

Abstract
Mutations in the PTEN gene are associated with a broad spectrum of disorders, including Cowden syndrome (CS), Bannayan–Riley–Ruvalcaba syndrome, Proteus syndrome, and Lhermitte–Duclos disease. In addition, PTENmutations have been described in a few patients with autism spectrum disorders (ASDs) and macrocephaly. In this study, we screened the PTEN gene for mutations and deletions in 88 patients with ASDs and macrocephaly (defined as ≥2 SD above the mean). Mutation analysis was performed by direct sequencing of all exons and flanking regions, as well as the promoter region. Dosage analysis of PTEN was carried out using multiplex ligation-dependent probe amplification (MLPA). No partial or whole gene deletions were observed. We identified a de novo missense mutation (D326N) in a highly conserved amino acid in a 5-year-old boy with autism, mental retardation, language delay, extreme macrocephaly (+9.6 SD) and polydactyly of both feet. Polydactyly has previously been described in two patients with Lhermitte–Duclos disease and CS and is thus likely to be a rare sign of PTEN mutations. Our findings suggest that PTEN mutations are a relatively infrequent cause of ASDs with macrocephaly. Screening of PTEN mutations is warranted in patients with autism and pronounced macrocephaly, even in the absence of other features of PTEN-related tumor syndromes.
Keywords: Cowden syndrome, Bannayan–Riley–Ruvalcaba syndrome, polydactyly, sequence analysis, multiplex ligation-dependent probe amplification
INTRODUCTION
The phosphatase and tensin homolog (PTEN) gene has been implicated in a family of related tumor syndromes with autosomal dominant inheritance and highly variable expression, including Cowden syndrome (CS), Bannayan–Riley–Ruvalcaba syndrome (BRRS), and Proteus syndrome. CS is a familial cancer syndrome characterized by multiple hamartomas, particularly of the skin, and by a high risk for breast, thyroid, and endometrial cancers. BRRS is characterized by macrocephaly, developmental delay, lipomatosis and pigmented macules of the penis. Proteus syndrome is characterized by asymmetrical limb overgrowth, macrocephaly, connective tissue nevi, and lipomatosis. The common feature of these disorders is a predisposition to hamartomas, and they are thus collectively referred to as PTEN hamartoma-tumor syndrome [Zbuk et al., 2006]. Several reports have also described PTEN mutations in children with autism spectrum disorders (ASDs), mental retardation, and macrocephaly [Zori et al., 1998; Goffin et al., 2001; Parisi et al., 2001; Delatycki et al., 2003; Butler et al., 2005]. In four cases, the children inherited the PTEN mutation from a parent affected with CS or BRRS [Zori et al., 1998; Goffin et al., 2001; Parisi et al., 2001; Delatycki et al., 2003]. A recent mutation analysis of PTEN in 18 subjects with ASD and macrocephaly found three males with previously unidentified germline PTEN mutations [Butler et al., 2005].
The PTEN gene, on 10q23.3, encodes a dual specificity phosphatase that negatively regulates the phosphatidylinositol 3-kinase (PI3K)/AKT (protein kinase B) pathway [Waite and Eng, 2002]. Although initially discovered as a tumor suppressor, more recent studies have highlighted an important role for PTEN in brain development. The PI3K/AKT pathway has been implicated in neuronal survival, neurite outgrowth, synaptic plasticity, learning, and memory [Dudek et al., 1997; Lin et al., 2001; Chen et al., 2005; Jaworski et al., 2005]. Conditional transgenic mice lacking PTEN in the cerebral cortex and hippocampus exhibit macrocephaly, neuronal hypertrophy and abnormal dendritic arborization, with increased dendritic thickness and spine density [Kwon et al., 2006]. These mice also display abnormal social interactions and heightened responses to sensory stimuli, analogous to what is observed in autism.
Autism is a neurodevelopmental syndrome characterized by social deficits, communication impairments and restricted and repetitive behaviors and interests, with onset before the age of 3 years. Males are affected four times more frequently than females. Autism has a strong genetic component with complex inheritance [Veenstra-Vanderweele et al., 2004]. Monogenic disorders and chromosomal abnormalities are identified in at least 10% of patients, but the underlying cause remains unknown in the majority of patients. Linkage and association studies have failed to identify any definite genes that confer a significant risk for autism [Veenstra-Vanderweele et al., 2004]. It is likely that the etiological heterogeneity involved in autism is hindering progress in the identification of susceptibility variants, because many of the patients included in these studies, considered to have “idiopathic” autism, have in fact monogenic or genomic disorders that have gone undetected.
Autism is associated with macrocephaly in approximately 20% of cases [Fombonne et al., 1999; Miles et al., 2000]. Brain overgrowth is usually not present at birth but develops during childhood [Lainhart et al., 1997]. Although macrocephaly is one of the most widely replicated neurobiological findings in autism, its pathogenesis remains unknown. The discovery of PTEN mutations in patients with ASD and macrocephaly [Zori et al., 1998; Goffin et al., 2001; Parisi et al., 2001; Delatycki et al., 2003; Butler et al., 2005] suggests that PTEN could be involved in other cases of autism. Thus, the aim of this study was to assess the frequency of PTEN mutations in cases of ASD associated with macrocephaly, defined as an occipitofrontal head circumference (HC) ≥2 SD above the mean. We screened the PTEN gene by direct sequencing in 88 subjects with ASD and macrocephaly. Furthermore, as interstitial deletions of the 10q23 region encompassing PTEN and partial gene deletions have been identified in individuals with PTEN-related syndromes [Marsh et al., 1999; Hendriks et al., 2003; Zhou et al., 2003], we screened for PTEN deletions using multiplex ligation-dependent probe amplification (MLPA).
MATERIALS AND METHODS
Patients
A total of 88 patients with ASD and macrocephaly (HC ≥2 SD) were included in the study. Among these, 49 were recruited by the Paris Autism Research International Sibpair (PARIS) study at specialized clinical centers in seven countries (France, Sweden, Norway, Italy, Belgium, Austria, and the United States) and 39 were collected by the Mount Sinai School of Medicine and/or the Autism Genetic Resource Exchange (AGRE) [Geschwind et al., 2001]. Patients were selected from a larger pool of 462 families for which HC measures were available. We studied all subjects with HC ≥2 SD; in families with multiple affected subjects with macrocephaly, only one individual per family was included in the mutation screening.
The patients from the PARIS study included 45 males and 4 females from 16 multiplex families (with two or more affected siblings) and 33 simplex families (sporadic cases), with a mean age at the last evaluation of 10.5 ± 5.8 years (range 3.5–26). All patients were evaluated by experienced psychiatrists or child neurologists. Diagnoses were based on clinical evaluation and DSM-IV criteria; in addition, patients were assessed with the Autism Diagnostic Interview-Revised (ADI-R) [Lord et al., 1994] or the Asperger Syndrome Diagnostic Interview [Gillberg et al., 2001]. Forty-five individuals met criteria for autism, 2 for pervasive developmental disorder not otherwise specified (PDD-NOS), and 2 for Asperger syndrome. Thirty-five patients had mental retardation and 22 had limited or no language. Laboratory tests to rule-out medical causes of autism included standard karyotyping, fragile X testing, and metabolic screening; brain imaging and EEG were performed when possible. Patients diagnosed with medical disorders such as fragile X syndrome or chromosomal abnormalities were excluded. There were 41 individuals of Caucasian origin, 2 Black, 1 Asian, and 5 of mixed ethnicity.
Thirty-nine patients were recruited by the Seaver Autism Research Center (SARC) at Mount Sinai, New York, co-recruited by the SARC and AGRE, or obtained from AGRE. The patients included 31 males and 8 females from 35 multiplex families and 4 simplex families, with a mean age at the last evaluation of 10.5 ± 4.2 years (range 5–24). The ADI-R was used to assess affected subjects. There were 35 individuals with autism, 1 with borderline autism, 2 with Asperger syndrome, and 1 with PDD-NOS. Seventeen patients had mental retardation and 16 had limited or no language. Subjects with co-morbid genetic disorders were excluded. There were 32 individuals of Caucasian origin, 1 Black, 1 Asian, and 5 of mixed/unknown ethnicity. Informed consent was obtained from all families. The study was approved by the research ethics boards of the collaborating institutions.
PTEN Mutation Analyses
The nine exons of the PTEN gene, intron–exon boundaries, the 5′ untranslated region and the proximal promoter [Zhou et al., 2003] were screened by bidirectional sequencing using standard procedures. When variants not described previously were detected, the DNA of parents and siblings was analyzed by direct sequencing. Rare variants were also studied in a control sample of 150 Caucasians, 50 African-Americans, 10 Chinese, and 10 Japanese obtained from the Coriell repository.
PTEN Microdeletion Analyses
We performed MLPA of the PTEN gene using a commercially available kit (P105; MRC-Holland, Amsterdam, The Netherlands). The kit contains two PTEN probes in the promoter, four probes in the 3′ regions of exons 2, 3, 4, and 8, and three probes within exons 5, 6, and 7. Electrophoresis of PCR products was performed on an ABI 3730 sequencing platform and the resultant traces were analyzed using the software GeneMarker (SoftGenetics, State College, PA).
RESULTS
PTEN Mutation Analyses
Among the 88 patients screened, we identified seven heterozygous variants, four in the promoter region, and three intragenic variants, including two missense alterations, as detailed in Table I. Two of the promoter variants, −1026C→A (found in one patient) and −903G→A (found in two patients), had been described before in control subjects, and were thus assumed to be polymorphisms [Zhou et al., 2003].
TABLE I.
Sequence Variants Identified in the PTEN Gene in 88 Patients With Autism and Macrocephaly
| Location and nucleotide change | Protein change | Frequency | Head circumference | Inheritance | Siblings | Controls (n =220) |
|---|---|---|---|---|---|---|
| Promoter/5′UTR | ||||||
| −1088C→T | … | 1 | +2.5 SD | NA | NA | 0 |
| −1084C→Ta | … | 1 | +2.2 SD | Maternal | Present in 1 sib with autism and 1 unaffected sib | 2 |
| −1026C→Ab | … | 1 | +3.5 SD | Maternal | Present in 1 unaffected sib | 5 |
| −903G→Ab | … | 2 | +2.6 and +9.6 SDc | ND | ND | ND |
| Exon 1 | ||||||
| c.66C→G | D22E | 1 | +2.1 SD | Paternal | Present in 1 sib with autism | 1 |
| Intron 3 | ||||||
| IVS3-9T→C | … | 1 | +2.2 SD | Maternal | Absent in 1 unaffected sib | 0 |
| Exon 8 | ||||||
| c.976G→A | D326N | 1 | +9.6 SDc | De novo | No siblings | 0 |
NA, DNA not available for analysis; ND, not determined.
Described initially as a pathogenic mutation [Zhou et al., 2003] and then as a “variant of unknown significance” [Sarquis et al., 2006].
Described elsewhere as a polymorphism [Zhou et al., 2003] or a “variant of unknown significance” [Sarquis et al., 2006].
The patient with the de novo c.976G→A mutation in exon 8 and the largest head circumference (+9.6 SD) of all the study group, also had a 903G→A variant in the promoter.
A C→T promoter variant at −1088, found in a 21-year-old male with Asperger syndrome from Sweden, has not been described previously and was not detected in 220 controls. Parental samples were not available to evaluate the inheritance of this variant. There was no personal or family history of skin lesions or malignancies. The absence of the typical manifestations of PTEN-related syndromes in this adult patient suggests that this change is a rare variant, but further studies would be needed to clarify its significance.
A −1084C→T variant was detected in a girl from a multiplex family of Caucasian origin. This alteration had been described previously as a pathogenic mutation, on the basis of its absence among 186 controls, aberrant migration pattern of the PTEN immunoreactive protein and increased phosphorylation of the PTEN target protein AKT in the patient’s lymphoblastoid cell lines [Zhou et al., 2003]. Segregation analysis revealed that the mother, who was reportedly healthy, was heterozygous for the −1084C→T variant, as were the two sisters of the proband, one of which was also affected with autism. The father did not carry the variant. This sequence variant was also found in 2 out of 150 Caucasian controls, but was absent in 50 African-American and 20 Asian control samples. This high frequency in controls, compared to the estimated incidence of CS of 1:200,000 [Eng, 2003], suggests that the −1084C→T promoter variant is likely a polymorphism.
In exon 1, we identified a C→G transition (c.66C→G), resulting in a previously undescribed amino acid substitution, D22E, in a Caucasian boy with autism. The variant was inherited from his healthy father and was also present in his autistic sister but not in the mother. Screening of 220 controls showed the D22E variant in one Caucasian individual, suggesting that it could be a rare variant.
An IVS3-9T→C variant in intron 3, found in a female with autism, was transmitted by the healthy mother from Iran, but was not present in an unaffected brother. This variant was not detected in any of the 220 controls. Given the ethnic origin of the mother, this could be a rare variant, not found in our controls because they were not geographically matched.
Finally, we found a missense variant in exon 8 (c.976G→A, D326N) in a boy with autism. The substitution was not detected in the unaffected parents, indicating a de novo mutation in the proband. To confirm this result, a second DNA extraction from new blood samples was performed for the patient and both parents, confirming the initial results. Furthermore, to exclude non-paternity, we genotyped the proband and the parents with a standard panel of 15 highly informative microsatellite markers on multiple chromosomes. The results confirmed paternity and maternity. None of the 440 normal control chromosomes were found to carry this missense alteration.
The D326N mutation occurs within the C-terminal C2 domain (Fig. 1a), in an evolutionarily conserved residue found in PTEN orthologues from human to Drosophila melanogaster and Caenorhabditis elegans (Fig. 1b), implying an important functional role for this residue in the PTEN protein.
Fig. 1.
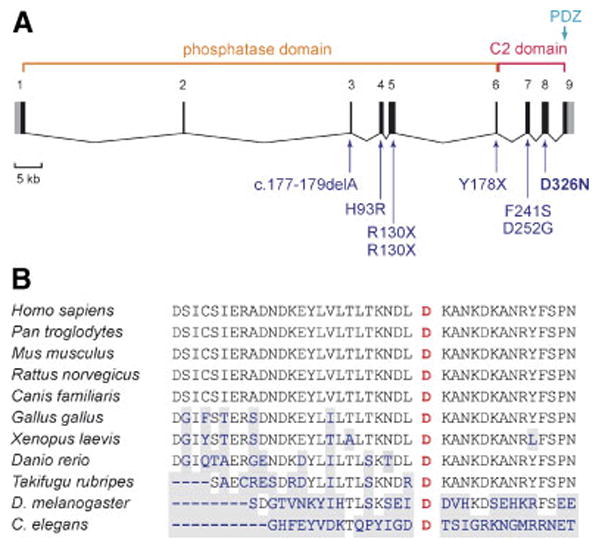
a: Exon/intron structure of the PTEN gene, with main functional domains and mutations described in patients with autism spectrum disorder reported previously [Zori et al., 1998; Goffin et al., 2001; Parisi et al., 2001; Delatycki et al., 2003; Butler et al., 2005]; the mutation identified in the present study (D326N) is indicated in bold. Non-coding sequences in exons 1 and 9 are indicated in gray. b: Alignment of the PTEN gene with homologous genes of other species shows the conservation of residue D326 in exon 8 implicated in our patient. Amino acids that are not conserved are shaded in gray. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
PTEN Microdeletion Analyses
There was no evidence for a deletion or duplication of any portion of the PTEN gene in any of the patients screened with MLPA.
Clinical Description of the Patient With the de Novo PTEN Mutation (D326N)
The proband is a 5-year-old boy, the only child of healthy parents of Caucasian and African origin. He was born after an uneventful pregnancy at 39 weeks gestation by caesarian section for fetal macrosomia. There was no history of gestational diabetes. His weight was 5,260 g (+4.2 SD) and his length was 54 cm (+2.6 SD). HC at birth was unavailable; HC at 20 days was 44 cm (+8 SD). He was born with postaxial hexadactyly of both feet, removed surgically. He sat at 6 months and walked at 18 months. He had delayed cognitive development; he said his first words at 18 months but used three word sentences only at 4.6 years. At 20 months, HC was 58.5 cm (+8.7 SD), height was 83.5 cm (+1.1 SD), and weight was 15.3 kg (+3.5 SD).
He was assessed at 3 years 9 months for his developmental delay and was diagnosed as having autistic disorder and mental retardation according to DSM-IV criteria. He fulfilled ADI-R criteria for autism [Lord et al., 1994]. His developmental age was 2.2 years. In the Vineland Adaptive Behavior Scales he achieved a level of 2 years for communication, 2.8 years for daily skills, 2.1 years for socialization, and 2.5 years for motor skills. Laboratory investigations, including karyotype, FISH for 15q11-q13, 22q11 and 22q13, fragile X testing, metabolic screening and EEG were normal. A brain MRI at 18 months was reported as normal, except for a discrete hypersignal in the posterior periventricular white matter.
When last evaluated at the age of 5 years 4 months, his HC was 63 cm (+9.6 SD), his height was 114 cm (+1.7 SD), and his weight was 29.3 kg (+6 SD). Besides his large head, he had no other dysmorphic facial features. He had two small café au lait spots, in an ankle and an arm, and a mongolian blue patch in the lumbosacral area. He had a 1 mm flesh-colored papule on the right shoulder, which had appeared recently according to his mother. His dermatological examination was otherwise unremarkable. In particular, he had no subcutaneous lipomas and no penile macules. He had echolalic speech and frequent unintelligible verbalizations, but he was able to say some clear single words and short sentences occasionally. Neurological examination was normal.
His family history was significant for the presence of two paternal cousins affected one with autism and the other with hyperactivity. There was no history of malignancies. The father’s HC was 59.5 cm (+1.8 SD) and his height was 187 cm (+2.2 SD). The mother’s HC was 56.5 cm (1.2 SD) and her height was 162 cm (mean).
DISCUSSION
PTEN gene mutation screening in 88 unrelated individuals with ASD and macrocephaly revealed one de novo missense mutation, D326N in exon 8, in a patient with autism and developmental delay. No PTEN deletions were identified in any of the patients. The patient with the PTEN mutation has manifestations of both CS and BRRS, including macrocephaly and mental retardation. Other findings reported in BRRS and present in our patient include prenatal and postnatal overgrowth, motor delay, language and speech delay, and café au lait macules [Parisi et al., 2001]. He did not show other manifestations of BRRS such as penile macules, lipomas, hemangiomas, or symptomatic gastrointestinal polyps. Clinical assessment showed no evidence of the mucocutaneous lesions pathognomonic of CS (trichilemmomas, acral keratoses, and papillomatous lesions). Many of the features/complications of CS, including skin lesions and cancers, do not develop until adulthood, making it difficult to diagnose this condition in children. Approximately 50% of the cases of CS are familial [Eng, 2003]. Thus, a positive family history of malignancies and characteristic skin lesions may help orient the diagnosis.
The significance of the polydactyly observed in our proband is not straightforward. Polydactyly is commonly observed in Subsaharian populations, and this finding may thus be fortuitous in our patient. However, there are some arguments in the literature that indicate that PTEN mutations may be associated with distal limb anomalies. Polydactyly has previously been described in two patients with PTEN-related syndromes. The first patient is a female with Lhermitte–Duclos disease (dysplastic gangliocytoma of the cerebellum), born with postaxial polydactyly in both hands [Bielschowsky and Simons, 1930]. Although a PTEN mutation was not diagnosed in this case, adult onset Lhermitte–Duclos disease is considered pathognomonic of CS [Zbuk et al., 2006]. The second patient is a boy with ASD, macrocephaly, and preaxial polydactyly of the right hand, who inherited a PTEN mutation from his father, affected with Lhermitte–Duclos disease and CS [Delatycki et al., 2003]. Interestingly, a patient with BRRS and autism carrying a PTEN mutation lacked the second toes [Zori et al., 1998]. Other distal limb abnormalities, including broad thumbs and halluces [Parisi et al., 2001], and radial ray hypoplasia [Reardon et al., 2001], have been described in patients with PTEN mutations. Thus, digital abnormalities may be a rare manifestation of PTEN haploinsufficiency.
Several case reports of patients with PTEN mutations have described the presence of ASDs. Their clinical features, together with those of our patient, are summarized in Table II. Zori et al. [1998] described a PTEN mutation (R130X) in a mother with CS and her son with BRRS and autistic behavior. The same mutation was described in a family in which the father and four of his five children had BRRS [Parisi et al., 2001]. They all had cognitive impairment, speech delay, and marked macrocephaly (+3.5–5.2 SD); in addition, one of the sons also exhibited autistic behavior. Similarly, Goffin et al. [2001] reported a mother with CS who transmitted a PTEN mutation to her son; the mother had normal cognitive function and social interactions, whereas the son had mental retardation and autism. In another family, the father with CS and Lhermitte–Duclos disease transmitted a PTEN mutation to his son, who had developmental delay and ASD [Delatycki et al., 2003]. In all these cases, the discovery of a mutation in the child was prompted by the diagnosis of CS or BRRS in a parent. Except for the patient described by Zori et al. [1998], none of the other children with autism fulfilled diagnostic criteria for any of the PTEN-related syndromes, reflecting the age-related penetrance and the variable expression of PTEN mutations. A recent mutation screening of PTEN in 18 subjects with ASD and macrocephaly identified three males carrying germline PTEN missense mutations [Butler et al., 2005]. The patients had pronounced macrocephalies (+4.5–8 SD), and except for one patient with freckles on the penis, no other clinical findings associated with PTEN-related disorders were noted. Although all the patients with autism and PTEN mutations described so far were macrocephalic, it should be noted that macrocephaly is not a universal finding in PTEN hamartomatumor syndromes. Macrocephaly is observed in only 38% of patients with CS [Eng, 2003], but it is the most consistent finding in BRRS, frequently associated with developmental delay [Parisi et al., 2001; Hendriks et al., 2003]. Notably, the eight patients with PTEN mutations and ASD identified so far are all males, similar to the male preponderance observed in idiopathic autism.
TABLE II.
Clinical Findings in Our Patient and in Other Patients With Autism Spectrum Disorders Carrying PTEN Mutations Described in the Literature
| Clinical finding | Present case | Zori et al. [1998] | Parisi et al. [2001] | Goffin et al. [2001] | Delatycki et al. [2003] |
Butler et al. [2005] |
||
|---|---|---|---|---|---|---|---|---|
| Patient 1 | Patient 2 | Patient 3 | ||||||
| Sex | Male | Male | Male | Male | Male | Male | Male | Male |
| Age (y) | 5 | 11 | 8 | 9 | 4 | 4 | 3.5 | 2.5 |
| Neonatal | ||||||||
| Birth weight (g) | 5,260 (+4.2 SD) | 3,430 (50th centile) | ? | 3,000 (50th centile) | 3,160 (20th centile) | 4,100 (90th centile) | 5,000 (+3 SD) | 3,400 (97th centile) |
| Birth length (cm) | 54 (+2.6 SD) | ? | 90–97th centile | 49 (50th centile) | 49.5 (50th centile) | ? | ? | 53.8 (95th centile) |
| Head circumference | 44 (+8 SD) | ? | 90–97th centile | 34 (50th centile) | 36 (50th centile) | ? | “Large head” | ? |
| Macrocephaly (cm) | 63 (+9.6 SD) | 59 (+3.7 SD) | 58.8 (+4.6 SD) | 59.9 (+5.2 SD) | 59.3 (+7 SD) | 62 (+8 SD) | 61.1 (+7 SD) | 56.5 (+4.5 SD) |
| Height (cm) | 114 (+1.7 SD) | Normal | 95th centile | 139 (+1.7 SD) | 107 (80th centile) | 109.8 (90th centile) | ? | 96 (97th centile) |
| Weight (kg) | 29.3 (+6 SD) | Normal | 95th centile | 34.1 (+1.7 SD) | 18 (75th centile) | 27.4 (+4 SD) | ? | ? |
| Neurological | ||||||||
| Hypotonia | − | + | ? | + | ? | − | + | − |
| ASD | Autism | Autistic behavior | Autistic behavior | Autism | ASD | Autism | Autism | Autism |
| Mental retardation | + | + | + | + | + | ? | + | + |
| Motor delay | + | + | + | + | + | ? | + | ? |
| Speech delay | + | + | + | + | + | + | + | ? |
| Seizures | − | − | − | − | − | − | − | Possible |
| MRI/CT | Normal | Normal | ND | Normal | Venous angioma in frontal lobe, dilated perivascular spaces | Marked dilatation of perivascular spaces | Normal | Normal |
| Hands/feet | Bilateral feet polydactyly | Lacked 2nd toes | − | − | Preaxial polydactyly of the right hand | − | − | − |
| Skin | ||||||||
| Hyperpigmented lesions | 2 café au lait spots, mongolian blue patch | Multiple nevi | − | − | 1 café au lait spot, mongolian blue patch | − | − | − |
| Lipomas | − | − | 1 lipoma in the back | − | − | − | − | − |
| Penile macules | − | + | + | − | − | − | − | + |
| Gastrointestinal polyps | − | Rectal polyps | − | − | − | − | − | − |
| Thyroid disease | − | Goiter and thyroid carcinoma at 18 years | − | − | − | − | − | − |
| Other | − | Inguinal hernias | Downslanting palpebral fissures | Scrotal tongue, open ductus Botallis, pulmonary valvular stenosis | Wormian bones | Epicanthal folds, flattened nasal bridge | Joint laxity, bilateral plantar creases, hydroceles | High arched palate, long ears |
| PTEN mutation | D326N | R130X | R130X | Y178X | c.177-179delA | H93R | D252G | F241S |
| Inheritance | De novo | Maternal | Paternal | Maternal | Paternal | De novo | (Father not tested) | ? |
| Familial macrocephaly | − | Maternal (CS) and paternal | Paternal (BRRS) and 3 affected sibs | Maternal (CS) | Paternal (CS +LDD) | Paternal | Paternal | ? |
ASD, Autism spectrum disorder; BRRS, Bannayan–Riley–Ruvalcaba syndrome; CD, Cowden syndrome; LDD, Lhermitte–Duclos disease; ND, not done.
Our results suggest that PTEN mutations are a relatively rare cause of autism. For this study, we selected among the subjects of the PARIS study and the Mount Sinai datasets with HC measures (n =462 families), all the patients with HC (2 SD (n =88). The proband with the PTEN mutation had the largest HC (+9.6 SD) among all the study subjects. Of the other 87 patients screened, 68 had a HC between +2 and +2.9 SD, 18 had a HC between +3 and +4 SD, and only 1 subject had a HC >+4 SD. This is in agreement with previous findings indicating that the macrocephaly commonly observed in autism is moderate [Miles et al., 2000]. This might explain why the mutation rate in our study (1 in 88) is much lower than that reported recently by Butler et al. [2005], who found three PTEN mutations among 18 patients with ASD and macrocephaly. In their study, six subjects had a HC greater than +4 SD, including the three subjects carrying PTEN mutations. These data suggest that the likelihood of identifying PTEN mutations is higher in individuals with marked macrocephaly. Our findings also indicate that although PTEN can play a role in rare cases of autism and pronounced macrocephaly, it does not appear to be involved in the abnormal regulation of brain growth seen in about 20% of patients with autism that present macrocephaly [Fombonne et al., 1999; Miles et al., 2000]. Several other syndromes associated with macrocephaly carry an increased risk for the development of autism (Table III), including fragile X syndrome [Hagerman, 2006], Sotos syndrome [Morrow et al., 1990; Zappella, 1990], neurofibromatosis type I [Williams and Hersh, 1998], and 22q13 deletions [Manning et al., 2004], but they too account for only a small number of patients with autism and macrocephaly.
TABLE III.
Syndromes With Macrocephaly Sometimes Associated With Autism Spectrum Disorders
| Syndrome | Gene | Chromosome | References |
|---|---|---|---|
| Sotos syndrome | NSD1 | 5q35 |
Morrow et al. [1990] Zappella [1990] |
| Fragile X syndrome | FMR1 | Xq27.3 | Hagerman [2006] |
| Neurofibromatosis type 1 | NF1 | 17q11.2 | Williams and Hersh [1998] |
| Terminal 22q deletion syndrome | SHANK3 | 22q13 | Manning et al. [2004] |
| PTEN hamartoma tumor syndromea | PTEN | 10q23.31 |
Butler et al. [2005] Delatycki et al. [2003] Goffin et al. [2001] Parisi et al. [2001] Zori et al. [1998] |
| Cole–Hughes syndrome | ? | ? | Naqvi et al. [2000] |
| Orstavik syndromeb | ? | ? |
Orstavik et al. [1997] Steiner et al. [2003] |
The PTEN hamartoma-tumor syndrome includes Cowden syndrome, Bannayan–Riley–Ruvalcaba syndrome, Proteus syndrome, and Lhermitte–Duclos disease.
The Orstavik syndrome was described by Orstavik et al. [1997] and is characterized by macrocephaly, epilepsy, autism, mental retardation, and dysmorphic features.
Interestingly, the patient with the PTEN mutation reported in this study had a father with a large head (+1.8 SD), even though the mutation appeared de novo in the child. Similarly, another patient with a de novo PTEN mutation and autism had a father with macrocephaly [Butler et al., 2005], while an individual with BRRS and autism, who inherited the PTEN mutation from his mother, also had a macrocephalic father [Zori et al., 1998] (see Table II). These findings suggest that other factors, independent of PTEN, are also involved in the cases of familial macrocephaly. Indeed, familial macrocephaly appears to be a common finding in autism, present in 45–62% of the parents of the macrocephalic subjects and in 37% of the normocephalic probands [Stevenson et al., 1997; Miles et al., 2000]. Macrocephaly has also been shown to be highly familial in non-autistic populations [Weaver and Christian, 1980].
The D326 residue mutated in our patient lies within the C2 domain, implicated in phospholipid membrane binding [Lee et al., 1999] and the regulation of cell migration [Raftopoulou et al., 2004]. The C2 domain recruits PTEN to the membrane and helps orient the catalytic domain appropriately to facilitate interaction with the membrane-bound substrate [Georgescu et al., 2000]. The D326 residue is involved in the hydrogen bond network that forms the phosphatase-C2 interface [Lee et al., 1999]. The 16 interface residues are among the best conserved across species and the majority have been found mutated in cancer [Lee et al., 1999]. Therefore, the c.976G→A transition observed in our patient is likely to disrupt PTEN function. Previous in vitro studies show that 90% of PTEN missense mutations, including several in the C2 domain, eliminate or reduce lipid-phosphatase activity, without affecting the ability of the protein to bind to the plasma membrane [Han et al., 2000].
Although our study implicates PTEN as an etiological factor in autism and macrocephaly, the nature of the underlying cellular events is largely unknown. In mice, the PTEN null mutation is lethal in the embryonic stage. Conditional knockout mice with selective inactivation of Pten in limited populations of hippocampal and cerebral cortex neurons display behavioral abnormalities reminiscent of autism, including decreased social interactions, exaggerated reaction to sensory stimuli, anxiety-like behaviors, seizures, and decreased learning [Kwon et al., 2006]. Pten mutant mice also exhibit progressive macrocephaly, with increased soma size, axonal and dendritic hypertrophy, and increased spine density [Kwon et al., 2006]. These findings are particularly interesting in view of previous studies showing abnormal development of dendritic spines in various neurodevelopmental disorders associated with autism, including fragile X syndrome, Rett syndrome, Angelman syndrome, tuberous sclerosis, Down syndrome, and William’s syndrome [Fiala et al., 2002; Ethell and Pasquale, 2005].
The major substrate of the lipid phosphatase activity of PTEN is phosphatidylinositol-3,4,5 triphosphate (PIP3). By dephosphorylating PIP3, PTEN opposes the action of PI3K, inhibiting the phosphorylation of AKT. Inactivation of PTEN results in increased levels of phosphorylated AKT, promoting cell growth, proliferation, survival and migration through multiple downstream effectors [Rodgers and Theibert, 2002]. PI3K and AKT have been shown to regulate dendritic arborization in neurons through the mammalian target of rapamycin (mTOR) signaling pathway [Jaworski et al., 2005]. AKT is an important negative regulator of the tuberous sclerosis complex gene products TSC1 and TSC2. Interestingly, mutations in TSC1 or TSC2 cause tuberous sclerosis, a neurocutaneous disorder associated with an increased risk for autism [Curatolo et al., 2004].
Through its protein phosphatase activity, PTEN dephosphorylates the signaling and scaffolding protein focal adhesion kinase (FAK) and the adapter protein Shc to inhibit cell migration and formation of focal adhesions induced by integrins and growth factors [Tamura et al., 1998; Gu et al., 1999]. Thus, PTEN could also alter cell adhesion and migration in the brain through similar mechanisms. FAK regulates dendrite outgrowth in developing neurons [Beggs et al., 2003] and controls extension and pruning of axons [Rico et al., 2004] through activation of Rho GTPases and assembly of actin filaments. Actin cytoskeleton dynamics play an essential role in the regulation of dendritic spine morphology and cognitive functions, as indicated by reports of mutations in several X-linked genes encoding components of the Rho family signaling pathway in families with mental retardation [Ethell and Pasquale, 2005].
In conclusion, the present findings confirm PTEN mutations as a cause of autism and macrocephaly, adding to the increasing list of single gene disorders underlying the developmental pathology of ASDs. Our results suggest that patients with autism and pronounced macrocephaly (≥4 SD) should be screened for PTEN mutations, even in the absence of other features of CS or BRRS. Ideally, PTEN screening should also be performed in patients with HC between +3 and +4 SD, but given the limited number of mutations in this category, molecular screening may be restricted to patients with personal or family history of features pointing to PTEN hamartoma-tumor syndrome. Furthermore, we suggest that polydactyly or oligodactyly may be considered uncommon manifestations of PTEN mutations. Because the diagnosis of a PTEN mutation implies a significant risk for malignancy, patients should be screened regularly for PTEN-related tumors. In addition, relatives should be offered genetic counseling, carrier detection and, if positive, oncologic surveillance.
Acknowledgments
We are grateful to all the families that participated in this research. We thank the clinicians of the Paris Autism Research International Sibpair (PARIS) Study for recruiting the families, the Autism Genetic Resource Exchange (AGRE), the DNA and cell bank of the INSERM U679 (Hôpital Pitié-Salpêtrière) and the Centre d’Investigations Cliniques, Hôpital Robert Debré, for obtaining and processing the samples from the French families.
Grant sponsor: Seaver Autism Research Center; Grant sponsor: NIH; Grant numbers: MH066673, NS-042165; Grant sponsor: INSERM; Grant sponsor: Assistance Publique-Hôpitaux de Paris; Grant sponsor: Fondation pour la Recherche Médicale; Grant sponsor: Fondation France Télécom; Grant sponsor: Fondation de France; Grant sponsor: Swedish Science Council.
References
- Beggs HE, Schahin-Reed D, Zang K, Goebbels S, Nave KA, Gorski J, Jones KR, Sretavan D, Reichardt LF. FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron. 2003;40:501–514. doi: 10.1016/s0896-6273(03)00666-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielschowsky M, Simons A. Über diffuse Hamartome (Ganglioneurome) des Kleinhirns und ihre Genese. J Psychol Neurol. 1930;41:50–75. [Google Scholar]
- Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, Eng C. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Garelick MG, Wang H, Lil V, Athos J, Storm DR. PI3 kinase signaling is required for retrieval and extinction of contextual memory. Nat Neurosci. 2005;8:925–931. doi: 10.1038/nn1482. [DOI] [PubMed] [Google Scholar]
- Curatolo P, Porfirio MC, Manzi B, Seri S. Autism in tuberous sclerosis. Eur J Paediatr Neurol. 2004;8:327–332. doi: 10.1016/j.ejpn.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Delatycki MB, Danks A, Churchyard A, Zhou XP, Eng C. De novo germline PTEN mutation in a man with Lhermitte-Duclos disease which arose on the paternal chromosome and was transmitted to his child with polydactyly and Wormian bones. J Med Genet. 2003;40:e92. doi: 10.1136/jmg.40.8.e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Eng C. PTEN: One gene, many syndromes. Hum Mutat. 2003;22:183–198. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- Ethell IM, Pasquale EB. Molecular mechanisms of dendritic spine development and remodeling. Prog Neurobiol. 2005;75:161–205. doi: 10.1016/j.pneurobio.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: Cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Fombonne E, Roge B, Claverie J, Courty S, Fremolle J. Microcephaly and macrocephaly in autism. J Autism Dev Disord. 1999;29:113–119. doi: 10.1023/a:1023036509476. [DOI] [PubMed] [Google Scholar]
- Georgescu MM, Kirsch KH, Kaloudis P, Yang H, Pavletich NP, Hanafusa H. Stabilization and productive positioning roles of the C2 domain of PTEN tumor suppressor. Cancer Res. 2000;60:7033–7038. [PubMed] [Google Scholar]
- Geschwind DH, Sowinski J, Lord C, Iversen P, Shestack J, Jones P, Ducat L, Spence SJ the AGRE Steering Committee. The Autism Genetic Resource Exchange: a resource for the study of autism and related neuropsychiatric conditions. Am J Hum Genet. 2001;69:463–466. doi: 10.1086/321292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillberg C, Gillberg C, Rastam M, Wentz E. The Asperger syndrome (and high-functioning autism) Diagnostic Interview (ASDI): A preliminary study of a new structured clinical interview. Autism. 2001;5:57–66. doi: 10.1177/1362361301005001006. [DOI] [PubMed] [Google Scholar]
- Goffin A, Hoefsloot LH, Bosgoed E, Swillen A, Fryns JP. PTEN mutation in a family with Cowden syndrome and autism. Am J Med Genet. 2001;105:521–524. doi: 10.1002/ajmg.1477. [DOI] [PubMed] [Google Scholar]
- Gu J, Tamura M, Pankov R, Danen EH, Takino T, Matsumoto K, Yamada KM. Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J Cell Biol. 1999;146:389–403. doi: 10.1083/jcb.146.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ. Lessons from fragile X regarding neurobiology, autism, and neurodegeneration. J Dev Behav Pediatr. 2006;27:63–74. doi: 10.1097/00004703-200602000-00012. [DOI] [PubMed] [Google Scholar]
- Han SY, Kato H, Kato S, Suzuki T, Shibata H, Ishii S, Shiiba K, Matsuno S, Kanamaru R, Ishioka C. Functional evaluation of PTEN missense mutations using in vitro phosphoinositide phosphatase assay. Cancer Res. 2000;60:3147–3151. [PubMed] [Google Scholar]
- Hendriks YM, Verhallen JT, van der Smagt JJ, Kant SG, Hilhorst Y, Hoefsloot L, Hansson KB, van der Straaten PJ, Boutkan H, Breuning MH, Vasen HF, Brocker-Vriends AH. Bannayan-Riley-Ruvalcaba syndrome: Further delineation of the phenotype and management of PTEN mutation-positive cases. Fam Cancer. 2003;2:79–85. doi: 10.1023/a:1025713815924. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci. 2005;25:11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lainhart JE, Piven J, Wzorek M, Landa R, Santangelo SL, Coon H, Folstein SE. Macrocephaly in children and adults with autism. J Am Acad Child Adolesc Psychiatry. 1997;36:282–290. doi: 10.1097/00004583-199702000-00019. [DOI] [PubMed] [Google Scholar]
- Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP. Crystal structure of the PTEN tumor suppressor: Implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Lin CH, Lu KT, Leu TH, Chang WC, Gean PW. A role for the PI-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron. 2001;31:841–851. doi: 10.1016/s0896-6273(01)00433-0. [DOI] [PubMed] [Google Scholar]
- Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24:659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- Manning MA, Cassidy SB, Clericuzio C, Cherry AM, Schwartz S, Hudgins L, Enns GM, Hoyme HE. Terminal 22q deletion syndrome: A newly recognized cause of speech and language disability in the autism spectrum. Pediatrics. 2004;114:451–457. doi: 10.1542/peds.114.2.451. [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Kum JB, Lunetta KL, Bennett MJ, Gorlin RJ, Ahmed SF, Bodurtha J, Crowe C, Curtis MA, Dasouki M, Dunn T, Feit H, Geraghty MT, Graham JM, Jr, Hodgson SV, Hunter A, Korf BR, Manchester D, Miesfeldt S, Murday VA, Nathanson KL, Parisi M, Pober B, Romano C, Eng C, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8:1461–1472. doi: 10.1093/hmg/8.8.1461. [DOI] [PubMed] [Google Scholar]
- Miles JH, Hadden LL, Takahashi TN, Hillman RE. Head circumference is an independent clinical finding associated with autism. Am J Med Genet. 2000;95:339–350. [PubMed] [Google Scholar]
- Morrow JD, Whitman BY, Accardo PJ. Autistic disorder in Sotos syndrome: A case report. Eur J Pediatr. 1990;149:567–569. doi: 10.1007/BF01957694. [DOI] [PubMed] [Google Scholar]
- Naqvi S, Cole T, Graham JM., Jr Cole-Hughes macrocephaly syndrome and associated autistic manifestations. Am J Med Genet. 2000;94:149–152. doi: 10.1002/1096-8628(20000911)94:2<149::aid-ajmg7>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Orstavik KH, Stromme P, Ek J, Torvik A, Skjeldal OH. Macrocephaly, epilepsy, autism, dysmorphic features, and mental retardation in two sisters: a new autosomal recessive syndrome? J Med Genet. 1997;34:849–851. doi: 10.1136/jmg.34.10.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi MA, Dinulos MB, Leppig KA, Sybert VP, Eng C, Hudgins L. The spectrum and evolution of phenotypic findings in PTEN mutation positive cases of Bannayan-Riley-Ruvalcaba syndrome. J Med Genet. 2001;38:52–58. doi: 10.1136/jmg.38.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftopoulou M, Etienne-Manneville S, Self A, Nicholls S, Hall A. Regulation of cell migration by the C2 domain of the tumor suppressor PTEN. Science. 2004;303:1179–1181. doi: 10.1126/science.1092089. [DOI] [PubMed] [Google Scholar]
- Reardon W, Zhou XP, Eng C. A novel germline mutation of the PTEN gene in a patient with macrocephaly, ventricular dilatation, and features of VATER association. J Med Genet. 2001;38:820–823. doi: 10.1136/jmg.38.12.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico B, Beggs HE, Schahin-Reed D, Kimes N, Schmidt A, Reichardt LF. Control of axonal branching and synapse formation by focal adhesion kinase. Nat Neurosci. 2004;7:1059–1069. doi: 10.1038/nn1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers EE, Theibert AB. Functions of PI 3-kinase in development of the nervous system. Int J Dev Neurosci. 2002;20:187–197. doi: 10.1016/s0736-5748(02)00047-3. [DOI] [PubMed] [Google Scholar]
- Sarquis MS, Agrawal S, Shen L, Pilarski R, Zhou XP, Eng C. Distinct expression profiles for PTEN transcript and its splice variants in Cowden syndrome and Bannayan–Riley–Ruvalcaba syndrome. Am J Hum Genet. 2006;79:23–30. doi: 10.1086/504392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner CE, Guerreiro MM, Marques-de-Faria AP. On macrocephaly, epilepsy, autism, specific facial features, and mental retardation. Am J Med Genet A. 2003;120:564–565. doi: 10.1002/ajmg.a.20032. [DOI] [PubMed] [Google Scholar]
- Stevenson RE, Schroer RJ, Skinner C, Fender D, Simensen RJ. Autism and macrocephaly. Lancet. 1997;349:1744–1745. doi: 10.1016/S0140-6736(05)62956-X. [DOI] [PubMed] [Google Scholar]
- Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280:1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- Veenstra-Vanderweele J, Christian SL, Cook EH., Jr Autism as a paradigmatic complex genetic disorder. Annu Rev Genomics Hum Genet. 2004;5:379–405. doi: 10.1146/annurev.genom.5.061903.180050. [DOI] [PubMed] [Google Scholar]
- Waite KA, Eng C. Protean PTEN: Form and function. Am J Hum Genet. 2002;70:829–844. doi: 10.1086/340026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver DD, Christian JC. Familial variation of head size and adjustment for parental head circumference. J Pediatr. 1980;96:990–994. doi: 10.1016/s0022-3476(80)80623-8. [DOI] [PubMed] [Google Scholar]
- Williams PG, Hersh JH. Brief report: The association of neurofibromatosis type 1 and autism. J Autism Dev Disord. 1998;28:567–571. doi: 10.1023/a:1026012414193. [DOI] [PubMed] [Google Scholar]
- Zappella M. Autistic features in children affected by cerebral gigantism. Brain Dysfunct. 1990;3:241–244. [Google Scholar]
- Zbuk KM, Stein JL, Eng C. PTEN hamartoma tumor syndrome (PHTS): Gene Reviews. 2006 http://www.genetests.org.
- Zhou XP, Waite KA, Pilarski R, Hampel H, Fernandez MJ, Bos C, Dasouki M, Feldman GL, Greenberg LA, Ivanovich J, Matloff E, Patterson A, Pierpont ME, Russo D, Nassif NT, Eng C. Germline PTEN promoter mutations and deletions in Cowden/Bannayan-Riley-Ruvalcaba syndrome result in aberrant PTEN protein and dysregulation of the phosphoinositol-3-kinase/Akt pathway. Am J Hum Genet. 2003;73:404–411. doi: 10.1086/377109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zori RT, Marsh DJ, Graham GE, Marliss EB, Eng C. Germline PTEN mutation in a family with Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome. Am J Med Genet. 1998;80:399–402. [PubMed] [Google Scholar]