

Abstract
Microglia are the resident macrophages of the central nervous system (CNS). In physiological conditions, resting microglia maintain tissue integrity by scanning the entire CNS parenchyma through stochastic and complex movements of their long processes to identify minor tissue alterations. In pathological conditions, over-activated microglia contribute to neuronal damage by releasing harmful substances, including inflammatory cytokines, reactive oxygen species, and proteinases, but they can provide tissue repair by releasing anti-inflammatory cytokines and neurotrophic factors. The reasons for this apparent paradox are unknown. In this paper, we first review the physiological role as well as both detrimental and beneficial actions of microglial during acute CNS disorders. Further, we discuss the possible reasons for this microglial dual role following CNS insults, considering that the final microglial phenotype is a direct consequence of both noxious and beneficial stimuli released into the extracellular space during the pathological insult. The nature of these micro-glial ligands is unknown, but we hypothesize that harmful and beneficial stimuli may be preferentially located at specific anatomical niches along the pathological environment triggering both beneficial and deleterious actions of these glial cells. According to this notion, there are no natural populations of detrimental microglia, but is the pathological environment that determines the final microglial phenotype.
Keywords: Beneficial, CNS damage, detrimental, glial cells, pattern recognition receptors, spinal cord injury, stroke
Introduction
Microglia are believed to derive from monocytes that invade the developing central nervous system (CNS) and persist over the adult life as resident macrophages (Alliot et al. 1999). A recent study using fate-mapping analysis confirmed that these glial cells derive from primitive myeloid progenitors that arise before embryonic day 8 (Ginhoux et al. 2010) and that postnatal hematopoietic progenitors do not contribute to microglia homeostasis in the adult brain. These cells perform a multitude of physiological roles in normal adult CNS (Nimmerjahn et al. 2005; Ransohoff and Perry 2009) and are believed to perform both detrimental and beneficial actions during acute and chronic neural disorders (Block et al. 2007; Perry et al. 2010). In physiological conditions, they stochastically move their processes in several directions in a complex way and scanning for minor tissue alterations for maintaining tissue integrity (Stence et al. 2001; Davalos et al. 2005; Nimmerjahn et al. 2005). Nevertheless, there is experimental evidence suggesting that activated microglia perform both beneficial and detrimental actions after CNS disorders including spinal cord injury (SCI), stroke, multiple sclerosis, amyotrophic lateral sclerosis, prion, Parkinson, Huntington, and Alzheimer diseases (Block et al. 2007; Ekdahl et al. 2009; Perry et al. 2010).
Why do microglia have a dual role after CNS diseases? There is not a definitive answer to this question. In this paper, we first review the dual role of microglia during acute CNS disorders. Further, we discuss the possible reasons for this duality under pathological conditions. We hypothesize that both harmful and beneficial stimuli are released upon injury into specific anatomical niches along the damaged areas triggering both beneficial and deleterious actions of microglia. Depending on the CNS-affected area and disease's etiology, both noxious and beneficial microglial phenotypes might coexist along the pathological environment. According to this notion, there are no natural populations of deleterious microglia, but is the pathological environment that determines the microglial phenotype.
The Physiological Roles of Microglia
Microglia patrol the adult CNS environment in physiological conditions
In the mature CNS, microglia adopt a highly ramified morphology under physiological conditions (Nimmerjahn et al. 2005). A study using confocal time-lapse analysis in hippocampal slices first has shown that microglia branches are highly dynamic structures upon activation (Stence et al. 2001). Further, two-photon laser scanning microscopy allowed visualization of fluorescent resting microglia in the brain of alive animals, showing that these glial cells continuously patrol the CNS parenchyma several times a day through stochastic movements of their long and fine branches maintaining tissue integrity (Davalos et al. 2005; Nimmerjahn et al. 2005). Under physiological conditions, there exist mechanisms assuring that microglial cells do not develop patterns of activation with undesirable consequences for CNS integrity (Bessis et al. 2007; Ransohoff and Perry 2009).
Neurons control microglial function by physical contact or by releasing neurotransmitters, peptides and/or growth factors including gamma-aminobutyric acid (GABA), glutamate, catecholamines, CD22, CCL21, fraktalkine, which act on receptors present on microglia membrane (Bessis et al. 2007). It has been shown that in organotypic hippocampal cultures active neurons release neurotrophins, such as neural growth factor (NGF), which control the expression of major histocompatibility class II (MHC-II) in microglia by acting, at least partially, on the p75 neurotrophin receptor (Neumann et al. 1998). Neuronal secretion of CD22 inhibits microglial release of pro-inflammatory cytokines by acting on CD45 receptor (Mott et al. 2004) and expression of CD200 may be important for controlling tumor necrosis factor-α (TNF-α) released by these glial cells (Broderick et al. 2002; Lyons et al. 2007). Finally, physical interaction between neuronal CD200 and CD200R present on microglia membrane likely represents an alternative way by which neurons can control microglial function (Broderick et al. 2002; Lyons et al. 2007). Microglia also express a number of neurotransmitter receptors indicating that activity-related release of neurotransmitters by neurons contribute for microglial control in physiological conditions (Bessis et al. 2007). These data clearly illustrate the necessity of controlling both inflammatory and immune microglial functions in physiological conditions in order to maintain the integrity of CNS circuits.
Microglia and Adult Neurogenesis
New neurons are generated in the adult brain from neural stem/progenitor cells present in the subventricular zone (SVZ) (Doetsch et al. 1997; Alvarez-Buylla and Garcia-Verdugo 2002) and subgranular zone of hippocampal dentate gyrus (Seri et al. 2001). Microglia seem to play an important physiological role of controlling adult neurogenesis in normal conditions (Aarum et al. 2003; Walton et al. 2006; Ekdahl et al. 2009). In vitro, mouse-derived microglia release soluble factors, which contribute to migration and differentiation of neural progenitors (Aarum et al. 2003; Walton et al. 2006). Microglia instruct neurogenesis in adult SVZ in culture (Walton et al. 2006). In adherent culture systems, there is a normal senescence and decrease in the number of progenitor cells (Walton et al. 2006). This is coincident with decreased numbers of microglial cells (Walton et al. 2006). Xanthosine treatment results in extended microglia lifespan, concomitant with increased neurogenic potential of SVZ-derived cells (Walton et al. 2006). In this experimental condition, a MAC-1-saporin antibody, which depletes microglia, decreases neurogenic potential, while microglia-conditioned medium restores neurogenesis (Walton et al. 2006). A recent in vivo study suggests that microglia contribute to hippocampal neurogenesis in adrenalectomized rats (Battista et al. 2006). In this study, the number of activated microglia displaying a more ramified morphology, not full phagocytes, correlated with increased neurogenesis and number of nestin-positive cells (Battista et al. 2006). Nevertheless, the role of microglia on adult neurogenesis is an open question. Experimental depletion of SVZ microglia using a Mac-1 antibody conjugated to saporin did not affect numbers and proliferation of migrating neuroblasts in the SVZ in nonpathological conditions or migration of neuroblasts after striatal stroke (Heldmann et al. 2011).
Several other functions are performed by microglia. A detailed discussion of microglial functions can be obtained in Ransohoff and Perry (2009).
Microglia Activation and Acute CNS Disorders
Morphological and molecular correlates of microglia activation
Microglia are extremely sensible to minor alterations on the CNS microenvironment, even ionic disbalance and stress (Kreutzberg 1996; Sugama et al. 2007; Ransohoff and Perry 2009). These cells are activated in pathological conditions, which is reflected in both morphological and biochemical alterations on their structure (Streit et al. 1999; Ransohoff and Perry 2009). Microglia activation involves a conspicuous change in their ramified morphology to an intermediate and amoeboid form culminating in a round morphological profile of full phagocytes (Morioka et al. 1993; Lehrmann et al. 1997; Thored et al. 2009). Concomitant with morphological alterations, microglial cells change their genetic machinery and upregulate several transcription factors (for example, NF-κB), cytoplasmic and surface molecules including MHC classes I and II, complement C3, Fc, thrombin, scavenger receptors (i.e., CD36, SR-A, CD204, SR-BI), cytokine, chemokine, CD4 and CD8 receptors, toll-like receptors, and several oxidative enzymes, such as NADPH oxidase (Perry and Gordon 1987; Schroeter et al. 1994; Jander et al. 1998; Streit et al. 1999; Husemann et al. 2002; Block et al. 2007; Ransohoff and Perry 2009).
An important question is which signals activate microglia in the event of tissue damage. These mechanisms are not completely clear. Nevertheless, there is experimental evidence suggesting that the release of purine nucleotides, including ATP, ADP, and UTP, by injured neurons is an important mechanism by which microglia are informed of tissue injury (Davalos et al. 2005; Nimmerjahn et al. 2005). These nucleotides seem to act on P2Y receptors in both microglia- and astrocytes-mediating chemotactic response of microglia branches (Davalos et al. 2005; Nimmerjahn et al. 2005). In addition, activation of connexin hemichannels in astrocytes seems to mediate amplification of the phenomenon by inducing a continuous release of purine nucleotides by these glial cells, which is fundamental for directing microglial processes to the injury site (Davalos et al. 2005; Nimmerjahn et al. 2005).
Beneficial actions of microglia after CNS diseases
Activated microglia can be beneficial in several experimental models of CNS diseases (Neumann et al. 2006; Schwartz et al. 2006; Lalancette-Hebert et al. 2007; Thored et al. 2009). After experimental axotomy of facial nerve, there is microglial activation inside facial motor nucleus (Graeber et al. 1988; Streit 2002). Microglia clearly play a beneficial role contributing to reinnervation of target muscles (Graeber et al. 1988; Streit 2002). In this experimental paradigm, a regeneration program involving microglial proliferation and activation is triggered resulting in the most conspicuous example of a neuro-protective role of microglia after nervous system damage (Streit 2002).
Microglia play a proregenerative role after SCI. Engraftment of cultured microglial cells into lesioned spinal cord induces axonal sprouting (Rabchevsky and Streit 1997). In this experimental condition, transplanted microglial cells seem to release growth factors, which contribute to axonal regeneration (Rabchevsky and Streit 1997). It has been proposed by Schwartz and colleagues that microglia/macrophages can be highly neuroprotective after SCI and other CNS diseases (Rapalino et al. 1998; Bomstein et al. 2003; Schwartz et al. 2006). According to these authors, neuroprotective microglia can express markers of antigen-presenting cells, including MHC-class II and B7.2, which allow interactions with lymphocyte with subsequent release of growth factors rendering a more amenable environment for neural regeneration (Butovsky et al. 2001; Bomstein et al. 2003; Schwartz et al. 2006).
Recent experimental evidences suggest that activated microglia may be highly neuroprotective after stroke (Neumann et al. 2006, 2008; Lalancette-Hebert et al. 2007). Application of BV2 microglia is neuroprotective after oxygen–glucose deprivation in organotypic hippocampal slice cultures (Neumann et al. 2006). Using three-dimensional (3D) two-photon microscopy, these authors have shown that transplanted microglia engage in physical contact with neurons in damaged area, protecting them in the experimental circumstances. In addition, blockage of microglia activation with minocycline impairs the neuroprotection afforded (Neumann et al. 2006). Using transgenic mice in which selective ablation of proliferating microglia is feasible, Lalancette-Hebert et al. (2007) have shown that microglia is clearly neuroprotective after middle cerebral artery occlusion (MCAO). After microglial ablation, there was an increase in the number of apoptotic cells concomitant with increased proinflammatory molecules (Lalancette-Hebert et al. 2007). In this study, Mac-2+ resident microglia release the proneurogenic molecule insulin-like growth factor (IGF-1), which likely contribute to the microglia-induced neuroprotection (Lalancette-Hebert et al. 2007). Recent studies suggest that microglia may be beneficial by engulfing neutrophils (Neumann et al. 2008) and releasing TNF-α (Lambertsen et al. 2009) after ischemia. In addition, microglia may be beneficial through their phagocytic functions. Some studies suggest that phagocytosis of injured tissue is important for remodeling and may limit secondary damage following brain hemorrhage (Zhao et al. 2007). Recent studies suggest that microglia may shape hippocampal adult neurogenesis by clearing out apoptotic newborn cells, which illustrates the important phagocytic function of microglia (Sierra et al. 2010).
Recently, we have shown that there is long-lasting microglial activation with a proneurogenic phenotype in SVZ after stroke (Thored et al. 2009). These glial cells release IGF-1 in late survival times after stroke, which has been confirmed by affymetrix analysis and quantitative polymerase chain reaction (PCR) (Thored et al. 2009). The results indicate that long-term activation of microglia in SVZ after stroke is important for regulating the previously described long-lasting neurogenesis in SVZ (Thored et al. 2006).
Detrimental actions of microglia after CNS diseases
There is clear experimental evidence suggesting that overactivated microglia may be extremely detrimental following acute neural disorders, including SCI (Popovich et al. 1999, 2002; Gomes-Leal et al. 2005; Kigerl et al. 2009) and stroke (Yrjanheikki et al. 1999; Yong et al. 2004; Hewlett and Corbett 2006; Hayakawa et al. 2008; Schabitz et al. 2008; Wu et al. 2009; Fagan et al. 2010).
Depletion of hematogenous macrophages with clodronate induces partial hindlimb recovery and neuroprotection after acute SCI (Popovich et al. 1999). Microglia/macrophage activation seems to contribute to axonal damage following experimental injection of N-methyl-d-Aspartate (NMDA) (Gomes-Leal et al. 2005) and zymosan (Popovich et al. 2002) into the rat spinal cord. The semisynthetic tetra-cycline minocycline, an inhibitor of microglial activation, reduces secondary oligodendrocyte and axonal degeneration as well as modulates apoptosis after SCI (Stirling et al. 2004) and cell cycle inhibition attenuates microglia-induced inflammatory response and decreases cell death after SCI (Tian et al. 2007). In addition, mild hypothermic treatment reduces spinal cord motor dysfunction by decreasing microglia activation (Morino et al. 2008).
Blockage of microglial activation with minocycline induces conspicuous neuroprotection in both cortex and striatum after experimental rat MCAO (Yrjanheikki et al. 1999). Recent studies have confirmed that minocycline has a potent neuroprotective effect after ischemia by inhibiting a cytokine-like mediator (high-mobility group box 1) in microglia (Hayakawa et al. 2008). Nevertheless, it is important to bear in mind that minocycline has other pleiotropic actions, including matrix metalloproteinases-9 (MMP-9) inhibition, PARP or NFκB, scavenging of peroxynitrite, upregulation of bcl-2, and may affect cells other than microglia (Yong et al. 2004; Kim and Suh 2009). Other microglial inhibitors render neuroprotection after stroke. PJ34, a potent poly(ADP-ribose) polymerase inhibitor, abolishes microglial activation and reduces hippocampal neuronal death by 84% if administered at 8 h after 10 min of global forebrain ischemia in rats (Hamby et al. 2007). Patients with acute stroke had a better neurological outcome with minocycline treatment (Lampl et al. 2007; Schabitz et al. 2008; Fagan et al. 2010). Recent in vitro and in vivo studies suggest that the neurotoxic actions of microglia during ischemia are mediated by microglial type II metabotropic receptors, TNF-α overproduction, NF-κB activation (Kaushal and Schlichter 2008), and Nox-1-dependent NADPH oxidase (Cheret et al. 2008). In addition, it has been reported that caspase activation is an important mechanism underlying the deleterious functions of microglia (Burguillos et al. 2011).
There are experimental evidences that microglial activation is detrimental for adult neurogenesis in different models of CNS injury (Ekdahl et al. 2003; Monje et al. 2003; Hoehn et al. 2005; Liu et al. 2007). Microglial activation impairs basal hippocampal adult neurogenesis induced by tissue damage associated with status epilepticus or lipopolysaccharide (LPS) infusion (Ekdahl et al. 2003). The impaired adult neuro-genesis is restored after microglial blockage using minocycline (Ekdahl et al. 2003). Minocycline or indomethacin treatment also enhances adult neurogenesis after MCAO in rodents (Hoehn et al. 2005; Liu et al. 2007).
How to Explain the Dual Role of Microglia after CNS Diseases?
We have seen that microglia have important physiological functions on the normal CNS and a dual role after neural disorders. How to explain this apparent paradox? In the following paragraphs, based on experimental evidences, including our own data, we propose a hypothesis to explain the dual role of microglia after CNS diseases.
Microglia are fundamental components of brain innate immune system responsible for protecting neural tissue against infections (Olson and Miller 2004; Town et al. 2005; Lehnardt 2010). To perform such a role, microglia monitor the CNS environment using membrane molecules called “pattern recognition receptors” (PRRs) (Akira et al. 2006). These PRRs include toll-like receptors (Olson and Miller 2004; Downes and Crack 2010), scavenger receptors (Husemann et al. 2002), and the complement receptor 3 Mac1 (Ross 2000).
During CNS infection, PRRs interact with pathogen-associated molecular patterns (PAMPs) in the membrane of infectious agents, which triggers the release of pathogen-killer molecules by microglia, including hydrogen peroxide, NO, O2−, peroxynitrite, and a range of pro-inflammatory molecules such as prostaglandin E2, IL-1β, and TNF-α (Olson and Miller 2004; Block et al. 2007; Buchanan et al. 2010; Downes and Crack 2010; Lehnardt 2010). A main downstream event following pathogen recognition by microglia is the activation of NADPH-oxidase, which is a fundamental step for free radical release and other pro-inflammatory microglial actions (Block and Hong 2005; Qin et al. 2005; Cheret et al. 2008). This is a physiological and an appropriate response of microglia in order to eliminate pathogens and preserve tissue integrity. Nevertheless, recent studies suggest that in noninfectious diseases, such as stroke, SCI, and chronic neurodegenerative diseases, microglia might mistake noninfectious disease-associated stimuli for the ones associated with infectious diseases, activating their killing mechanisms, which normally would be used to kill pathogens, but are unintentionally used to kill neurons (Block et al. 2007; Griffiths et al. 2007).
It has been suggested that PRR activation underlies the mechanisms of cell demise in a number of noninfectious neural and nonneural diseases (Karin et al. 2006; Town et al. 2006; Griffiths et al. 2007; Lehnardt et al. 2007; Ziegler et al. 2007). For example, TLR-2 mediates CNS injury in focal cerebral ischemia (Lehnardt et al. 2007) and activation of TLR-4 by a specific type of heat shock protein may be an endogenous molecular pathway common to many forms of neuronal injury (Lehnardt et al. 2008). TLR4 and CD14 are the primary LPS receptors in microglia (Lien et al. 2000) and in several experimental circumstances LPS induces microglia activation with neurotoxic consequences (Ling et al. 2006; Pei et al. 2007; Qin et al. 2007). Activation of other microglial PRRs is also involved in neurodegeneration (Cho et al. 2005; Block et al. 2007; Pei et al. 2007). These and other results suggest that noninfectious stimuli might bind microglial PRRs activating their killing mechanisms, which normally would be used to kill pathogens, but are unintentionally used to kill neurons in noninfectious CNS diseases (Block et al. 2007; Griffiths et al. 2007).
From the experimental data described above, it is possible to infer that microglia are beneficial neuroimmune cells that might become detrimental in pathological conditions. It is likely that during neural disorders, a mosaic of detrimental and beneficial stimuli are released by altered neurons, glia, blood vessels, and other sources into the extracellular space, and microglial cells interpret them by using their surface receptors. We hypothesize that these harmful and beneficial stimuli are released into specific anatomical niches along damaged areas triggering both beneficial and deleterious actions of microglia. Depending on the CNS-affected area and disease's etiology, both noxious and beneficial microglial phenotypes might coexist along the pathological environment. According to this notion, there are no natural populations of deleterious microglia, but is the pathological environment that determines the final microglial phenotype. We have reasons to suppose that this phenomenon occurs after stroke and other neural disorders. We will present experimental evidences to substantiate this hypothesis in the following paragraphs.
In a recent study, we have described different patterns of microglial activation over weeks after MCAO in both SVZ and striatum (Thored et al. 2009). In SVZ, microglia exhibited a more ramified or intermediate morphology, signifying a downregulated inflammatory profile, whereas amoeboid or round phagocytic microglia were frequent in the periinfarct striatum (Thored et al. 2009). In this study, SVZ microglia seem to be proneurogenic as they upregulate expression of IGF-1, a growth factor known to be neurogenic in different experimental conditions (Yan et al. 2006).
In the striatum of the same experimental animals, activated microglia were more activated and often amoeboid and round at two weeks after MCAO (Thored et al. 2009). These results suggest that microglia activation is differentially regulated in SVZ and striatum. The neurogenic niche seems to modulate microglia function toward a more neuroprotective/neurogenic phenotype. Gradients of both anti-inflammatory cytokines and growth factors inside SVZ might be involved. These results illustrate well how microglial phenotype may be influenced by the molecular constitution of different anatomical niches along the ischemic environment.
We have reasons to believe that both beneficial and detrimental microglia are present in different anatomical niches after MCAO. In an ongoing investigation, we have observed that clustered SVZ microglia were spatially associated with clusters of neuroblasts several weeks after MCAO (Fig. 1). In addition, we have observed zones of abnormal aggregate (clustering) of hyperactivated microglia/macrophages (round and/amoeboid cells) as well as nonoverlapping regions of microglia displaying an intermediate morphology in the ischemic striatum weeks after MCAO (Fig. 2). Double immunofluorescence for Iba1 (a microglia marker) and DCX (a neuroblast marker) revealed a surprising spatial correlation between these two cell populations in both SVZ and ischemic striatum. A few neuroblasts were present inside abnormal striatal microglial aggregations (Fig. 2A–C), which suggest that this anatomical niche is comprised by detrimental microglia/macrophages contributing to neuroblast death or impairing their survival. Nevertheless, in the striatal regions outside aggregations, which contain microglia with a more intermediate morphology, neuroblasts were intermingled with microglia (Fig. 2D–I) prompting us to believe in a proneurogenic role for microglia in these anatomical niches.
Figure 1.
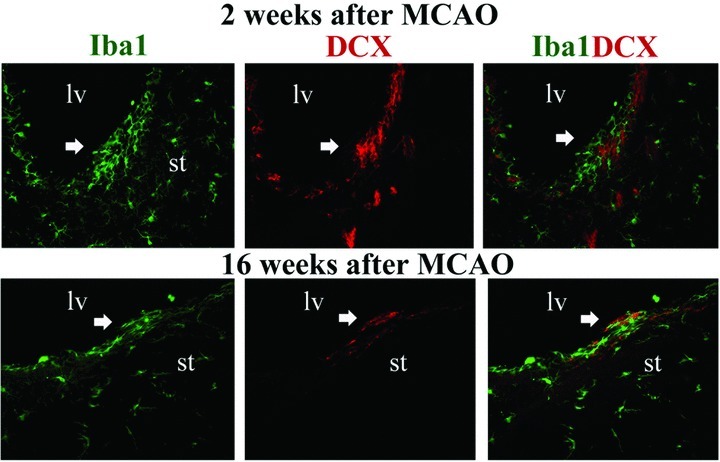
Spatial correlation between activated microglia and migrating neuroblasts in the subventricular zone (SVZ) after middle cerebral artery occlusion (MCAO). Microglia were labeled by anti-Iba1 (green) and migrating neuroblasts by antidoublecortin (red) double immunofluorescence. Ramified microglia were spatially associated with migrating neuroblasts in early (A–C) and late survival times (D–F) after MCAO. lv, lateral ventricle; st, striatum. Scale bar = 200 μm.
Figure 2.
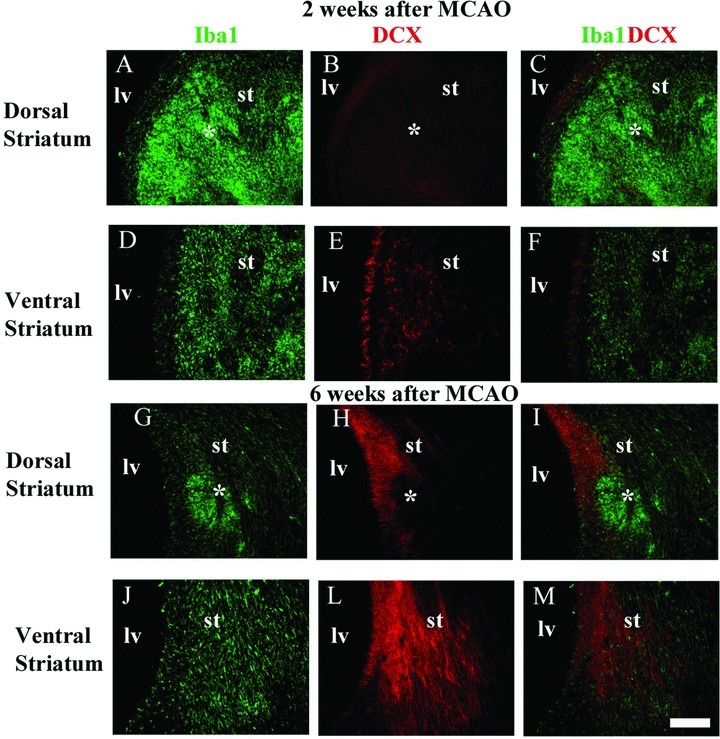
Spatial correlation between activated microglia and migrating neuroblasts in the striatum after middle cerebral artery occlusion (MCAO). Microglia were labeled by anti-Iba1 (green) and migrating neuroblasts by antidoublecortin (red) double immunofluorescence. Aggregations (*clustering) of overactivated microglia/macrophages are present in the dorsal striatum at 2 (A–C) and 6 weeks (G–I) after MCAO. Neuroblasts were less frequent in the ischemic striatal regions containing overactivated microglia/macrophages clustering (dorsal striatum), but intermingled with moderately activated microglia in striatal regions outside microglia/macrophages clustering (ventral striatum). lv, lateral ventricle; st, striatum. Scale bar = 100 μm.
Based on the experimental evidence described, we propose that detrimental (overactivated) and beneficial (intermediately activated) microglia might be present in discrete anatomical niches along the ischemic environment. Inhibition of stroke-induced microglia clustering formation, without avoiding intermediate (more physiological) levels of microglia activation can be a promising experimental approach for future investigations.
Microglia with different phenotypes in discrete anatomical niches along the pathological environment seem to be present in other experimental conditions, including chronic neurodegenerative diseases (Block et al. 2005; Battista et al. 2006; Fendrick et al. 2007). Activated microglia displaying a more ramified morphological profile (not amoeboid or full phagocytes) were reported to modulate hippocampal neurogenesis in adrenalectomized rats (Battista et al. 2006). Microglial/macrophages aggregates were also suggested to be neurotoxic in a mouse model of amyotrophic lateral sclerosis (ALS) (Fendrick et al. 2007). In these experimental circumstances, formation of multinucleated giant cells seems to be highly detrimental. Normal appearing microglia were present in other anatomical regions displaying less tissue damage (Fendrick et al. 2007). It is possible that microglia become multinucleate giant cells, fusing their membranes and releasing neurotoxins exacerbating tissue loss, when in aggregation. Macrophage aggregations are sites of overactivated and potentially neurotoxic microglia and a feature of several CNS diseases, including stroke, trauma, HIV infection associated dementia, and ALS (Block and Hong 2005). Nevertheless, studies using a model of prion disease (Perry et al. 2007) have indicated that microglia can switch to a phenotype contributing to neuronal damage without morphological changes (Perry et al. 2007). Thus, in some experimental models of CNS disease there is no direct correlation between morphological profile and functional phenotype.
Different stimuli acting on different microglial receptors may render different microglial phenotypes after CNS diseases. Schwartz and colleagues have shown that it is the type of stimulus that determines the microglial phenotype (Butovsky et al. 2005; Schwartz et al. 2006). LPS or amyloid-β induces a detrimental microglial phenotype by inducing these cells to release great amounts of TNF-α and NO, while low doses of interferon gamma or the anti-inflammatory cytokine IL-4 induces a beneficial profile in microglia manifested by release of growth factors, including brain-derived neurotrophic factor (BDNF) and IGF-1 (Butovsky et al. 2005; Schwartz et al. 2006). In vivo, macrophages stimulated by tissues with known regenerative capacity, for example sciatic nerve (Rapalino et al. 1998) or skin (Bomstein et al. 2003), acquire a neuroprotective profile. In these experimental conditions, the environmental stimuli, such as growth factors, might bind to surface microglial receptors, activating intracellular biochemical pathways favoring physiological-neuroprotective actions. This has similarities to what happens in peripheral tissues, in which macrophages can be phenotypically polarized by the microenvironment to perform different functions (Martinez et al. 2008). In peripheral tissues, macrophages can be classified in two main groups: classically activated macrophages (M1) and alternatively activated macrophages (M2). M1 macrophages are mainly activated by interferon gamma and LPS, while M2 after exposure to IL-4, IL-13, TGF beta or glucocorticoids (Martinez et al. 2008). In noninfectious conditions, M2-polarized macrophages play a role in resolution of inflammation through phagocytic mechanisms and by releasing growth factors, accompanied by reduced pro-inflammatory cytokine secretion (Martinez et al. 2008). It is possible that specific ligands can polarize microglia to different phenotypes like in the periphery (Durafourt et al. 2012). The presence of alternative microglia in the CNS is supported by recent investigations (Schwartz et al. 2006; Thored et al. 2009).
The ideas discussed above suggest that a beneficial or detrimental microglial phenotype might be a direct consequence of which kind of PRRs are activated in a determined CNS disease. This idea raises a clear therapeutic implication. Which microglial receptors are activated to induce neurodegeneration? Could they be experimentally blocked on microglia? Recent studies suggest that specific blockage of PRRs (for example TLR4) and/or NADPH oxidase can be a promising therapeutic approach for acute and chronic neural disorders (Block et al. 2007; Skaper 2011). In addition, activation of NADPH oxidase seems to be a very important event underlying the deleterious actions of microglia and experimental inhibition of this enzyme induces significant neuro-protection (Block et al. 2007). Investigations on the intracellular biochemical pathways responsible for both detrimental and beneficial actions of microglia are needed for development of drugs, which are able to maximize microglial beneficial functions and antagonize the deleterious ones.
The ligands triggering the paradoxical actions of microglia after CNS diseases are unknown. Nevertheless, neuro-melanin, α-synuclein, fibrillar Aβ, Aβ, prion may play a detrimental role on chronic neurodegenerative diseases (Block et al. 2007). The nature of these ligands remains to be determined after acute neural disorders, such as stroke and brain/spinal cord trauma. Purine nucleotides (Davalos et al. 2005), anti-inflammatory cytokines (Butovsky et al. 2005), and growth factors (Neumann et al. 1998) might be potential candidates as beneficial ligands.
Conclusion and Future Perspectives
In this paper, we shed light on the possible reasons by which microglia can be both detrimental and beneficial after CNS diseases. We face microglia as the guardians of CNS, which contribute to maintenance of its integrity in physiological conditions. In pathological conditions, some microglial cells might be affected by the disease process becoming overactivated contributing to neuronal damage, whereas others might maintain an intermediate (more physiological) level of activation contributing to neuronal rescue and repair processes. This might be a consequence of the fact that both harmful and beneficial stimuli are released upon injury into specific anatomical niches along the damaged areas triggering both beneficial and deleterious actions of microglia. Depending on the CNS-affected area and disease's etiology, both noxious and beneficial microglial phenotypes might coexist along the pathological environment.
Further studies are necessary to characterize, both morphologically and molecularly, the different anatomical niches of microglial activation after stroke and other neural disorders. These studies must unravel the ligands that render harmful and beneficial microglial phenotypes as well as the molecules released by activated microglia in both circumstances. In addition, these new experimental studies must investigate the effects of drugs that do not completely abolish microglia activation, but rather modulate this phenomenon, for example, avoiding clustering formation without interfering with physiological (beneficial) levels of activation after CNS diseases. It is also fundamental to find out which microglia receptors are specifically activated to induce beneficial or detrimental actions after a CNS disease. Experimental manipulation of these receptors, and/or pharmacological application of their beneficial ligands, may be promising therapeutic approaches used in the future for human neural disorders.
Acknowledgments
The author thanks to Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPQ), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and PROPESP UFPA for financial support and to Professor Victor Hugh Perry (CNS Inflammation Group of Southampton University) for helpful comments on the manuscript. Author is also grateful to Professor Olle Lindvall (University of Lund, Sweden) for allowing the facilities of his laboratory for MCAO experiments and immunofluorescence analysis.
Conflict of Interest
The authors declare no conflict of interest.
References
- Aarum J, Sandberg K, Haeberlein SL, Persson MA. Migration and differentiation of neural precursor cells can be directed by microglia. Proc. Natl. Acad. Sci. U.S.A. 2003;100:15983–15988. doi: 10.1073/pnas.2237050100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Alliot F, Godin I, Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res. Dev. Brain Res. 1999;117:145–152. doi: 10.1016/s0165-3806(99)00113-3. [DOI] [PubMed] [Google Scholar]
- Alvarez-Buylla A, Garcia-Verdugo JM. Neurogenesis in adult subventricular zone. J. Neurosci. 2002;22:629–634. doi: 10.1523/JNEUROSCI.22-03-00629.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battista D, Ferrari CC, Gage FH, Pitossi FJ. Neurogenic niche modulation by activated microglia: transforming growth factor beta increases neurogenesis in the adult dentate gyrus. Eur. J. Neurosci. 2006;23:83–93. doi: 10.1111/j.1460-9568.2005.04539.x. [DOI] [PubMed] [Google Scholar]
- Bessis A, Bechade C, Bernard D, Roumier A. Microglial control of neuronal death and synaptic properties. Glia. 2007;55:233–238. doi: 10.1002/glia.20459. [DOI] [PubMed] [Google Scholar]
- Block F, Dihne M, Loos M. Inflammation in areas of remote changes following focal brain lesion. Prog. Neurobiol. 2005;75:342–365. doi: 10.1016/j.pneurobio.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog. Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Bomstein Y, Marder JB, Vitner K, Smirnov I, Lisaey G, Butovsky O, Fulga V, Yoles E. Features of skin-coincubated macrophages that promote recovery from spinal cord injury. J. Neuroimmunol. 2003;142:10–16. doi: 10.1016/s0165-5728(03)00260-1. [DOI] [PubMed] [Google Scholar]
- Broderick C, Hoek RM, Forrester JV, Liversidge J, Sedgwick JD, Dick AD. Constitutive retinal CD200 expression regulates resident microglia and activation state of inflammatory cells during experimental autoimmune uveoretinitis. Am. J. Pathol. 2002;161:1669–1677. doi: 10.1016/S0002-9440(10)64444-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan MM, Hutchinson M, Watkins LR, Yin H. Toll-like receptor 4 in CNS pathologies. J. Neurochem. 2010;114:13–27. doi: 10.1111/j.1471-4159.2010.06736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burguillos MA, Deierborg T, Kavanagh E, Persson A, Hajji N, Garcia-Quintanilla A, Cano J, Brundin P, Englund E, Venero JL, et al. Caspase signalling controls microglia activation and neurotoxicity. Nature. 2011;472:319–324. doi: 10.1038/nature09788. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Hauben E, Schwartz M. Morphological aspects of spinal cord autoimmune neuroprotection: colocalization of T cells with B7–2 (CD86) and prevention of cyst formation. FASEB J. 2001;15:1065–1067. doi: 10.1096/fj.00-0550fje. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Talpalar AE, Ben-Yaakov K, Schwartz M. Activation of microglia by aggregated beta-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-gamma and IL-4 render them protective. Mol. Cell. Neurosci. 2005;29:381–393. doi: 10.1016/j.mcn.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Cheret C, Gervais A, Lelli A, Colin C, Amar L, Ravassard P, Mallet J, Cumano A, Krause KH, Mallat M. Neurotoxic activation of microglia is promoted by a nox1-dependent NADPH oxidase. J. Neurosci. 2008;28:12039–12051. doi: 10.1523/JNEUROSCI.3568-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Park EM, Febbraio M, Anrather J, Park L, Racchumi G, Silverstein RL, Iadecola C. The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J. Neurosci. 2005;25:2504–2512. doi: 10.1523/JNEUROSCI.0035-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Garcia-Verdugo JM, Alvarez-Buylla A. Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J. Neurosci. 1997;17:5046–5061. doi: 10.1523/JNEUROSCI.17-13-05046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downes CE, Crack PJ. Neural injury following stroke: are Toll-like receptors the link between the immune system and the CNS? Br. J. Pharmacol. 2010;160:1872–1888. doi: 10.1111/j.1476-5381.2010.00864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durafourt BA, Moore CS, Zammit DA, Johnson TA, Zaguia F, Guiot MC, Bar-Or A, Antel JP. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia. 2012;60:717–727. doi: 10.1002/glia.22298. [DOI] [PubMed] [Google Scholar]
- Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O. Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13632–13637. doi: 10.1073/pnas.2234031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekdahl CT, Kokaia Z, Lindvall O. Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience. 2009;158:1021–1029. doi: 10.1016/j.neuroscience.2008.06.052. [DOI] [PubMed] [Google Scholar]
- Fagan SC, Waller JL, Nichols FT, Edwards DJ, Pettigrew LC, Clark WM, Hall CE, Switzer JA, Ergul A, Hess DC. Minocycline to improve neurologic outcome in stroke (MINOS): a dose-finding study. Stroke. 2010;41:2283–2287. doi: 10.1161/STROKEAHA.110.582601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendrick SE, Xue QS, Streit WJ. Formation of multinucleated giant cells and microglial degeneration in rats expressing a mutant Cu/Zn superoxide dismutase gene. J. Neuroinflammation. 2007;4:9. doi: 10.1186/1742-2094-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes-Leal W, Corkill DJ, Picanco-Diniz CW. Systematic analysis of axonal damage and inflammatory response in different white matter tracts of acutely injured rat spinal cord. Brain Res. 2005;1066:57–70. doi: 10.1016/j.brainres.2005.10.069. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Streit WJ, Kreutzberg GW. Axotomy of the rat facial nerve leads to increased CR3 complement receptor expression by activated microglial cells. J. Neurosci. Res. 1988;21:18–24. doi: 10.1002/jnr.490210104. [DOI] [PubMed] [Google Scholar]
- Griffiths M, Neal JW, Gasque P. Innate immunity and protective neuroinflammation: new emphasis on the role of neuroimmune regulatory proteins. Int. Rev. Neurobiol. 2007;82:29–55. doi: 10.1016/S0074-7742(07)82002-2. [DOI] [PubMed] [Google Scholar]
- Hamby AM, Suh SW, Kauppinen TM, Swanson RA. Use of a poly(ADP-ribose) polymerase inhibitor to suppress inflammation and neuronal death after cerebral ischemia-reperfusion. Stroke. 2007;38:632–636. doi: 10.1161/01.STR.0000250742.61241.79. [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Mishima K, Nozako M, Hazekawa M, Mishima S, Fujioka M, Orito K, Egashira N, Iwasaki K, Fujiwara M. Delayed treatment with minocycline ameliorates neurologic impairment through activated microglia expressing a high-mobility group box1-inhibiting mechanism. Stroke. 2008;39:951–958. doi: 10.1161/STROKEAHA.107.495820. [DOI] [PubMed] [Google Scholar]
- Heldmann U, Mine Y, Kokaia Z, Ekdahl CT, Lindvall O. Selective depletion of Mac-1-expressing microglia in rat subventricular zone does not alter neurogenic response early after stroke. Exp. Neurol. 2011;229:391–398. doi: 10.1016/j.expneurol.2011.03.005. [DOI] [PubMed] [Google Scholar]
- Hewlett KA, Corbett D. Delayed minocycline treatment reduces long-term functional deficits and histological injury in a rodent model of focal ischemia. Neuroscience. 2006;141:27–33. doi: 10.1016/j.neuroscience.2006.03.071. [DOI] [PubMed] [Google Scholar]
- Hoehn BD, Palmer TD, Steinberg GK. Neurogenesis in rats after focal cerebral ischemia is enhanced by indomethacin. Stroke. 2005;36:2718–2724. doi: 10.1161/01.STR.0000190020.30282.cc. [DOI] [PubMed] [Google Scholar]
- Husemann J, Loike JD, Anankov R, Febbraio M, Silverstein SC. Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. Glia. 2002;40:195–205. doi: 10.1002/glia.10148. [DOI] [PubMed] [Google Scholar]
- Jander S, Schroeter M, D’Urso D, Gillen C, Witte OW, Stoll G. Focal ischaemia of the rat brain elicits an unusual inflammatory response: early appearance of CD8+ macrophages/microglia. Eur. J. Neurosci. 1998;10:680–688. doi: 10.1046/j.1460-9568.1998.00078.x. [DOI] [PubMed] [Google Scholar]
- Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–835. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- Kaushal V, Schlichter LC. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J. Neurosci. 2008;28:2221–2230. doi: 10.1523/JNEUROSCI.5643-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Suh YH. Minocycline and neurodegenerative diseases. Behav. Brain Res. 2009;196:168–179. doi: 10.1016/j.bbr.2008.09.040. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci. 2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambertsen KL, Clausen BH, Babcock AA, Gregersen R, Fenger C, Nielsen HH, Haugaard LS, Wirenfeldt M, Nielsen M, Dagnaes-Hansen F, et al. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J. Neurosci. 2009;29:1319–1330. doi: 10.1523/JNEUROSCI.5505-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampl Y, Boaz M, Gilad R, Lorberboym M, Dabby R, Rapoport A, Anca-Hershkowitz M, Sadeh M. Minocycline treatment in acute stroke: an open-label, evaluator-blinded study. Neurology. 2007;69:1404–1410. doi: 10.1212/01.wnl.0000277487.04281.db. [DOI] [PubMed] [Google Scholar]
- Lehnardt S. Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll-like receptor-mediated neuronal injury. Glia. 2010;58:253–263. doi: 10.1002/glia.20928. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Lehmann S, Kaul D, Tschimmel K, Hoffmann O, Cho S, Krueger C, Nitsch R, Meisel A, Weber JR. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J. Neuroimmunol. 2007;190:28–33. doi: 10.1016/j.jneuroim.2007.07.023. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Schott E, Trimbuch T, Laubisch D, Krueger C, Wulczyn G, Nitsch R, Weber JR. A vicious cycle involving release of heat shock protein 60 from injured cells and activation of Toll-like receptor 4 mediates neurodegeneration in the CNS. J. Neurosci. 2008;28:2320–2331. doi: 10.1523/JNEUROSCI.4760-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrmann E, Christensen T, Zimmer J, Diemer NH, Finsen B. Microglial and macrophage reactions mark progressive changes and define the penumbra in the rat neocortex and striatum after transient middle cerebral artery occlusion. J. Comp. Neurol. 1997;386:461–476. [PubMed] [Google Scholar]
- Lien E, Means TK, Heine H, Yoshimura A, Kusumoto S, Fukase K, Fenton MJ, Oikawa M, Qureshi N, Monks B, et al. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J. Clin. Invest. 2000;105:497–504. doi: 10.1172/JCI8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling Z, Zhu Y, Tong C, Snyder JA, Lipton JW, Carvey PM. Progressive dopamine neuron loss following supra-nigral lipopolysaccharide (LPS) infusion into rats exposed to LPS prenatally. Exp. Neurol. 2006;199:499–512. doi: 10.1016/j.expneurol.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Liu Z, Fan Y, Won SJ, Neumann M, Hu D, Zhou L, Weinstein PR, Liu J. Chronic treatment with minocycline preserves adult new neurons and reduces functional impairment after focal cerebral ischemia. Stroke. 2007;38:146–152. doi: 10.1161/01.STR.0000251791.64910.cd. [DOI] [PubMed] [Google Scholar]
- Lyons A, Downer EJ, Crotty S, Nolan YM, Mills KH, Lynch MA. CD200 ligand receptor interaction modulates microglial activation in vivo and in vitro: a role for IL-4. J. Neurosci. 2007;27:8309–8313. doi: 10.1523/JNEUROSCI.1781-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front. Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- Morino T, Ogata T, Takeba J, Yamamoto H. Microglia inhibition is a target of mild hypothermic treatment after the spinal cord injury. Spinal Cord. 2008;46:425–431. doi: 10.1038/sj.sc.3102163. [DOI] [PubMed] [Google Scholar]
- Morioka T, Kalehua AN, Streit WJ. Characterization of microglial reaction after middle cerebral artery occlusion in rat brain. J. Comp. Neurol. 1993;327:123–132. doi: 10.1002/cne.903270110. [DOI] [PubMed] [Google Scholar]
- Mott RT, Ait-Ghezala G, Town T, Mori T, Vendrame M, Zeng J, Ehrhart J, Mullan M, Tan J. Neuronal expression of CD22: novel mechanism for inhibiting microglial proinflammatory cytokine production. Glia. 2004;46:369–379. doi: 10.1002/glia.20009. [DOI] [PubMed] [Google Scholar]
- Neumann H, Misgeld T, Matsumuro K, Wekerle H. Neurotrophins inhibit major histocompatibility class II inducibility of microglia: involvement of the p75 neurotrophin receptor. Proc. Natl. Acad. Sci. U.S.A. 1998;95:5779–5784. doi: 10.1073/pnas.95.10.5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann J, Gunzer M, Gutzeit HO, Ullrich O, Reymann KG, Dinkel K. Microglia provide neuroprotection after ischemia. FASEB J. 2006;20:714–716. doi: 10.1096/fj.05-4882fje. [DOI] [PubMed] [Google Scholar]
- Neumann J, Sauerzweig S, Ronicke R, Gunzer F, Dinkel K, Ullrich O, Gunzer M, Reymann KG. Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J. Neurosci. 2008;28:5965–5975. doi: 10.1523/JNEUROSCI.0060-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Pei Z, Pang H, Qian L, Yang S, Wang T, Zhang W, Wu X, Dallas S, Wilson B, Reece JM, et al. MAC1 mediates LPS-induced production of superoxide by microglia: the role of pattern recognition receptors in dopaminergic neurotoxicity. Glia. 2007;55:1362–1373. doi: 10.1002/glia.20545. [DOI] [PubMed] [Google Scholar]
- Perry VH, Gordon S. Modulation of CD4 antigen on macrophages and microglia in rat brain. J. Exp. Med. 1987;166:1138–1143. doi: 10.1084/jem.166.4.1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat. Rev. Immunol. 2007;7:161–167. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat. Rev. Neurol. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Guan Z, Wei P, Huitinga I, van Rooijen N, Stokes BT. Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp. Neurol. 1999;158:351–365. doi: 10.1006/exnr.1999.7118. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Guan Z, McGaughy V, Fisher L, Hickey WF, Basso DM. The neuropathological and behavioral consequences of intraspinal microglial/macrophage activation. J. Neuropathol. Exp. Neurol. 2002;61:623–633. doi: 10.1093/jnen/61.7.623. [DOI] [PubMed] [Google Scholar]
- Qin L, Block ML, Liu Y, Bienstock RJ, Pei Z, Zhang W, Wu X, Wilson B, Burka T, Hong JS. Microglial NADPH oxidase is a novel target for femtomolar neuroprotection against oxidative stress. FASEB J. 2005;19:550–557. doi: 10.1096/fj.04-2857com. [DOI] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabchevsky AG, Streit WJ. Grafting of cultured microglial cells into the lesioned spinal cord of adult rats enhances neurite outgrowth. J. Neurosci. Res. 1997;47:34–48. [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu. Rev. Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Rapalino O, Lazarov-Spiegler O, Agranov E, Velan GJ, Yoles E, Fraidakis M, Solomon A, Gepstein R, Katz A, Belkin M, et al. Implantation of stimulated homologous macrophages results in partial recovery of paraplegic rats. Nat. Med. 1998;4:814–821. doi: 10.1038/nm0798-814. [DOI] [PubMed] [Google Scholar]
- Ross GD. Regulation of the adhesion versus cytotoxic functions of the Mac-1/CR3/alphaMbeta2-integrin glycoprotein. Crit. Rev. Immunol. 2000;20:197–222. [PubMed] [Google Scholar]
- Schabitz WR, Schneider A, Laage R. Minocycline treatment in acute stroke: an open-label, evaluator-blinded study. Neurology. 2008;71:1461. doi: 10.1212/01.wnl.0000338449.84851.97. [DOI] [PubMed] [Google Scholar]
- Schroeter M, Jander S, Witte OW, Stoll G. Local immune responses in the rat cerebral cortex after middle cerebral artery occlusion. J. Neuroimmunol. 1994;55:195–203. doi: 10.1016/0165-5728(94)90010-8. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Butovsky O, Bruck W, Hanisch UK. Microglial phenotype: is the commitment reversible? Trends Neurosci. 2006;29:68–74. doi: 10.1016/j.tins.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Seri B, Garcia-Verdugo JM, McEwen BS, Alvarez-Buylla A. Astrocytes give rise to new neurons in the adult mammalian hippocampus. J. Neurosci. 2001;21:7153–7160. doi: 10.1523/JNEUROSCI.21-18-07153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, Tsirka SE, Maletic-Savatic M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7:483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaper SD. Ion channels on microglia: therapeutic targets for neuroprotection. CNS Neurol. Disord. Drug Targets. 2011;10:44–56. doi: 10.2174/187152711794488638. [DOI] [PubMed] [Google Scholar]
- Stence N, Waite M, Dailey ME. Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia. 2001;33:256–266. [PubMed] [Google Scholar]
- Stirling DP, Khodarahmi K, Liu J, McPhail LT, McBride CB, Steeves JD, Ramer MS, Tetzlaff W. Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J. Neurosci. 2004;24:2182–2190. doi: 10.1523/JNEUROSCI.5275-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40:133–139. doi: 10.1002/glia.10154. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Prog. Neurobiol. 1999;57:563–581. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- Sugama S, Fujita M, Hashimoto M, Conti B. Stress induced morphological microglial activation in the rodent brain: involvement of interleukin-18. Neuroscience. 2007;146:1388–1399. doi: 10.1016/j.neuroscience.2007.02.043. [DOI] [PubMed] [Google Scholar]
- Thored P, Arvidsson A, Cacci E, Ahlenius H, Kallur T, Darsalia V, Ekdahl CT, Kokaia Z, Lindvall O. Persistent production of neurons from adult brain stem cells during recovery after stroke. Stem Cells. 2006;24:739–747. doi: 10.1634/stemcells.2005-0281. [DOI] [PubMed] [Google Scholar]
- Thored P, Heldmann U, Gomes-Leal W, Gisler R, Darsalia V, Taneera J, Nygren JM, Jacobsen SE, Ekdahl CT, Kokaia Z, et al. Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia. 2009;57:835–849. doi: 10.1002/glia.20810. [DOI] [PubMed] [Google Scholar]
- Tian DS, Xie MJ, Yu ZY, Zhang Q, Wang YH, Chen B, Chen C, Wang W. Cell cycle inhibition attenuates microglia induced inflammatory response and alleviates neuronal cell death after spinal cord injury in rats. Brain Res. 2007;1135:177–185. doi: 10.1016/j.brainres.2006.11.085. [DOI] [PubMed] [Google Scholar]
- Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J. Neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Jeng D, Alexopoulou L, Tan J, Flavell RA. Microglia recognize double-stranded RNA via TLR3. J. Immunol. 2006;176:3804–3812. doi: 10.4049/jimmunol.176.6.3804. [DOI] [PubMed] [Google Scholar]
- Walton NM, Sutter BM, Laywell ED, Levkoff LH, Kearns SM, Marshall GP, 2nd, Scheffler B, Steindler DA. Microglia instruct subventricular zone neurogenesis. Glia. 2006;54:815–825. doi: 10.1002/glia.20419. [DOI] [PubMed] [Google Scholar]
- Wu J, Yang S, Xi G, Fu G, Keep RF, Hua Y. Minocycline reduces intracerebral hemorrhage-induced brain injury. Neurol. Res. 2009;31:183–188. doi: 10.1179/174313209X385680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan YP, Sailor KA, Vemuganti R, Dempsey RJ. Insulin-like growth factor-1 is an endogenous mediator of focal ischemia-induced neural progenitor proliferation. Eur. J. Neurosci. 2006;24:45–54. doi: 10.1111/j.1460-9568.2006.04872.x. [DOI] [PubMed] [Google Scholar]
- Yong VW, Wells J, Giuliani F, Casha S, Power C, Metz LM. The promise of minocycline in neurology. Lancet Neurol. 2004;3:744–751. doi: 10.1016/S1474-4422(04)00937-8. [DOI] [PubMed] [Google Scholar]
- Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc. Natl. Acad. Sci. U.S.A. 1999;96:13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, Grotta JC, Aronowski J. Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann. Neurol. 2007;61:352–362. doi: 10.1002/ana.21097. [DOI] [PubMed] [Google Scholar]
- Ziegler G, Harhausen D, Schepers C, Hoffmann O, Rohr C, Prinz V, Konig J, Lehrach H, Nietfeld W, Trendelenburg G. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem. Biophys. Res. Commun. 2007;359:574–579. doi: 10.1016/j.bbrc.2007.05.157. [DOI] [PubMed] [Google Scholar]