleotides. We were also unable to lower substantially the PrPC levels throughout the brain after ICV infusion of ASO 771; whether this approach can be modified into an effective therapeutic regimen remains to be determined.
Materials and Methods
ASO synthesis. Twelve oligonucleotides corresponding to regions within the 3′ UTR were synthesized and purified as previously described.17 Oligonucleotides were PS-modified, chimeric oligonucleotides composed of five 2′-O-(2-methoxy)ethyl modifications on both the 5′ and 3′ ends, and 10 oligodeoxynucleotides in the center to support RNase H activity.18 Oligonucleotide sequences were as follows: ASO 742 (TATATTCTTATTGGCCCGGT), ASO 747 (GCCTATGCTAAGTTACATGT), and ASO 771 (CCAAGGGTCACACGGTAAGC). ASO 923 (CCTTCCCTGAAGGTTCCTCC) was used as a control oligonucleotide because it shares identical chemistry and length to the PrP-targeted ASOs, is not predicted to hybridize to any known human or rat genes, and was previously shown not to have detectable effects in tissue culture or in mouse models.19
Quantitative RT-PCR. Total RNA was isolated using an RNeasy Mini prep kit (QIAGEN, Valencia, CA) according to the manufacturer's protocol. We combined 5–10 ng total RNA with 100 nmol/l of each of the gene-specific, dual-labeled probes, and forward and reverse primers in a buffered solution consisting of 1× TaqMan Buffer A (Applied Biosystems, Foster City, CA), 5.5 mmol/l MgCl2, 200 mmol/l concentrations of each dNTP (Amersham Biosciences, Piscataway, NJ), 2 U RNase inhibitor, 0.625 U AmpliTaq Gold, and 6.25 U murine leukemia virus reverse transcriptase. Except for dNTP solutions, all reagents above were obtained from Applied Biosystems. Quantitative RT-PCR reactions were conducted and analyzed on an ABI Prism 7700 Sequence Detector (Applied Biosystems). Glyceraldehyde 3-phosphate dehydrogenase mRNA levels were used as an internal reference for normalization among samples. Primer probe set sequences for PrP were: forward, 5′-TCTGTGTCCCCCATAGGCTAA-3′ reverse, 5′-AGAGCAACTGGTCTACTGTACATTTCC-3′ probe, 5′-CCCCTGGCACTGATGGGCCC-3.
Cell culture. For b.END, N2a and GT1 lines, cells were grown in 6-cm dishes in minimal essential medium until attaining 90–95% confluence. Cells were trypsinized and diluted tenfold into 60-mm plates containing 4 ml of minimal essential medium. On the following day, ASOs were added to the cells at various concentrations in their normal growth media and incubated for variable periods of time. For b.END cells, cells were washed once with PBS and were transfected using the lipofectamine transfection reagent (Invitrogen, Carlsbad, CA) using 3 µl lipofectin/1 ml Optimem/100 nmol/l ASO. The transfection mix was removed after 4 hours and replaced with normal growth media. All incubations were performed at 37 °C. Cells were harvested in 0.5 ml cold lysis buffer (10 mmol/l Tris-HCl, pH 8; 100 mmol/l NaCl; 0.5% NP-40; and 0.5% deoxycholate). RNA was harvested at different time points; for b.END cells, RNA was harvested 24 hours after transfection.
Stock b.END and N2a cells were maintained in minimal essential medium. The cells were harvested using 4 ml of 0.5% trypsin and plated in a 1:20 dilution, fed on day 4, and trypsinized again in a 1:3 dilution onto 10-cm plates on day 6. All media were supplemented with 10% fetal bovine serum, 2 mmol/l Glutamax (GIBCO BRL, Carlsbad, CA), 100 units/ml penicillin, and 100 units/ml streptomycin in a humidified 37 °C incubator with 5% CO2.
Immunoblotting. Confluent 6-cm diameter plates (~4 × 106 cells) were washed in PBS and then lysed by the addition of 500 µl of lysis buffer. The nuclear pellet was removed, and the protein concentration was determined by bicinchoninic acid assay as recommended by the manufacturer (Pierce, Rockford, IL). Proteinase K was added (1:50, PK:total protein), and the lysate was incubated at 37 °C for 1 hour. The reaction was stopped by the addition of 2 mmol/l phenylmethanesulfonylfluoride. Insoluble material was precipitated by ultracentrifugation at 48,000g for 1 hour at 4 °C. The pellet was resuspended in loading buffer, boiled for 5 minutes, then run on a 15% Tris·HCl gel for western blot analysis by using D18 as the detection antibody. Capture enzyme-linked immunosorbent assays with D18 were performed as previously described.20
Quantification of PrPC levels. PrPC and PrPSc levels were quantified by either enzyme-linked immunosorbent assay or performing densitometry of scanned western blots films using NIH Image J software and expressed as percentages relative to that measured in cells treated with PBS.
Inoculation and animal studies. All animal protocols were approved by the University of California San Francisco Institutional Animal Care and Use Committee and met ethical standards for animal experimentation. Ip inoculations were performed in the abdomen. For therapeutic studies, FVB mice were anesthetized with isoflurane, then inoculated with 30 µl of mouse-adapted RML prions. Inocula were injected directly into the right thalamus. Mice were monitored thrice weekly. When they showed signs of prion disease, mice were euthanized. Brain cannulation and Alzet pump insertion in the subcutaneous pocket were performed on anesthetized animals.
Surgical placement of osmotic pumps and harvesting tissues for analysis. FVB mice weighing at least 20 g were placed in a chamber for the induction of anesthesia using 3–5% isoflurane in an air mixture. The animal was placed in a stereotaxic apparatus (Kopf 942, two-channel digital small animal stereotax; David Kopf Instruments, Tujunga, CA); a surgical plane of anesthesia was maintained with 2.5% isoflurane by a nose cone fitted to the stereotaxic instrument. A 1.5–2 cm midline incision was made in the scalp from the posterior of the occipital plate to the line connecting the eyes. A subcutaneous pocket measuring 5–6 cm deep was made with blunt dissection posterior from the incision over the left flank. The preloaded Alzet 2002 pump with tubing attached to the infusion cannula was inserted pump-first into the subcutaneous pocket, leaving the cannula outside of the incision. The top tab of the infusion cannula was placed in a cannula-holding apparatus (Plastics One, Roanoke, VA). The tubing of the cannula was aligned with the hole in the skull and advanced into the hole until the base was against the skull. The incision was closed by sutures.
Histoblot. Histoblotting was performed as described previously.21 Coronal cryosections of 10-µm thickness were collected at the level of the septum (Bregma 0 mm). For prion-infected FVB mice, additional cryosections were taken from the hippocampus (Bregma −1.64 mm), midbrain 1 (Bregma −2.92 mm), midbrain 2 (Bregma −4.20 mm), pons (Bregma −5.46 mm), and cerebellum (Bregma −5.88 mm). Sections were placed on glass slides, then transferred to nitrocellulose membranes that were wetted in lysis buffer. Nitrocellulose strips were immersed in 100 mmol/l NaOH solution, incubated for 1 hour at room temperature, rinsed in tris-buffered saline and Tween-20 (TBST), immersed again in 100 mmol/l NaOH solution, incubated for 1 hour at room temperature, rinsed in TBST, then immersed in 3 mol/l guanidine isothiocyanate solution for 10 minutes at room temperature, and rinsed in TBST. Strips were blocked in 5% nonfat milk (made in TBST) for 30 minutes at room temperature, stained with the anti-PrP recombinant antibody D18 followed by alkaline phosphatase–conjugated, goat anti-human secondary antibody and detection with 5-bromo-4-chloro-3-indolyl-phosphate/nitro blue tetrazolium (BCIP/NBT).
Neuropathology. Brains were removed rapidly from euthanized animals and either immersion-fixed in 10% buffered formalin or frozen on dry ice for neuropathological analysis. For evaluation of neurodegeneration, paraffin-embedded brains sections (8 µm) were stained with hematoxylin and eosin. For immunohistochemistry, PrPSc was detected on formalin-fixed, paraffin-embedded tissue sections by the hydrolytic autoclaving method and with recFab HuM-P against PrP.22 For evaluation of reactive astrocytic gliosis, we used a rabbit antiserum to glial fibrillary acidic protein (Dako, Carpinteria, CA) with peroxidase immunohistochemistry.23
Acknowledgments
This work was supported by grants from the National Institutes of Health (AG031220, AG10770, and AG021601) as well as by gifts from the Sherman Fairchild Foundation, Schott Foundation for Public Education, and Rainwater Charitable Foundation. The authors thank the staff at the Hunters Point animal facility.
References
- Tilly G, Chapuis J, Vilette D, Laude H., and, Vilotte JL. Efficient and specific down-regulation of prion protein expression by RNAi. Biochem Biophys Res Commun. 2003;305:548–551. doi: 10.1016/s0006-291x(03)00805-2. [DOI] [PubMed] [Google Scholar]
- Daude N, Marella M., and, Chabry J. Specific inhibition of pathological prion protein accumulation by small interfering RNAs. J Cell Sci. 2003;116 Pt 13:2775–2779. doi: 10.1242/jcs.00494. [DOI] [PubMed] [Google Scholar]
- Pfeifer A, Eigenbrod S, Al-Khadra S, Hofmann A, Mitteregger G, Moser M.et al. (2006Lentivector-mediated RNAi efficiently suppresses prion protein and prolongs survival of scrapie-infected mice J Clin Invest 1163204–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MD, Farmer M, Mirabile I, Brandner S, Collinge J., and, Mallucci GR. Single treatment with RNAi against prion protein rescues early neuronal dysfunction and prolongs survival in mice with prion disease. Proc Natl Acad Sci USA. 2008;105:10238–10243. doi: 10.1073/pnas.0802759105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocisko DA, Vaillant A, Lee KS, Arnold KM, Bertholet N, Race RE.et al. (2006Potent antiscrapie activities of degenerate phosphorothioate oligonucleotides Antimicrob Agents Chemother 501034–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpuj MV, Giles K, Gelibter-Niv S, Scott MR, Lingappa VR, Szoka FC.et al. (2007Phosphorothioate oligonucleotides reduce PrP levels and prion infectivity in cultured cells Mol Med 13190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallucci G, Dickinson A, Linehan J, Klöhn PC, Brandner S., and, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302:871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- Safar JG, DeArmond SJ, Kociuba K, Deering C, Didorenko S, Bouzamondo-Bernstein E.et al. (2005Prion clearance in bigenic mice J Gen Virol 86Pt 102913–2923. [DOI] [PubMed] [Google Scholar]
- Bennett CF., and, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- Wu H, Lima WF, Zhang H, Fan A, Sun H., and, Crooke ST. Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J Biol Chem. 2004;279:17181–17189. doi: 10.1074/jbc.M311683200. [DOI] [PubMed] [Google Scholar]
- Tamgüney G, Francis KP, Giles K, Lemus A, DeArmond SJ., and, Prusiner SB. Measuring prions by bioluminescence imaging. Proc Natl Acad Sci USA. 2009;106:15002–15006. doi: 10.1073/pnas.0907339106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi S, Lipford G, Wagner H., and, Kretzschmar H. Postexposure prophylaxis against prion disease with a stimulator of innate immunity. Lancet. 2002;360:229–230. doi: 10.1016/S0140-6736(02)09513-2. [DOI] [PubMed] [Google Scholar]
- Smith RA, Miller TM, Yamanaka K, Monia BP, Condon TP, Hung G.et al. (2006Antisense oligonucleotide therapy for neurodegenerative disease J Clin Invest 1162290–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB, Groth D, Serban A, Koehler R, Foster D, Torchia M.et al. (1993Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies Proc Natl Acad Sci USA 9010608–10612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büeler H, Raeber A, Sailer A, Fischer M, Aguzzi A., and, Weissmann C. High prion and PrPSc levels but delayed onset of disease in scrapie-inoculated mice heterozygous for a disrupted PrP gene. Mol Med. 1994;1:19–30. [PMC free article] [PubMed] [Google Scholar]
- Sandberg MK, Al-Doujaily H, Sharps B, Clarke AR., and, Collinge J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature. 2011;470:540–542. doi: 10.1038/nature09768. [DOI] [PubMed] [Google Scholar]
- Cheruvallath ZS, Cole DL., and, Ravikumar VT. A novel solid support for synthesis of oligonucleotide 3'-phosphorothioate monoesters. Bioorg Med Chem Lett. 2003;13:281–284. doi: 10.1016/s0960-894x(02)00922-8. [DOI] [PubMed] [Google Scholar]
- McKay RA, Miraglia LJ, Cummins LL, Owens SR, Sasmor H., and, Dean NM. Characterization of a potent and specific class of antisense oligonucleotide inhibitor of human protein kinase C-alpha expression. J Biol Chem. 1999;274:1715–1722. doi: 10.1074/jbc.274.3.1715. [DOI] [PubMed] [Google Scholar]
- Drygin D, Barone S., and, Bennett CF. Sequence-dependent cytotoxicity of second-generation oligonucleotides. Nucleic Acids Res. 2004;32:6585–6594. doi: 10.1093/nar/gkh997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Phuan PW, Perkins B, Ullman J, May BC, Cohen FE.et al. (2007Cell division modulates prion accumulation in cultured cells Proc Natl Acad Sci USA 10417971–17976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraboulos A, Jendroska K, Serban D, Yang S-L, DeArmond SJ., and, Prusiner SB. Regional mapping of prion proteins in brains. Proc Natl Acad Sci USA. 1992;89:7620–7624. doi: 10.1073/pnas.89.16.7620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramoto T, Kitamoto T, Tateishi J., and, Goto I. The sequential development of abnormal prion protein accumulation in mice with Creutzfeldt-Jakob disease. Am J Pathol. 1992;140:1411–1420. [PMC free article] [PubMed] [Google Scholar]
- Muramoto T, DeArmond SJ, Scott M, Telling GC, Cohen FE., and, Prusiner SB. Heritable disorder resembling neuronal storage disease in mice expressing prion protein with deletion of an alpha-helix. Nat Med. 1997;3:750–755. doi: 10.1038/nm0797-750. [DOI] [PubMed] [Google Scholar]