

Abstract
Functionalization of the β-pyrrolic positions of the corrole macrocycle with –NO2 groups is limited at present to metallocorrolates due to of the instability exhibited by corrole free bases under oxidizing conditions. A careful choice of the oxidant can limit the transformation of corroles into decomposition products or isocorrole species, preserving the corrole aromaticity, and thus allowing the insertion of nitro groups onto the corrole framework. Here we report results obtained by reacting 5,10,15-tritolylcorrole (TTCorrH3) with the AgNO2/NaNO2 system, to give mono- and di-nitrocorrole derivatives when stoichiometry is carefully controlled. Reactions were found to be regioselective, affording the 3-NO2TTCorrH3 and 3,17-(NO2)2TTCorrH3 isomers as the main products in the case of mono- and di-substitution, in 53 and 20% yields, respectively. In both cases, traces of other mono- and di-substituted isomers were detected, which were structurally characterized by X-ray crystallography. The influence of the β-nitro substituents on the corrole properties is studied in detail by UV-visible, electrochemical, and spectroelectrochemical characterization of these functionalized corroles. Density Functional Theory (DFT) and time-dependent DFT (TDDFT) calculations of the ground and excited state properties of these β-nitrocorrole derivatives also afforded significant information, closely matching the experimental observations. It is found that the β-NO2 substituents conjugate with the π-aromatic system of the macrocycle, which initiates significant changes in both the spectroscopic and redox properties of the so functionalized corroles. This effect is more pronounced when the nitro group is introduced at the 2-position, because in this case the conjugation is, for steric reasons, more efficient than in the 3-nitro isomer.
Introduction
The establishment of efficient synthetic methodologies for the preparation of corrole1-4 has allowed a more detailed investigation of its chelating properties as a ligand, and of its unique behavior in several reactions.5 The investigation of corrole functionalization reactions is fundamental for the potential exploitation of this macrocyle in practical uses, since the manipulation of the peripheral positions can deeply affect the corrole properties, as shown by the first seminal examples reported in the literature.6
Compared with porphyrins, the peripheral functionalization of the corrole macrocycle is possible only in a limited number of cases, but recently some successful substitutions have been reported in the literature.2,7 In particular we have been interested in the nitration reaction, because the insertion of one or more –NO2 groups onto the β-pyrrole positions represents one of the most valuable modifications for researchers interested in functionalization of the corrole skeleton; for example, the potential of the nitro group in activating nucleophilic substitution on the adjacent β-carbons of the pyrrole rings is well known. Furthermore, it is also interesting to evaluate the influence of the strong electron withdrawing character of the nitro group on the behavior of the corrole ring, which is characterized by high electron density and its non-innocent character as ligand.8 In this connection, we recently reported the first successful example of a vicarious nucleophilic substitution performed on corrole nitro-derivatives by reacting copper and germanium β-(NO2) corrolates with 4-amino-4H-1,2,4-triazole.9 The reaction afforded β-amino-β-nitro derivatives, which can have great synthetic value for the elaboration of new chromophores, as well as extended aromatic β-fused architectures. To date β-nitrocorrole derivatives of copper,9 germanium,10 gallium,11 silver,12 cobalt,13 and iron14 have been prepared by using a variety of nitrating systems and reaction conditions, while all attempts to nitrate the corrole free-base failed, mainly because of the lability of corroles under these substitution conditions. This was evidenced under a variety of reaction conditions with the formation of isocorrole species. The recent development of demetalation protocols for the effective removal of silver15 and copper16 from the corresponding corrole complexes has allowed the preparation of nitrocorrole free bases from the application of the reductive DBU/THF system to a silver 3-NO2 corrolate (3-NO2TtBuCorrAg), obtained by using a large excess of AgNO2.15 Although this approach allowed circumvention of the instability of corrole under the nitrating systems tested to date, it opened the way to preparation of otherwise unavailable 3-NO2 corroles. Such preparations of functionalized free bases in a single step is highly desirable, particularly in the preparation of polynitrated free bases. Prompted by the recent results obtained in the case of nitration of copper corrole complexes9 carried out with an optimized ratio of silver salt and sodium nitrite mixture as the nitrating system, we decided to apply the same reaction conditions to corrole free bases, surmising that minimizing the oxidant stoichiometry could preserve corrole aromaticity during the reaction, but still allow nucleophilic attack by the NO2- ion upon the macrocycle.
We report here the first successful example of nitration of the corrole free base, obtained by reacting 5,10,15-tritolylcorrole (TTCorrH3) with the AgNO2/NaNO2 system. Variation of the molar ratio of reagents allowed us to obtain 3-nitro- and 3,17-dinitro-derivatives of corrole in satisfying yield, together with traces of other isomers, namely 2-NO2-, 2,3-(NO2)2 and 3,12-(NO2)2 corroles (Chart 1). While the substitution pattern of 2-NO2TTCorrH3 was established structurally by X-ray crystallography, in the case of 2,3-(NO2)2TTCorrH3 and 3,12-(NO2)2TTCorrH3 isomers, the substitution pattern was unambiguously confirmed by preparation of the corresponding Co triphenylphosphine complexes, which were also structurally characterized.
Chart 1.
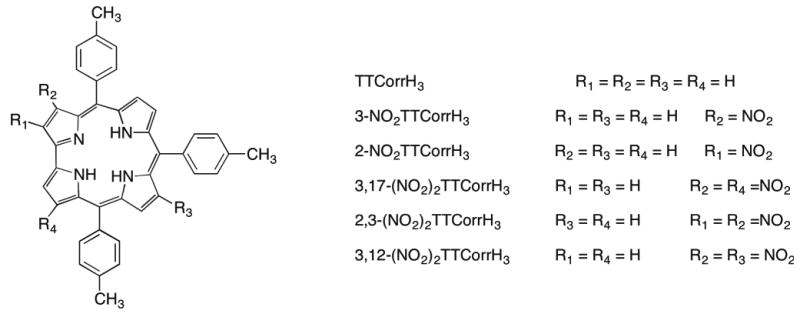
Molecular structures of β-nitrocorroles presented in this work.
The definition of the synthetic protocols for the preparation of free base nitrocorroles prompted us to study in detail the influence of the introduction of the nitro group at the corrole β-positions. This has been accomplished through a detailed UV-visible, electrochemical, and spectroelectrochemical characterization of these functionalized corroles. The changes in the UV-visible spectra and in the first reduction potential following β-nitration have been rationalized by Density Functional Theory (DFT) and time-dependent DFT (TDDFT) calculations of the ground and excited states of the TTCorrH3, 2-NO2TTCorrH3, 3-NO2TTCorrH3, and 3,17-(NO2)2TTCorrH3 series of free base corroles. Taken together, the experimental and theoretical results provide informative details on the behavior of these macrocycles.
Experimental Section
General
Silica gel 60 (70-230 mesh, Sigma Aldrich) was used for column chromatography. Reagents and solvents (Aldrich, Merck or Fluka) were of the highest grade available and were used without further purification, except for benzonitrile (PhCN), which was distilled over P2O5 under vacuum prior to use, and tetra-n-butylammonium perchlorate (TBAP), which was dried under vacuum at 30° C for at least one week prior to use. 1H NMR spectra were recorded on a Bruker AV300 (300 MHz) spectrometer. Chemical shifts are given in ppm relative to residual CHCl3 (7.25 ppm). UV-visible spectra were measured on a Cary 50 spectrophotometer. Mass spectra (FAB mode) were recorded on a VGQuattro spectrometer in the positive-ion mode using m-nitrobenzyl alcohol (Aldrich) as a matrix.
Diffraction data for chloroform solvates of 2-NO2TTCorrH3 and 2,3-(NO2)2TTCorrCoPPh3 and for a dichloromethane solvate of 3,12-(NO2)2TTCorrCoPPh3 were collected at low temperature on a Bruker Kappa Apex-II CCD diffractometer with either CuKα (λ=1.54184 Å) or MoKα (λ=0.71073 Å) radiation and an Oxford Cryosystems Cryostream chiller. Refinement was by full-matrix least squares using SHELXL,17 with H atoms in idealized positions, except for those on N, for which coordinates were refined. Absorption corrections were applied with a multi-scan method (Bruker SADABS18).
Synthesis of β-nitrocorroles
Preparation of 3-(NO2)TTCorrH3
NaNO2 (75 mg, 1.1 mmol) and AgNO2 (19 mg, 0.12 mmol) were added to a refluxing solution of TTCorrH3 (70 mg, 0.12 mmol) in DMF (12 mL), and the progress of the reaction was followed by TLC analysis and UV-visible spectroscopy. The reaction was complete in 10 min, as indicated by the UV-visible spectrum of the reaction mixture that showed the spectral features of the desired product. The reaction mixture was cooled, quenched by addition of few drops of aqueous hydrazine solution, precipitated by adding distilled water and then filtered. The crude product was taken up in CH2Cl2 and applied to a silica gel column for chromatographic purification. Elution with CH2Cl2/hexane (7:3) afforded traces of the silver complex, TTCorrAg, as a first reddish band followed by a second fraction containing a complex mixture of green and brownish compounds that were not easy to separate using a column. A subsequent main green fraction was isolated and crystallized from CH2Cl2/MeOH giving the 3-nitroderivative as an emerald green powder (38 mg, 52% yield). The spectroscopic characterization of 3-NO2TTCorrH3 was fully in agreement with data in the literature.16
The second fraction was subjected to preparative TLC on silica gel plates (CH2Cl2/hexane 1:1 as eluant). A green fraction having 0.52 Rf was isolated and identified as 2-NO2TTCorrH3.
2-NO2TTCorrH3. Mp> 300 °C. UV-visible (CHCl3): λmax, nm (log ε) 408 (4.52), 460 (4.53), 588 (4.33), 715 (4.31). 1H NMR (300 MHz, CDCl3): 9.13 (d, 1H, J = 4.20 Hz β-pyrrole), 9.08 (s, 1H, β-pyrrole), 8.74 (d, 1H, J = 4.83 Hz β-pyrrole), 8.64 (d, 1H, J = 4.89 Hz β-pyrrole), 8.34 (d, 1H, J = 4.95 Hz β-pyrrole), 8.28 (m, 3H, β-pyrrole+phenyls), 8.03 (d, 2H, J = 7.92 Hz phenyls), 7.95 (d, 2H, J = 7.83 Hz phenyls), 7.67 (d, 2H, J = 7.86 Hz phenyls), 7.59 (m, 6H phenyls), 2.70 (s, 3H, p-CH3), 2.67 (s, 6H, p-CH3). Anal. Calcd for C40H31N5O2: C, 78.2; H, 5.1; N, 11.3%. Found: C, 78.3; H, 5.1; N, 11.4%. MS (FAB): m/z 613(M+). Crystal data: C40H31N5O2 . 1.47 CHCl3, triclinic space group P-1, a=9.6282(10), b=14.5927(14), c=15.2094(15) Å, α=64.348(5), β=83.229(5), γ=87.496(5)°, V=1912.8(3) Å3, T=90.0(5) K, Z=2, ρcalcd=1.370 g cm-3, μ(CuKα)=3.42 mm-1. A total of 19,520 data was collected at to θ=68.7°. R=0.059 for 6350 data with I>2σ(I) of 6674 unique data and 510 refined parameters, CCDC 847685.
Preparation of 3,17-(NO2)2TTCorrH3
TTCorrH3 (100 mg, 0.18 mmol) was dissolved in DMF (15 mL) and the solution was heated at reflux. Then, NaNO2 (99 mg, 1.44 mmol) and AgNO2 (55 mg, 0.36 mmol) were added and the progress of the reaction was followed by TLC analysis and UV-visible spectroscopy. The reaction was complete in 30 min. The reaction mixture was cooled, quenched by addition of a few drops of aqueous hydrazine solution, precipitated by adding distilled water, and then filtered. The crude product was taken up in CH2Cl2 and applied to a silica gel column for chromatographic purification. Elution with CH2Cl2 afforded the isocorrole 3-(NO2)-5-(OH)TTisoCorH2 (10 mg, 9% yield) as the first fraction, followed by two green bands corresponding to the dinitroisomers 2,3-(NO2)2TTCorrH3 (2 mg, 1.7% yield) and 3,12-(NO2)2TTCorrH3 (1 mg, 0.8% yield), respectively. The last brilliant green fraction was collected and crystallized from CH2Cl2/n-hexane giving the 3,17-(NO2)2TTCorrH3 as a brilliant green powder (24 mg, 20% yield).
3,17-(NO2)2-TTCorrH3. Mp> 300 °C. UV-visible (CHCl3): λmax, nm (log ε) 439 (4.54), 483 (4.63), 707 (4.52). 1H NMR (300 MHz, CDCl3): 8.94 (s, 2H, β-pyrrole), 8.52 (d, 2H, J = 4.80 Hz β-pyrrole), 8.23 (d, 2H, J = 4.86 Hz β-pyrrole), 7.95 (m, 6H, phenyls), 7.55 (m, 6H, phenyls), 2.68 (s, 3H, p-CH3), 2.63 (s, 6H, p-CH3). Anal. Calcd for C40H30N6O4: C, 72.9; H, 4.6; N, 12.8%. Found: C, 72.8; H, 4.6; N, 12.7%. MS (FAB): m/z 658 (M+).
Preparation of 2,3-(NO2)2-TTCorrCoPPh3
2,3-(NO2)2-TTCorrH3 (5 mg, 0.08 mmol) in CH2Cl2 (10 mL) and Cobalt acetate (60 mg, 0.24 mmol) and triphenylphosphine (63 mg, 0.24 mmol) in CH3OH (15 mL) were mixed and the resulting solution was heated at reflux for 1 hour. The solvent was evaporated and the residue dissolved in CH2Cl2 and filtered through a silica plug to afford the complex as a green-bluish fraction. Crystallization from CH2Cl2/CH3OH afforded the title compound in an almost quantitative yield.
2,3-(NO2)2-TTCorrCoPPh3. Mp> 300 °C. UV-visible (CHCl3): λmax, nm (log ε) 370 (3.92), 552 (3.47), 1H NMR (300 MHz, CDCl3): 9.06 (d, 1H, J = 4.50 Hz β-pyrrole), 8.35 (d, 1H, J = 4.89 Hz β-pyrrole), 8.15 (d, 1H, J = 4.89 Hz β-pyrrole), 8.10 (d, 1H, J = 5.07 Hz β-pyrrole), 8.02 (d, 1H, J = 5.07 Hz β-pyrrole), 7.98 (d, 1H, J = 4.47 Hz β-pyrrole), 7.58-7.36 (m, 15 H, phenyls), 7.22 (m, 3 H, p-phosphine), 6.94 (m, 6 H, m-phosphine), 5.43 (m, 6 H, o-phosphine), 2.60 (s, 3H, p-CH3), 2.59 (s, 6H, p-CH3). Anal. Calcd for C58H42CoN6O4P: C, 71.31; H, 4.33; N, 8.60%. Found: C, 71.28; H, 4.36; N, 8.63%. MS (FAB): m/z 715 (M+-PPh3). Crystal data: C58H42CoN6O4P . 0.81 CHCl3, orthorhombic space group Fdd2, a = 37.190(3), b = 37.996(6), c = 13.8041(10) Å, V=19,506(4) Å3, T=90.0(5) K, Z=16, Dx=1.463 g cm-3, μ(MoKα)=0.58 mm-1. A total of 36,267 data was collected to θmax=30.0°. R=0.059 for 10,933 data with I>2σ(I) of 12,276 unique data and 634 refined parameters, Flack19 parameter 0.022(12). Disordered solvent contribution was removed using the SQUEEZE procedure.20 CCDC 869740
Preparation of 3,12-(NO2)2-TTCorrCoPPh3
3,12-(NO2)2-TTCorrCoPPh3 was prepared as reported above for the 2,3-dinitro isomer.
3,12-(NO2)2-TTCorrCoPPh3. Mp> 300 °C. UV-visible (CHCl3): λmax, nm (log ε) 450 (4.31), 594 (4.08), 1H NMR (300 MHz, CDCl3): 8.98 (s, 1H, β-pyrrole), 8.67(s, 1H, β-pyrrole), 8.53 (d, 2H, J = 4.65 Hz β-pyrrole), 8.30 (d, 2H, J = 5.04 Hz β-pyrrole), 8.21 (d, 2H, J = 5.13 Hz β-pyrrole), 8.07 (d, 2H, J = 4.53 Hz β-pyrrole), 7.66-7.32 (m, 15 H, phenyls), 7.28 (m, 3 H, p-phosphine), 7.18 (m, 6 H, m-phosphine), 6.53 (m, 6 H, o-phosphine), 2.62 (s, 3H, p-CH3), 2.57 (s, 3H, p-CH3), 2.54 (s, 3H, p-CH3). Anal.Calcd for C58H42CoN6O4P: C, 71.31; H, 4.33; N, 8.60%. Found: C, 71.27; H, 4.35; N, 8.64%. MS (FAB): m/z 715 (M+-PPh3). Crystal data: C58H42CoN6O4P · 0.5 CH2Cl2, monoclinic space group P2/c, a = 18.225(3), b = 9.2487(14), c = 29.294(4) Å, β = 105.212(10)°, V = 4764.8(13) Å3, T = 120.0(5) K, Z = 4, Dx = 1.421 g cm−3, μ(CuKα)= 4.12 mm−1. A total of 19,491 data was collected to θmax = 64.9°, R=0.059 for 6277 reflections with I > 2σ(I) of 7795 unique data and 644 refined parameters. Disordered CH2Cl2 was modeled as a half-populated site near a twofold axis. CCDC 869741.
Electrochemical and spectroelectrochemical measurements
Cyclic voltammetry was carried out with an EG&G model 173 potentiostat/galvanostat. A homemade three-electrode electrochemistry cell was used and consisted of a platinum button or glassy carbon working electrode, a platinum wire counter electrode, and a saturated calomel reference electrode (SCE). The SCE was separated from the bulk of the solution by a fritted-glass bridge of low porosity which contained the solvent/supporting electrolyte mixture. All potentials are referenced to the SCE.
UV-visible spectroelectrochemical experiments were performed with an optically transparent platinum thin-layer electrode of the type described in the literature.21 Potentials were applied with an EG&G Model 173 potentiostat/galvanostat. Time-resolved UV-visible spectra were recorded with a Hewlett-Packard Model 8453 diode array rapid-scanning spectrophotometer.
Determination of equilibrium constants of free base corroles
In order to study the protonation or deprotonation process, microliter quantities of TFA/CH2Cl2 and piperidine/CH2Cl2 were added gradually to 6.0 mL CH2Cl2 solution of CorrH3 in a 1.0 cm home-made glass cell. Solutions of the corroles were freshly degassed by N2 flux before each measurement and the spectra recorded after each addition of titration reagents. The concentration of the corrole solutions were in the 10-6 M range, in order to avoid aggregation of the macrocycle.
The free base is protonated in acid and deprotonated in basic solutions. The spectroscopic data were analyzed by the Hill equation, which gives the equilibrium constant (K) for the reactions.
Quantum Chemical Calculations
All calculations were performed with Turbomole V6.3 201122 using both pure (BP86)23 and hybrid (B3LYP)24 functionals in combination with the extensively polarized basis sets of triple-ζ quality including high angular momentum polarization functions (def2-TZVPP).25 In the BP86 calculations, the resolution of the identity (density fitting) approach26 was used to save computer time in combination with the def2-TZVPP/J auxiliary basis set.27 The ground state molecular structures of TTCorrH3, 2-NO2TTCorrH3, 3-NO2TTCorrH3, 3,17-(NO2)2-TTCorrH3 and related monoanionic species were fully optimized (without constraints) in the gas-phase and in dichloromethane solution. For the optimization of the monoanions, the unrestricted formalism was used. Solvation effects on the optimized structures were modeled through a dielectric continuum model, which was chosen to be the COSMO model.28 The optimized structures were confirmed to be local minima by calculating the harmonic vibrational frequencies. The thermodynamic parameters for the one-electron reduction reaction of the investigated metal-free corroles were computed at BP86 level, at the geometries optimized in dichloromethane solution. The zero-point energies (ZPE), thermal corrections, and entropy terms for the optimized geometries of the neutral and monoanionic species were obtained from frequency calculations in dichloromethane. Vertical absorption energies and oscillator strengths of the lowest singlet excited states of TTCorrH3 and its nitro derivatives were computed in dichloromethane solution at TDDFT29 level using the hybrid B3LYP24 functional and the DFT/B3LYP ground state geometries optimized in dichloromethane. The calculated excitation energies contain, apart from the altered “solvated” orbitals (slow term), also the contributions from the “fast” solvent response term.30
Results and Discussion
Synthesis
In the last few years, we have been particularly interested in the elaboration of synthetic protocols for the synthesis of β-nitrocorrole derivatives. By using different nitrating systems, we have been able to establish diverse methodologies for the convenient preparation of mono- and dinitro-metallocorrolates, starting in most cases from metallocorroles, such as the Cu, Ge, Fe derivatives. Our attempts to introduce nitro groups directly onto the free base, to obtain the corresponding functionalized corrole failed, since the required β-substitution took place in combination with other structural modifications, thereby precluding the isolation of the pure nitrocorrole free base. For example, using a large excess of AgNO2, both functionalization and metalation of the macrocycle occurred simultaneously, affording the corresponding Ag(III) 3-nitrocorrole as the reaction product.12 In the case of TFA/NaNO2 or HCl/NaNO2 systems, the formation of β-nitroisocorroles was observed, but these were converted in a second step into the corresponding aromatic corrole complexes by cobalt insertion.13 Our hypothesis for the nitration reaction mechanism is that the first step requires an oxidant species for the generation of the corrole α-cation radical, which is then attacked by nitrite to afford the final product. However, corrole free bases demonstrate extreme sensitivity under oxidizing reaction conditions, leading to a quite facile conversion of the macrocycle into the corresponding isocorrole species.9,13,15,31-35 Therefore, the nitration reaction conditions made the preparation of aromatic nitro-compounds difficult, instead favoring the formation of isocorrole derivatives through nucleophile insertion onto a meso-carbon position, which was observed for the 3-NO2-5-OH-isocorroles.9,31 Since the oxidation step is crucial for the reaction outcome, we surmised that a careful choice of the oxidant species could potentially drive the reaction pathway towards nitrocorroles, inhibiting the further transformation to isocorroles.
We recently reported that when the nitration reaction is performed on corrole using an equimolar amount of an oxidizing agent, such as chloranil in combination with an excess of NaNO2, the 3-NO2-5-OH-isocorrole is rapidly obtained as the main reaction product, confirming the regioselectivity of both mononitration and the formation of the single 5-substituted isocorrole isomer.31 Our attention turned towards other oxidants and, prompted by the results reported recently for nitration of copper corrolates, we tested the AgNO2/NaNO2 system, which allows the preparation of mono- and di-nitro-derivatives in good yields just by varying the reagent stoichiometry.9
Thus, TTCorrH3 was reacted in refluxing DMF with a mixture of sodium and silver nitrites using a corrole/AgNO2/NaNO2 molar ratio of 1:1:9 (Scheme 1) with the intention of achieving mononitration of the corrole. The reaction proceeded very fast and in 10 minutes complete disappearance of the starting compound was observed by TLC analysis, together with the formation of a green major product. The UV-visible spectrum of the mixture changed rapidly even from the beginning of the reaction and showed at the end the typical spectral features of 3-NO2TTCorrH3,15 indicating it as the main product. Chromatographic purification afforded traces of the silver complex of the corrole as the first rose-red band together with a second fraction containing a mixture of brownish and greenish products, inseparable on the column. A third green fraction was the major reaction product and corresponded to 3-NO2TTCorrH3, obtained in 52% yield. It is worth mentioning that the formation of the 3-nitro-5-hydroxy-isocorrole was not observed, showing that the amount of silver nitrite was correctly chosen to allow the β-functionalization, and to preserve at the same time the integrity of the nitrocorrole free base formed in the reaction medium. The second band was further purified on a preparative silica gel TLC plate, using a mixture of CH2Cl2/hexane as eluant. A green band (Rf=0.52) was isolated in sufficient amount to be characterized using the usual spectroscopic techniques. This fraction showed an unusual UV-visible spectrum, characterized by a split Soret band (of similar intensity) at 408 and 460 nm, a less intense narrow band centered at 588 nm, and a broad band at about 715 nm. The FAB mass spectrum of this compound afforded a molecular peak at m/z 613, which is consistent with a mononitrocorrole. The 1H NMR spectrum and integral calculations confirmed it as a monosubstituted corrole showing a proton singlet at 9.08 ppm. It is noteworthy that the pattern of substitution is clearly different from that of the known 3-NO2TTCorrH3,, showing a pyrrolic doublet more deshielded compared with the singlet, just as observed for the 2-bromocorrole isomer obtained by reacting the TTCorrH3 with EtMgBr.33 The definitive identification of position C2 as the site of substitution for this compound was afforded by X-ray crystallographic analysis, carried out on a single crystal obtained by slow diffusion of n-hexane into a dilute dichloromethane solution. This unambiguously confirmed the new monosubstituted corrole as the 2-NO2TTCorrH3 isomer (Figure 1).
Scheme 1.
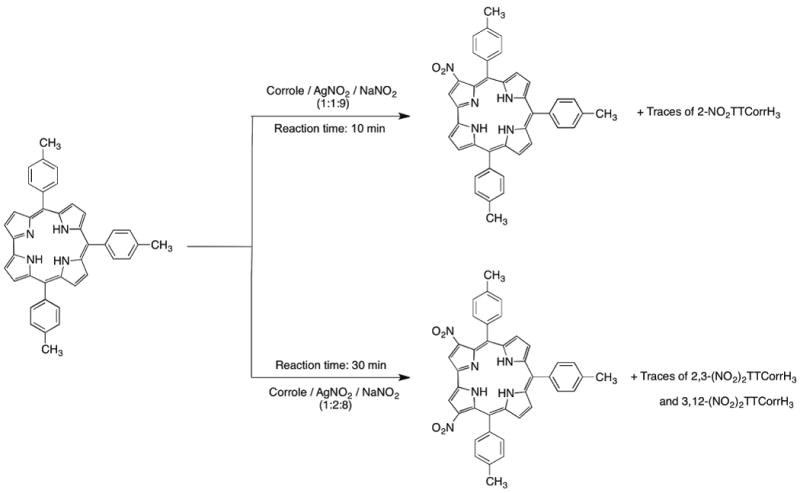
Synthesis of (NO2)xTTCorrH3.
Figure 1.
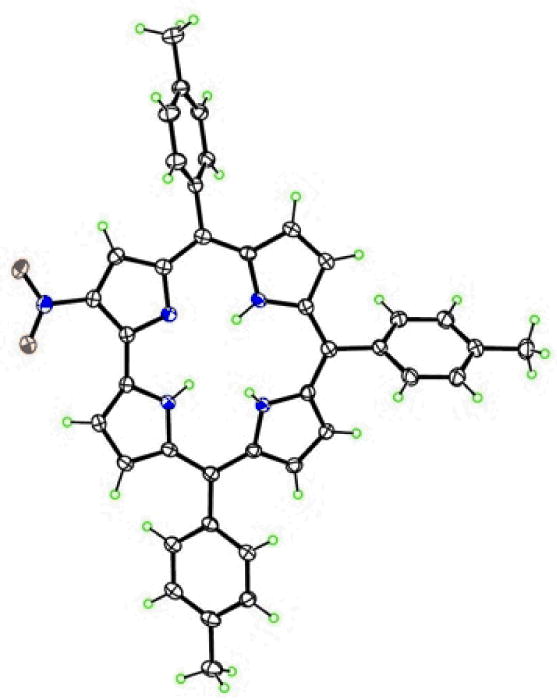
The molecular structure of 2-NO2TTCorrH3, with 50% ellipsoids.
The three protonated N atoms in the interior of the molecule force marked deviations of the corrole system from coplanarity. The mean deviation of its 23 atoms is 0.159 Å, with a maximum of 0.415(4) Å. The nitrated “A” pyrrole and the adjacent “B” pyrrole not directly bonded to it come nearest to being coplanar, forming a dihedral angle of 10.5(6)°. The main distortion of the corrole is a twist, in which the other two pyrroles “C” and “D” tip alternately above and below this plane, forming dihedral angles of 18.1(3) and 20.6(2)° with it and 23.8(3)° with each other. The N atoms of the two alternately tipped pyrroles are somewhat pyramidal, with their H atoms lying 0.14(4) and -0.30(4) Å out of the best planes of their respective pyrroles. The third N-H hydrogen atom is not tipped out of plane, but forms an N-H…N hydrogen bond (N…N 2.623(3) Å; N-H…N 134(3)°). The nitro group lies essentially in the plane of the pyrrole to which it is bonded, with a C1-C2-N-O torsion angle 1.2(4)°. The two directly-bonded pyrroles exhibit a N-C1-C19-N twist of 7.6(3)°.
With the aim of driving the reaction towards dinitrocorrole derivatives, we modified the amounts of nitrating agents by altering the corrole/AgNO2 molar ratio to 1:2 (Scheme 1). Of course, in this case we were aware that the increase of the oxidant reagent could induce the formation of the isocorrole, instead of forming dinitrocorroles. The reaction of TTCorrH3 with silver and sodium nitrites in a 1:2:8 molar ratio, afforded in 30 minutes a mixture of compounds observable from the TLC analysis as green and bluish-green bands together with a number of decomposition products.
Chromatographic purification on silica gel using dichloromethane as eluant afforded, after elution of small traces of the silver tritolylcorrolate, a first major fraction consisting of the 3-NO2-5-OH-TTisoCorrH2, obtained in 9% yield. Afterwards, a small quantity of two green fractions was isolated and subjected to spectroscopic identification. The band having the greater Rf value showed a UV-visible spectral profile quite similar to 3-NO2TTCorrH3, but generally red-shifted, while the second fraction displayed a strongly red-shifted Soret band at 472 nm together with two satellite bands at 613 and 666 nm. The mass spectrometry analysis afforded a molecular peak at m/z 658 for both compounds, tentatively identifying them as two different dinitrocorrole isomers. The proton NMR spectra of both compounds were of poor resolution, hampering definitive identification of the sites of substitution by the nitro groups.
The last green fraction obtained by column chromatographic purification was the main reaction product, obtained in 20% yield, and fully characterized as 3,17-(NO2)2TTCorrH3. The UV-visible spectrum was unusual, having a split Soret band at 439 and 483 nm and an intense broad absorbtion at ca. 700 nm.
To allow a more definitive identification of the other dinitrocorrole isomers, we decided to prepare the corresponding Co complexes, by reaction of these products with Co(OAc)2 and PPh3; the reaction provided cobalt complexes having well resolved 1H NMR spectra. In the case of the first compound, the lack of proton singlets together with the presence of a down-field shifted doublet above 9 ppm, clearly separated from the other five pyrrolic resonances at ca 8-8.5 ppm, led to the conclusion that for this compound the substitution occurred on the C2 and C3 positions of the corrole macrocycle. We were able to obtain crystals of this isomer suitable for X-ray crystallographic analysis, which helped to elucidate the structure of the corresponding free base as 2,3-(NO2)2TTCorrH3.
The structure of 2,3-(NO2)2TTCorrCoPPh3 is shown in Figure 2. The Co atom has square pyramidal coordination with Co-N distances in the range 1.869(3) - 1.886(3) Å (mean 1.880 Å), Co-P distance 2.2321(8) Å, and the Co is 0.2634(13) Å out of the best plane of the four pyrrole N atoms. The 23-atom C19N4 corrole core deviates only slightly from planarity, with a rms deviation 0.059 Å and maximum deviation 0.119(3) Å for C7. The small distortion from planarity is a slight saddle. One nitro group lies nearly in the corrole best plane, with C1-C2-N-O torsion angle -6.9(5)°, while the other is more nearly orthogonal to the corrole, with C2-C3-N-O torsion angle -73.1(4)°.
Figure 2.
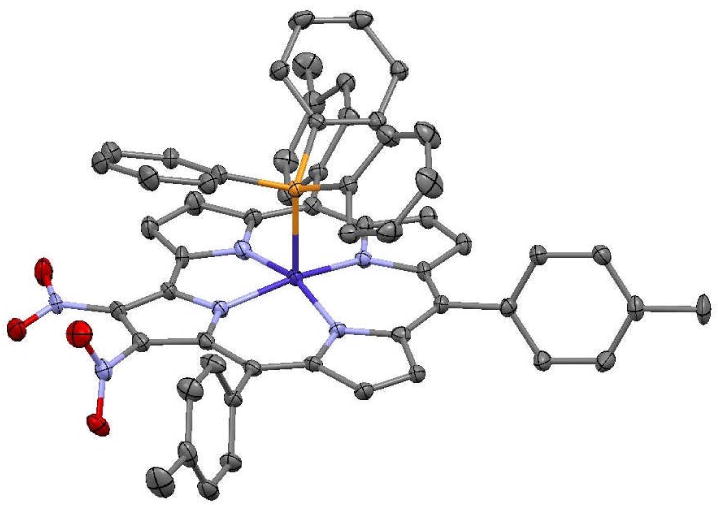
The molecular structure of 2,3-(NO2)2TTCorrCoPPh3, with 50% ellipsoids and H atoms not shown.
The second Co complex showed two proton singlets at 8.98 and 8.67 ppm, respectively, and three different proton signals of equal intensity at 2.62, 2.57 and 2.54 ppm, indicating it to be an asymmetric product. Also in this case the X-ray crystallographic characterization (Figure 3) led to identification of 3,12-(NO2)2TTCorrCoPPh3, confirming the corresponding corrole free base as the 3,12-(NO2)2TTCorrH3 isomer. It is interesting to note that this isomer is, to the best of our knowledge, the first example of a disubstituted corrole where pyrroles A and C are functionalized, instead the usual A and D pattern.
Figure 3.
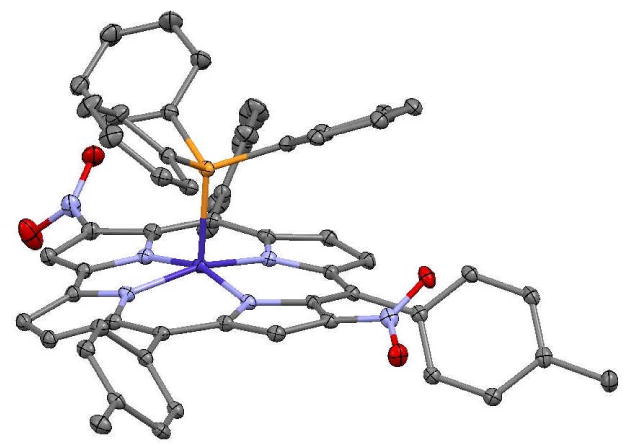
The molecular structure of 3,12-(NO2)2TTCorrCoPPh3, with 30% ellipsoids and H atoms not shown.
The structure of 3,12-(NO2)2TTCorrCoPPh3 is illustrated in Figure 3, and is quite similar to that of 2,3-(NO2)2TTCorrCoPPh3, with a somewhat more nonplanar corrole core. The Co atom has square pyramidal coordination with Co-N distances in the range 1.873(3) - 1.892(3) Å (mean 1.878 Å), Co-P distance 2.2113 (11) Å, and the Co is 0.2734(15) Å out of the best plane of the four pyrrole N atoms. The 23-atom C19N4 corrole core is approximately three times more distorted from planarity than that of 2,3-(NO2)2TTCorrCoPPh3, having a rms deviation of 0.159 Å and maximum deviation 0.339(4) Å for C8. The distortion is best described as a saddle, although all four N atoms lie on the same side of the 23-atom best plane by a mean distance of 0.118 Å. The two nitro groups are twisted out of the corrole best plane by similar amounts, forming torsion angles: C4-C3-N-O, −40.0 (6)° and C11-C12-N-O, 29.2 (5)°.
It is worth mentioning that in all cases the use of the AgNO2/NaNO2 system allowed the β-functionalization of the macrocycle, without inducing the concomitant metalation, which occurs when the reaction is carried out with an excess of silver nitrite. In this case the AgNO2 acts only as oxidant, and the proper amount chosen prevented the metalation of the macrocycle. The transient formation of the silver corrole complex, then demetalated by the final addition of hydrazine can be safely excluded, because hydrazine is not effective to promote silevr corrole demetalation and because the same products were again obtained when the nitration reaction was carried out avoiding the addition of this reagent.
Electrochemistry and Spectroelectrochemistry
The redox behavior of free base corroles is complicated in non-aqueous media by the facile gain or loss of protons, which can give in solution a mixture of the neutral corrole along with either its protonated [(Corr)H4]+ or deprotonated [(Corr)H2]- form. To investigate the effect of nitration and the solvent on both the overall oxidation/reduction mechanism and the electrochemically initiated protonation/deprotonation process, a series of three corroles, TTCorrH3, 3-NO2TTCorrH3 and 3,17-(NO2)2TTCorrH3, were selected for electrochemical and spectroelectrochemical characterization in pyridine, PhCN and CH2Cl2.
We earlier reported the electrochemistry and spectroelectrochemistry of several meso-substituted corroles in PhCN and pyridine, including TTCorrH3.36 It was demonstrated that the electroactive species in PhCN was the neutral corrole CorrH3, while in pyridine, the monoanion [(Corr)H2]- is the prevalent species,36 the deprotonation process being almost complete. A similar solvent effect on deprotonation is seen for the currently investigated corroles, as seen by the cyclic voltammograms in Fig. 4 for TTCorrH3 in CH2Cl2, PhCN and pyridine.
Figure 4.
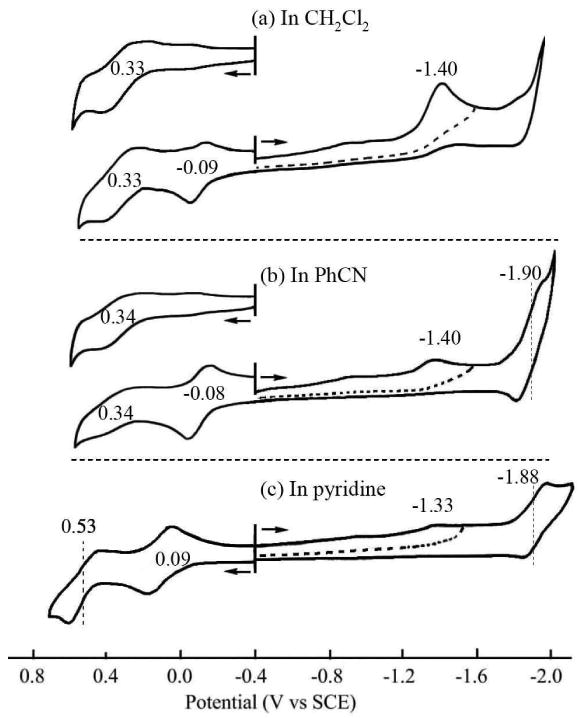
Cyclic voltammograms of TTCorrH3 in (a) CH2Cl2, (b) PhCN and (c) pyridine containing 0.1 M TBAP.
The first reduction of TTCorrH3 is irreversible in all three solvents and located at Ep = -1.33 to -1.40 V for a scan rate of 0.1 V/s. Only a residual amount of neutral CorrH3 is present in the basic pyridine solvent and a small peak current is seen for reduction of the fully protonated corrole. This contrasts with CH2Cl2, where CorrH3 is the main electroactive species in solution and the peak current is much higher for the first reduction. Sweeping the potential from -0.40 to values beyond the first reduction in PhCN or pyridine drives to completion the deprotonation of TTCorrH3, after which the deprotonated [(TTCorr)H2]- product undergoes a reversible one-electron ring-reduction, which is located at E1/2 = -1.90 V in PhCN and -1.88 V in pyridine.
As indicated above TTCorrH3 undergoes deprotonation in both CH2Cl2 and PhCN after the first one-electron reduction and this then generates [(Corr)H2]-. As a result, when reversing the scan at potential negative of the first reduction and then scanning in a positive direction, a new redox couple assigned to [(Corr)H2]-/(*orr)H2) can be seen in CH2Cl2 and PhCN. This process is located at E1/2 = -0.09 to -0.08 V and it is not observed in CH2Cl2 or PhCN, when sweeping the potential towards more positive values from the initial potential of -0.4 V. This oxidation of [(Corr)H2]- is however seen in pyridine on all scans, in a positive or negative direction (E1/2 = 0.09 V).
To investigate the effect of nitration on both the overall oxidation/reduction mechanism and the protonation/deprotonation processes, 3-NO2TTCorrH3 and 3,17-(NO2)2TTCorrH3, were also characterized electrochemically and spectroelectrochemically in pyridine, PhCN and CH2Cl2.
The addition of one or two nitro substituents to the β-pyrrole positions of the macrocycle leads to a positive shift in E1/2 for each oxidation or reduction of the nitro-substituted free base corroles, as compared to measured half wave potentials for the oxidation or reduction of TTCorrH3. This is as expected, due to the strong electron-withdrawing character of the NO2 substituents, with the magnitude of the positive shift in E1/2 varying as a function of both the number of the nitro groups on the macrocycle (0, 1 or 2), as well as the specific electrode reaction.
A summary of redox potentials for of TTCorrH3, 3-NO2TTCorrH3 and 3,17-(NO2)2TTCorrH3 in the different solvents is given in Table 1, while typical cyclic voltammograms are shown in Figure 5 for the three corroles in pyridine, containing 0.1 M TBAP. As shown in this figure, the addition of one or two nitro substituents to the β-pyrrole positions of the macrocycle leads to similar positive shifts in E1/2 for the first two oxidations. A positive shift in E1/2 of 130 ∼ 160 mV for each additional nitro group is at the 3 and 17 positions of the macrocycle. For example, the [(Corr)H2]-/(*Corr)H2) reaction in pyridine is located at E1/2 = 0.09 V for TTCorrH3 (with no NO2 group), while 3-NO2TTCorrH3 (with one NO2 group) is oxidized at E1/2 = 0.25 V and 3,17-(NO2)2TTCorrH3 (with two NO2 groups) is oxidized at E1/2 = 0.41 V. It is also worth mentioning that a much larger substituent effect of the nitro group is observed for the reductions than for the oxidations. For example, a 540 mV shift in E1/2 for the second reduction is seen upon going from TTCorrH3 to 3-NO2TTCorrH3. Although the magnitude of the shift in E1/2 for the second and third reductions is reduced to 290 and 310 mV respectively, upon going from 3-NO2TTCorrH3 to 3,17-(NO2)2TTCorrH3, the effect of NO2 on these processes is still much larger than that observed for the first oxidation. This trend in substituent effects was also observed for β-NO2 substituted Fe(III), Cu(III) and Ge(IV) corroles having similar structures9,10,37 and fits with the major effect of the nitro group being to lower the energy of the lowest unoccupied molecular orbital (LUMO), vide infra.
Table 1.
Half-wave or peak potentials (V vs SCE) for the different forms of (NO2)xTTCorrH3 in solvents containing 0.1 M TBAP.
| Solvent | cpd | ox | red | ||||
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| 2nd | 1st | 1st | 2nd | 3rd | |||
| pyridine | TTCorrH3 | 0.56a | 0.09 | -1.33a | -1.88 | ||
| 3-NO2TTCorrH3 | 0.70a | 0.25 | -0.87a | -1.34 | -1.71 | ||
| 3,17-(NO2)2TTCorrH3 | 0.83a | 0.41 | -0.75a | -1.05 | -1.40 | ||
| CH2Cl2 | TTCorrH3 | 0.33 | -0.09b | -1.40a | |||
| 3-NO2TTCorrH3 | 0.50a | 0.16b | -0.84a | -1.42a | -1.60a | ||
| 3,17-(NO2)2TTCorrH3 | -- | 0.33b | -0.62a | -1.06 | -1.39 | ||
| PhCN | TTCorrH3 | 0.34 | -0.08b | -1.40a | -1.90 | ||
| 3-NO2TTCorrH3 | -- | 0.14b | -0.84a | -1.39 | -1.76a | ||
| 3,17-(NO2)2TTCorrH3 | 0.94 | 0.35b | -0.62a | -1.08 | -1.38 | ||
Peak potential at scan rate of 0.1 V/s;
This redox couple can only be seen on the second positive potential sweep after scanning through the first reduction.
Figure 5.
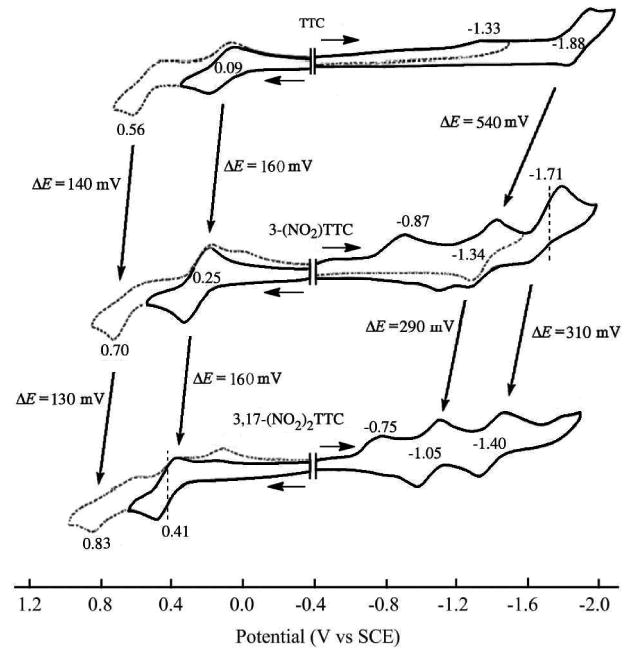
Cyclic voltammograms of TTCorrH3, 3-NO2TTCorrH3 and 3,17-(NO2)2TTCorrH3, in pyridine containing 0.1 M TBAP.
As mentioned above, the first reduction of the investigated corroles is irreversible in pyridine, as well as in CH2Cl2 and PhCN (see Table 1). In all three solvents, the reduction leads to formation of the deprotonated [(Corr)H2]- species as the final product. An example of the thin-layer UV-visible spectral changes observed during the first one-electron reduction of 3-NO2TTCorrH3 in CH2Cl2 are shown in Figure 6a. As seen in this figure, the initial spectrum of 3-NO2TTCorrH3 is characterized by three separated Soret bands at 396, 438 and 464 nm and two visible Q-like bands at 589 and 667 nm. Controlled potential reduction at -0.95 V in the thin-layer cell leads to a decreased intensity of the 396 and 464 nm bands, as well as to a complete disappearance of the 438 nm band, while the two initially separated visible bands become merged into a single peak at 630 ∼ 667 nm (Figure 6a). These spectral changes match perfectly spectral changes observed in the same solvent during a titration of 3-NO2TTCorrH3 with piperidine (Figure 6b), and gives further evidence for formation of the deprotonated species in the electrochemical reaction, where [(Corr)H2]- is characterized by absorption bands at 396, 463 and 666 nm. The diagnostic log-log plot of the titration data is shown in the inset of Figure 6b and has a slope of 1.0 with a log K = 3.5 for dissociation of one proton under the given solution condition.
Figure 6.
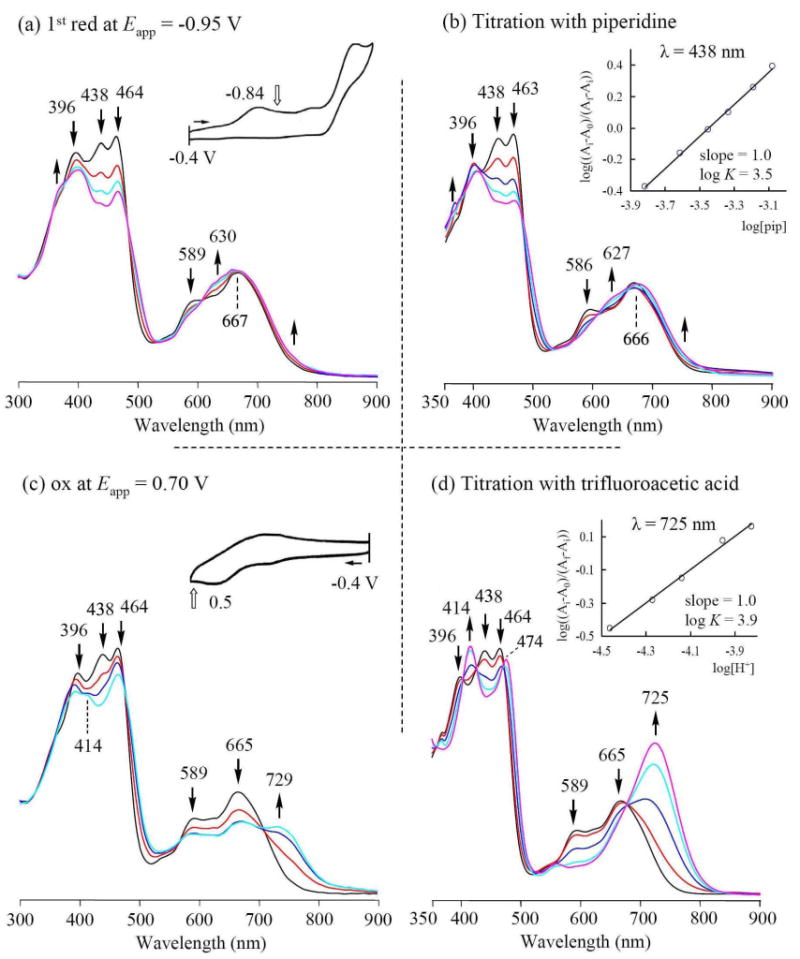
UV-visible spectral changes of 3-NO2TTCorrH3 in CH2Cl2 during (a) the controlled reduction at -0.95 V, (b) successive addition of piperidine (inset shows the Hill plot), (c) the controlled oxidation at 0.70 V and (d) successive addition of TFA (inset shows the Hill plot).
As shown by the cyclic voltammogram insert in Figure 6c, 3-NO2TTCorrH3 undergoes an irreversible to quasi-reversible one-electron oxidation at Ep = 0.50 V in CH2Cl2 at scan rate of 0.1 V/s. Similar irreversible oxidations have been observed for some free-base porphyrins, where the product of the first electron abstraction involves a protonation process,38 although in the case of free-base corroles the oxidation mechanism seems to be more complicated. Our previous studies36b suggest that the one-electron oxidation of triarylcorroles initially gives the corrole radical cation, [(Corr)H3]+, which then rapidly loses a proton after reaction with another molecule of the neutral corrole in solution to generate a mixture of the free-base neutral corrole radical, (Corr)H2, and the corrole monocation, [(Corr)H4]+. In Figure 6c and 6d it is possible to compare the spectral changes observed for 3-NO2TTCorrH3, when a controlled potential oxidation at 0.70 V was carried out (see Figure 6c), and when the same macrocycle was titrated with successive additions of TFA in CH2Cl2 solution. The final UV-visible spectra under these two conditions are quite similar to each other.
As expected, the introduction of one or two nitro groups at the β-positions of the corrole macrocycle influences the corresponding acid-base properties of the molecule. Spectral changes of the type reported in Figure 6b and 6d for 3-NO2TTCorrH3, were exploited to obtain equilibrium constants for deprotonation (log Ka) and protonation (log Kb) processes of the three investigated free-base corroles in CH2Cl2 and the corresponding protonation-deprotonation constants are reported in Table 2. The equilibrium constant for proton dissociation increased upon going from TTCorrH3 (log Ka = 1.1) to 3-NO2TTCorrH3 (log Ka = 3.5) and then to 3,17-(NO2)2TTCorrH3 (log Ka = 4.9), while for log Kb, a reverse trend is obtained (see Table 2), indicating that the acidity of free-base corroles increases, while the basicity decreases upon increasing the number of peripheral nitro substituents. It is interesting to note that the effect of the nitro groups is higher for the increase of the acidity constants than that observed for the corresponding Kb.
Table 2.
Equilibrium Constants for Deprotonation (log Ka) and Protonation (log Kb) of (NO2)xTTCorrH3 in CH2Cl2.
| cpd | log Ka | log Kb |
|---|---|---|
|
|
|
|
| reacting with piperidine | reacting with trifluoroacetic acid | |
| TTCorrH3 | 1.1 | 4.5 |
| 3-NO2TTCorrH3 | 3.5 | 3.9 |
| 3,17-(NO2)2TTCorrH3 | 4.9 | 3.3 |
Theoretical Studies
To rationalize the effects of introducing nitro groups at the periphery of TTCorrH3 the structural, electronic optical and electrochemical properties of TTCorrH3 and its nitro derivatives were theoretically investigated using DFT and TDDFT methods.
(a) Molecular structures
In metal-free corroles the inner hydrogen atoms can be distributed in different ways among the four nitrogen atoms39 which results in the formation of tautomers. In this study only one tautomeric form of TTCorrH3, 2-NO2TTCorrH3, 3-NO2TTCorrH3 and 3,17-(NO2)2TTCorrH3 was examined theoretically. In the choice of the tautomers of TTCorrH3 and its nitro derivatives we have been guided by the X-ray structural data available for TPCorrH3 (TPCorrH3 = 5,10,15-triphenylcorrole)39c and 2-NO2TTCorrH3, respectively. In both compounds the inner hydrogen atoms are located on B, C, and D pyrrole rings. The structural analysis was performed in the gas-phase and in dichloromethane solution, using both BP86 and B3LYP functionals. The optimized structures showed very little sensitivity to the used functional and solvation effects. Table 3 collects the relevant structural parameters computed at the B3LYP level, in the gas-phase, for TTCorrH3, 2-NO2TTCorrH3, 3-NO2TTCorrH3, and 3,17-(NO2)2TTCorrH3. For 2-NO2TTCorrH3, comparison is made between the theoretical and experimental data.
Table 3.
Selected Bond Lengths (Å), Bond Angles (deg) and Metrical Parameters Calculated for TTCorrH3 and its Nitro Derivatives, 2-NO2TTCorrH3, 3-NO2TTCorrH3, and 3,17-(NO2)2TTCorrH3.
| TTCorrH3 | 2-NO2TTCorrH3a | 3-NO2TTCorrH3 | 3,17-(NO2)2TTCorrH3 | |
|---|---|---|---|---|
| C–N(nitro)b | 1.430/1.421(3) | 1.445 | 1.447/1.441 | |
| N1–N2 | 2.621 | 2.639/2.623(3) | 2.623 | 2.627 |
| N2–H2…N1 | 132.2 | 131.5/134(3) | 130.4 | 129.8 |
| θ1c | 10.2 | 9.4/10.5(6) | 12.4 | 13.7 |
| θ3d | 22.2 | 22.4/23.8(3) | 23.6 | 27.9 |
| θ1e | 10.3 | 11.0/7.3(3) | 12.1 | 14.4 |
| θ2f | 2.8/1.2(4) | 31.4 | 32.9/20.0 |
Experimental values (this work) in italics.
C2–N5, C3–N5, and C3–N5/C17–N6 bond lengths for 2-NO2TTCorrH3, 3-NO2TTCorrH3, and 3,17-(NO2)2TTCorrH3, respectively.
Dihedral angle between the B and A pyrrole mean planes.
Dihedral angle between the C and D pyrrole mean planes.
N1–C5–C19–N4 torsional angle.
C1–C2–N5–O1, C4–C3–N5–O1, C4–C3–N5–O1/C16–C17–N6–O3 torsional angles for 2-NO2TTCorrH3, 3-NO2TTCorrH3, and 3,17-(NO2)2TTCorrH3, respectively.
As observed in other metal-free triarylcorroles,39c,40 in TTCorrH3 the corrole macrocycle exhibits a significant distortion from planarity to minimize the steric hindrance between the inner protons. The macrocycle adopts a puckered conformation in which the pyrrole rings turn either slightly up or down. According to the computed twist angle between the pyrrole mean planes, θ1, the pyrrolenine ring A and the adjacent pyrrole not directly bonded to it, B, are close to be coplanar, mostly due the presence of an intramolecular hydrogen bond between the N2–H group and the iminic N1 atom (N1–N2 = 2.621 Å, –H2···N1 = 132.2°). The D and C rings are tipped up and down with the respect to the 23 atom mean plane forming a dihedral angle (θ3) of 22.2° with each other. The two directly bonded pyrroles, A and D, show a N–C–C–N twist (ϕ1) of 10.3°. Comparison of the structural parameters of 2-NO2TTCorrH3 with those of TTCorrH3 shows that the introduction of a nitro group at the C2 position has very little impact on the geometry of the corrole macrocycle. As indicated by the C1–C2–N5–O1 angle (ϕ2), the NO2 group is substantially coplanar with the pyrrolenine ring A. This, together with a relatively short C2–N5 bond length (1.430 Å), suggests conjugation between the corrole and the nitro substituent. The geometrical data listed in Table 3 for 2-NO2TTCorrH3 show agreement between theory and experiment, thus confirming that the puckering of the corrole macrocycle is dictated by intrinsic electronic factors. Because of steric hindrance with the adjacent phenyl ring the nitro group in 3-NO2TTCorrH3 is no longer coplanar with the pyrrolenine ring A, the C4–C3–N5–O1 angle amounting to 31.4°. This results in decreased conjugation between the nitro group and the corrole macrocycle, as confirmed by the elongation of the C–N bond. In turn, the phenyl ring on the 5 position tilts towards the bipyrrolic block by 50.2°. This reflects in a slight increase of macrocycle puckering. In 3,17-(NO2)2TTCorrH3 the puckering of the corrole macrocycle is further enhanced relative to the mononitrated corroles, due to the cooperative effects of the nitro substituents. It is worth noting, however, that while the twisting of the nitro group on the 3 position is very much the same as in 3-NO2TTCorrH3 (32.9° vs 31.4°), that of the nitro group on the 17 position is only 20.0°, leading to a more effective conjugation with the pertinent pyrrole.
(b) Ground-state electronic structure and optical spectra
To provide an interpretation of the UV/vis spectral changes accompanying the nitration of TTCorrH3, TDDFT calculations of the electronic absorption spectra in dichloromethane solution were performed for 2-NO2TTCorrH3, 3-NO2TTCorrH3, 3,17-(NO2)2TTCorrH3 and the parent TTCorrH3. Before dealing with the excited states, we briefly discuss the ground-state electronic structure of the investigated metal-free corroles. To highlight the electronic effects of the nitro groups, the electronic structure of the unsubstituted free-base is taken as a reference. An energy level scheme of the highest occupied and lowest unoccupied Kohn-Sham orbitals of TTCorrH3 and its nitro derivatives is shown in Figure 7.
Figure 7.
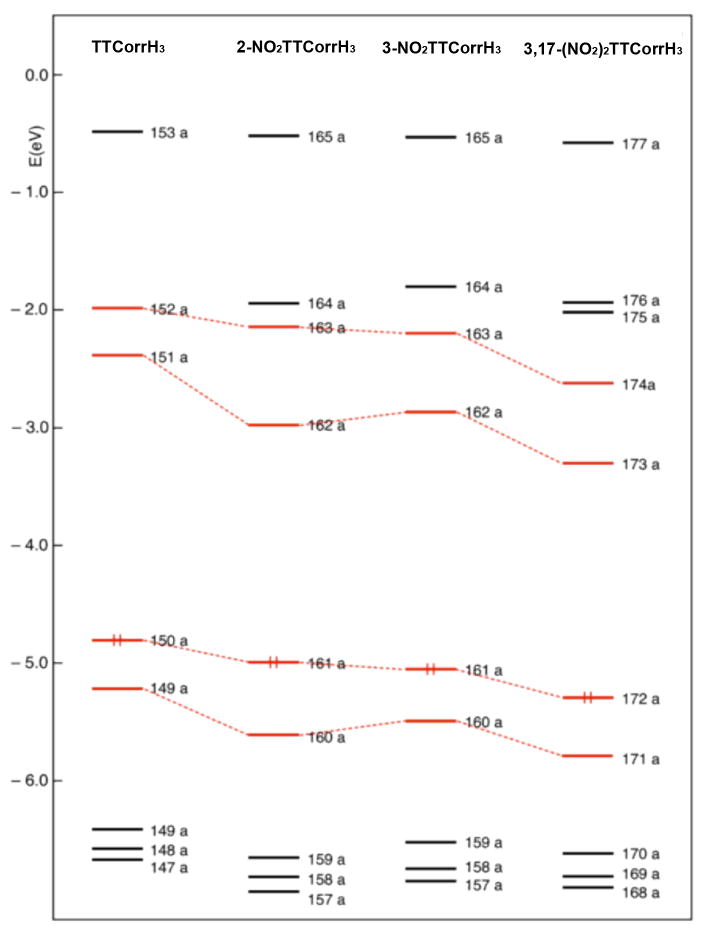
Energy level scheme for TTCorrH3 and its nitro derivatives. The Gouterman-derived MOs are indicated with red lines.
The plots of the two highest occupied and of the two lowest unoccupied MOs of these compounds are displayed in Figure 8. The HOMO–1 and HOMO of TTCorrH3 resemble the HOMO–1(au) and HOMO(b1u) of TPPH2 (D2h symmetry).41 Less pronounced is the similarity between the LUMO and LUMO+1 of TTCorrH3 and the LUMO(b2g) and LUMO+1(b3g) of TPPH2 (D2h symmetry).41 In the nitro derivatives and in the parent TTCorrH3, a quite large energy gap separates the HOMO-1 from the lower lying MOs, which are largely localized on the aryl groups and on the pyrrolenine ring. In the nitro derivatives one or two MOs having some amplitude on the nitro groups interpose between the LUMO+1 and the high-lying set of π-phenyl orbitals. According to the level scheme of Figure 7, the outstanding effect of introducing nitro groups into TTCorrH3 is the stabilization of the LUMO and, to a lesser extent, of the HOMO–1. As a result, the LUMO/LUMO+1 splitting increases dramatically on going from TTCorrH3 to the nitro derivatives. The large stabilization of the LUMO moving from TTCorrH3 to the nitro derivatives is due to the C–N(nitro) π-bonding character of this orbital well visible in the plots displayed in Figure 8. In 3-NO2TTCorrH3 the LUMO is slightly less stabilized than in 2-NO2TTCorrH3 because the nitro group is rotated by 31.4° with the respect to the pertinent pyrrole plane and hence the overlap between the C3 and N5 2pz orbitals is less effective than in 2-NO2TTCorrH3 where the nitro group is almost coplanar with the pertinent pyrrole. In the dinitro derivative both the LUMO and LUMO+1 experience a significant downward shift as compared to the parent TTCorrH3. This is because both these MOs have C–N(nitro) π-bonding character. Due to the presence of two C–N(nitro) π-bonding interactions, the stabilization of the LUMO and LUMO+1 is quite large in 3,17-(NO2)2-TTCorrH3. However, it should be noted that, for overlap reasons, the C–N(nitro) π-bonding interaction involving the nitro group on the 3 position is somewhat less effective than that involving the nitro group on the 17 position. As pointed out earlier, the nitro groups on the 3 and 17 positions are rotated with respect to the plane of the pertinent pyrrole by 32.9° and 20.0°, respectively.
Figure 8.
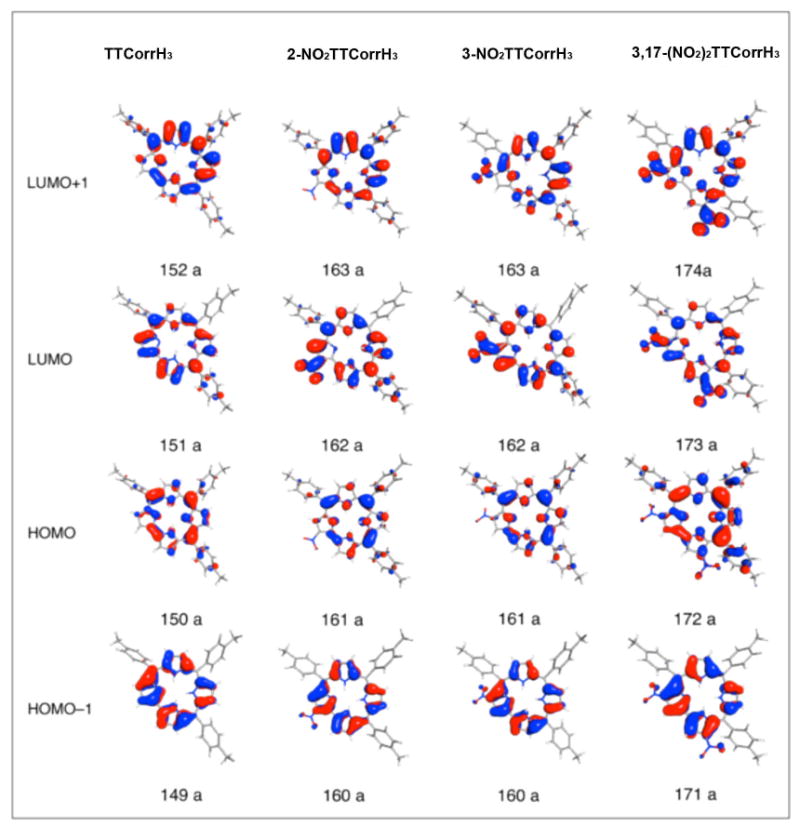
Plots of the frontier orbitals of TTCorrH3 and its nitro derivatives.
In summary, comparison between the one-electron levels of the nitro corroles and those of the parent free-base reveals that the most striking differences are the increased LUMO/LUMO+1 splitting and the diminished HOMO/LUMO energy gap, which are mainly due to the downshift of the LUMO in the nitro derivatives.
We see now how the previously discussed electronic structure changes accompanying the nitration of the corrole macrocycle reflect on the UV-visible spectroscopic properties of the investigated nitrocorroles. The excitation energies and oscillator strengths calculated for the lowest singlet excited states of TTCorrH3 and its nitro derivatives are reported in Tables 4–7 together with the major one-electron transitions contributing to the excited-state solution vectors.
Table 4.
Composition, Vertical Excitation Energies, E (eV/nm), and Oscillator Strengths, f, for the Lowest Optically Allowed Excited States of TTCorrH3 in CH2Cl2
| state | composition (%) | E | f |
|---|---|---|---|
| 11A | 150a→151a (78) 149a→152a (15) |
2.12/585 | 0.2905 |
| 21A | 150a→152a (47) 149a→151a (46) |
2.30/539 | 0.03580 |
| 31A | 149a→151a (48) 150a→152a (47) |
2.96/418 | 1.122 |
| 41A | 149a→152a (80) 150a→151a (14) |
3.05/406 | 1.439 |
Table 7.
Composition, Vertical Excitation Energies, E (eV/nm), and Oscillator Strengths, f, for the Lowest Optically Allowed Excited States of 3,17-(NO2)2TTCorrH3 in CH2Cl2
| state | composition (%) | E | f |
|---|---|---|---|
| 11A | 172a→173a (96) | 1.72/720 | 0.4537 |
| 21A | 171a→173a (59) 172a→174a (40) |
2.04/608 | 0.01098 |
| 31A | 172a→174a (54) 171a→173a (35) |
2.46/503 | 0.6455 |
| 41A | 171a→174a (71) 172a→176a (20) |
2.69/462 | 0.01524 |
| 51A | 170a→173a (68) 172a→175a (24) |
2.81/442 | 0.03678 |
| 61A | 172a→176a (47) 170a→173a (16) 171a→175a (14) |
2.90/427 | 0.1994 |
| 71A | 172a→175a (37) 170a→173a (20) 171a→174a (16) |
2.93/422 | 0.1740 |
In Figure 9, the computed and experimental absorption spectra of these compounds in CH2Cl2 are compared. Considering first TTCorrH3 as point of reference for the nitro derivatives, only two excited states are computed in the energy regime of the four Q bands, the 11A and 21A. As found in TPPH2,42 these states originate from out-of-phase combination of the Gouterman transitions. However, due to the lifting of the LUMO/LUMO+1 degeneracy induced by the appreciable distortion of the macrocycle in TTCorrH3, the mixing coefficients of the Gouterman transitions change significantly relative to TPPH2. As a matter of fact, the 11A excited state in TTCorrH3 is mostly described by the HOMO→LUMO transition, the transition out of the HOMO–1 into the LUMO+1 entering in this state with only a minor (15%) weight. The 21A is instead a nearly 50:50 mixture of the HOMO→LUMO+1 and HOMO–1→LUMO transitions. Due to a less effective mixing of the contributing transitions and, hence, to a less effective cancellation of their large dipole moments, the oscillator strength computed for the 11A state is significantly larger than that of the 21A. By analogy with the assignment previously proposed for TPPH2,42 the Q bands observed in the CH2Cl2 absorption spectrum of TTCorrH3 at 652 nm and 574 nm are assigned to the 11A and 21A states, respectively, although the computed absorption wavelengths are somewhat blue-shifted with respect to the experimental values. According to the TDDFT calculations, the Q bands at 616 nm and 465 nm are identified as vibrational in origin. The B band profile of TTCorrH3 is nicely accounted for by the nearly degenerate 31A and 41A excited states. These originate from in-phase combinations of the Gouterman type transitions and, hence, have large oscillator strength (see Table 4).
Figure 9.

Comparison between computed (TDDFT/B3LYP) and experimental absorption spectra of TTCorrH3 and its nitro derivatives in CH2Cl2.
Coming to the nitrocorroles, in the energy regime of the Q bands, TDDFT calculations predict, just as for the parent TTCorrH3, only two excited states, the 11A and 21A. As can be inferred from the composition of these states in Tables 4–7 for all three nitro derivatives, the 11A is a nearly pure (>90%) HOMO→LUMO state and the 21A is a mixture of the HOMO→LUMO+1 and HOMO–1→LUMO transitions. The lowest excited state nicely accounts for the energy and intensity of the longest wavelength Q band. The 21A excited state is responsible for the higher energy Q band appearing as a distinct peak at 588 nm in the UV-visible spectrum of 2-NO2TTCorrH3 and as a broad feature at around 600 nm in the spectra of the other nitro derivatives. The calculations also account for the observed red shift of this band on going from TTCorrH3 to the nitro derivatives. Distinct from TTCorrH3, the nitro corroles exhibit a complex B band system characterized by several overlapping features. The increased complexity of the B band system in nitrocorroles is related to the involvement of non-Gouterman transitions in the excited states accounting for this spectral region. The calculations indicate that the Gouterman-type transitions, which account for the nearly degenerate B bands of TTCorrH3, mix to a variable extent with transitions out of the HOMO–2 (a MO largely localized on the aryl groups and on the pyrrolenine ring) into the LUMO, or with transitions out of the HOMO into the LUMO+2 (a MO with sizable amplitude on the nitro groups). As for 2-NO2TTCorrH3, the two prominent B bands are well accounted for by the 41A and 51A excited states computed at 449 and 392 nm and with oscillator strength of 0.8068 and 0.7931, respectively. According to their composition (see Table 5), these states are largely composed of the Gouterman-type transitions with the abovementioned non-Gouterman transitions entering with a minor, although not negligible, weight. Four intense excited states are computed in the energy regime of the three B band system of 3-NO2TTCorrH3. On the basis of their energy, the 31A and 41A are assigned to the lowest energy features at 465 and 438 nm, respectively, and the nearly degenerate 51A and 61A states to the most prominent feature at 397 nm.
Table 5.
Composition, Vertical Excitation Energies, E (eV/nm), and Oscillator Strengths, f, for the Lowest Optically Allowed Excited States of 2-NO2TTCorrH3 in CH2Cl2
| state | composition (%) | E | f |
|---|---|---|---|
| 11A | 161a→162a (94) | 1.74/711 | 0.2655 |
| 21A | 160a→162a (72) 161a→163a (25) |
2.21/560 | 0.1042 |
| 31A | 161a→164a (82) 160a→163a (16) |
2.61/474 | 0.07460 |
| 41A | 161a→163a (59) 160a→162a (20) 160a→164a (16) |
2.76/449 | 0.8068 |
| 51A | 160a→163a (49) 159a→162a (31) |
3.16/392 | 0.7931 |
The B band system of the dinitrocorrole is characterized by an intense, sharp feature at 483 nm followed to the blue by a broad, less intense feature. The energy and oscillator strength computed for the 31A excited state leaves no doubt about the assignment of the longer wavelength B band to this state. It is noteworthy that the 31A state consists of the same Gouterman transitions as the 21A, HOMO–1→LUMO and HOMO→LUMO+1, with approximately reversed weights. The large transition dipoles of these transitions reinforce in the 31A and cancel in the 21A state with the result that the former is by far more intense than the latter.
In the energy regime of the broad B feature, the calculations predict four excited states, two of which, the nearly degenerate 61A and 71A, account for most of the intensity of this band.
(c) Reduction Potentials
The redox behavior of metal-free corroles is rather complex owing to the proton transfer equilibrium reaction CorrH3 ⇌ [CorrH2]–. As seen in the previous section, the first reduction potential of the three-protonated species, CorrH3, positively shifts when one or two nitro groups are introduced at the β-position of the macrocycle, with the addition of a second nitro group to the macrocycle decreasing the effect of nitration on the redox potential under the same experimental conditions. To rationalize the observed trend, we have computed the first reduction potential of 2-NO2TTCorrH3, 3-NO2TTCorrH3, 3,17-(NO2)2TTCorrH3 and the parent TTCorrH3 in dichloromethane solution, using a DFT/COSMO model.
The standard reduction potential, , for the reaction
| (1) |
is related to the standard free energy change relative to the standard hydrogen electrode (SHE) through the equation
| (2) |
In eq 2, n is the number of electrons consumed in the reduction reaction, 1 in the present case, and F is a constant. If the free energies are expressed in molar units, F is the Faraday constant (the negative of the charge on one mole of electrons), but if energies are expressed in eV per molecule, as in the present paper, then F is equal to the unit charge e–. is the free energy change associated with reaction (1), whereas is the free energy change of the following reaction
| (3) |
has been established in literature to be –4.28 eV.43
As the experimental half-wave potentials of the investigated metal-free corroles were measured using SCE as the reference electrode, the theoretical reduction potentials obtained from eq 2 were converted to SCE referenced potentials by subtracting 0.09 V, which was obtained by taking the literature value of the reduction potential of aqueous SCE relative to SHE at 298 K (0.24 V)44 and correcting it to its corresponding value in dichloromethane solution using the literature value of the liquid junction potential (ljp) for dichloromethane-water (0.15 V).45 The corrected one-electron reduction potentials of TTCorrH3, 2-NO2TTCorrH3, 3-NO2TTCorrH3, and 3,17-(NO2)2TTCorrH3 are compared with the experimental half-wave potentials in Table 8 where the thermodynamic parameters computed for the one-electron reduction process in eq 1 are also reported. The data in Table 8 indicate that there is an excellent agreement between the theoretically predicted one-electron reduction potentials and the available experimental values. The thermodynamic data reveal that the one-electron reduction process is energetically controlled by enthalpic factors, the entropic factors playing a negligible role. The ΔEsolv values indicate that the energetic contribution of solvation to the enthalpic term, though relevant, is substantially constant along the series. Thus, the trend is primarily determined by the electronic enthalpy that favors energetically the reduced form over the oxidized one in the order TTCorrH3 ≪ 2-NO2TTCorrH3 ≅ 3-NO2TTCorrH3 < 3,17-(NO2)2TTCorrH3. In nitrocorroles, the one-electron reduced form is much more stable than the oxidized form as compared to TTCorrH3 and this is consistent with the added electron entering a MO with C–N(nitro) π-bonding character in the former, as witnessed by the significant shortening of the C–N(nitro) bond. In 2-NO2TTCorrH3 and 3-NO2TTCorrH3 the C–N(nitro) bond shortens by 0.027 Å and 0.033 Å, respectively. In the dinitro derivative, the two C–N(nitro) bonds shorten, on the average, by 0.025 Å.
Table 8.
Thermodynamic Parameters (eV) for the One-electron Reduction Reaction 1, and One-electron Reduction Potentials (V) for TTCorrH3, 2-NO2TTCorrH3, 3-NO2TTCorrH3, and 3,17-(NO2)2TTCorrH3.
|
|
|
ΔEsolva |
|
vs SCE | E1/2bvs SCE | ||||
|---|---|---|---|---|---|---|---|---|---|
| TTCorrH3 | –2.93 | 0.02 | –1.41 | –2.91 | –1.46 | –1.40 | |||
| 2-(NO2)TTCorrH3 | –3.49 | 0.03 | –1.50 | –3.52 | –0.85 | – | |||
| 3-(NO2)TTCorrH3 | –3.44 | 0.03 | –1.47 | –3.47 | –0.90 | –0.84 | |||
| 3,17-(NO2)2TTCorrH3 | –3.78 | 0.04 | –1.31 | –3.82 | –0.55 | –0.62 |
is the difference between the dielectric energy of the reduced species and the dielectric energy of the oxidized species.
Experimental values in dichloromethane solution, this work.
Conclusions
We have reported the first example of efficient insertion of nitro groups into the β-pyrrole positions of corrole free bases, effectively overcoming the formation of isocorrole by controlling the amount and the nature of the oxidant used for the reaction. The regioselectivity usually observed in the functionalization of corrole ring is also demonstrated for the nitration reaction, producing the 3-nitro- and the 3,17-dinitro-corroles as the main reaction products of mono- and di-substitution; we have also been able to characterize some other minor regioisomers, such as the 2-nitro, 2,3- and the 3,12-dinitro derivatives. It is interesting to note that this last regioisomer is the first example of antipodal functionalization of corrole ring.
The introduction of nitro substituents at the β-pyrrole positions of the corrole ring strongly influence the chemical and spectroscopic behavior of the macrocycle. The strong electron-withdrawing character of the nitro group leads to a positive shift of the E1/2 of the redox processes of corrole and to an increase of the macrocycle acidity. It is noteworthy that the introduction of the first nitro group is more effective than the second substituent in changing the properties of the functionalized macrocycle. Optical absorption spectra of β-nitrocorroles are strongly influenced by the peripheral nitro groups, which increase the number of bands, characterized also by a significant red shift.
The theoretical results on these β-nitrocorrole derivatives also afforded significant information, closely matching the experimental observations. It was found that the β-NO2 substituents conjugate with the π-aromatic system of the macrocycle, which initiates significant changes in both the spectroscopic and redox properties of the so functionalized corroles. This effect is more pronounced when the nitro group is introduced at the 2-position, because in this case the conjugation is, for steric reasons, more efficient than in the 3-nitro isomer.
In closing, these nitro-functionalized corroles are also useful starting materials for further modifications of the corrole ring and their availability can open new opportunities for the preparation of functionalized corroles; these studies are now in progress in our laboratories.
Supplementary Material
Table 6.
Composition, Vertical Excitation Energies, E (eV/nm), and Oscillator Strengths, f, for the Lowest Optically Allowed Excited States of 3-NO2TTCorrH3 in CH2Cl2
| state | composition (%) | E | f |
|---|---|---|---|
| 11A | 161a→162a (91) | 1.86/667 | 0.3338 |
| 21A | 160a→162a (69) 161a→163a (27) |
2.19/565 | 0.1304 |
| 31A | 161a→164a (38) 161a→163a (37) 160a→162a (14) |
2.65/468 | 0.3584 |
| 41A | 160a→163a (32) 161a→164a (27) 161a→163a (23) |
2.73/455 | 0.3085 |
| 51A | 159a→162a (54) 161a→164a (17) 160a→163a (14) |
3.07/403 | 0.4981 |
| 61A | 159a→162a (36) 160a→163a (33) 161a→164a (14) |
3.13/396 | 0.5669 |
Synopsis Toc.
The regioselective synthesis of β-nitro substituted corrole free base has been investigated, allowing the preparation of the mono- and bis-derivatives in good yields, by the careful choice of the AgNO2/NaNO2 reagent ratio. The influence of the β-nitro substituents on the corrole properties is studied in detail by UV-visible, electrochemical, and spectroelectrochemical characterization of these functionalized corroles.
Acknowledgments
We gratefully acknowledge the support of MIUR, Italy (PRIN project 2009Z9ASCA), the US National Institutes of Health (K.M.S., grant CA132861), and the Robert A. Welch Foundation (K.M.K., grant E-680).
Footnotes
Supporting Information Available: CIF files of 2-NO2TTCorrH3, 2,3-(NO2)2TTCorrCoPPh3 and 3,12-(NO2)2TTCorrCoPPh3. This material is available free of charge via the Internet at http://pubs.acs.org
Contributor Information
Kevin M. Smith, Email: kmsmith@lsu.edu.
Karl M. Kadish, Email: kkadish@uh.edu.
Angela Rosa, Email: angela.rosa@unibas.i.
Roberto Paolesse, Email: roberto.paolesse@uniroma2.it.
References
- 1.Nardis S, Monti D, Paolesse R. Mini-Rev Org Chem. 2005;2:355. [Google Scholar]
- 2.Paolesse R. Synlett. 2008:2215. [Google Scholar]
- 3.Paolesse R. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 2. Academic Press; New York: 2000. pp. 201–232. [Google Scholar]
- 4.Gryko DT. J Porphyrins Phthalocyanines. 2008;12:906. [Google Scholar]
- 5.Gross Z, Aviv-Harel I. Coordination Chemistry Reviews. 2011;255:717–736. [Google Scholar]
- 6.(a) Gross Z, Aviv-Harel I. Chem Eur J. 2009;15:8382–8394. doi: 10.1002/chem.200900920. [DOI] [PubMed] [Google Scholar]; (b) Kupershmidt L, Okun Z, Amit T, Mandel S, Saltsman I, Mahammed A, Bar-Am O, Gross Z, Youdim MBH. J Neurochem. 2010;113:363–373. doi: 10.1111/j.1471-4159.2010.06619.x. [DOI] [PubMed] [Google Scholar]; (c) Kanamori A, Catrinescu MM, Mahammed A, Gross Z, Levin LA. J Neurochem. 2010;114:488–498. doi: 10.1111/j.1471-4159.2010.06781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lemon CM, Brothers PJ. J Porphyrins Phthalocyanines. 2011;15:809–834. [Google Scholar]
- 8.Walker FA, Licoccia S, Paolesse R. J Inorg Biochem. 2006;100:810. doi: 10.1016/j.jinorgbio.2006.01.038. and references therein. [DOI] [PubMed] [Google Scholar]
- 9.Stefanelli M, Mandoj F, Mastroianni M, Nardis S, Mohite P, Fronczek FR, Smith KM, Kadish KM, Xiao X, Ou Z, Chen P, Paolesse R. Inorg Chem. 2011;50:8281–8292. doi: 10.1021/ic2008073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mastroianni M, Zhu W, Stefanelli M, Nardis S, Fronczek FR, Smith KM, Ou Z, Kadish KM, Paolesse R. Inorg Chem. 2008;47:11680–11687. doi: 10.1021/ic801421a. [DOI] [PubMed] [Google Scholar]
- 11.Saltsman I, Mahammed A, Goldberg I, Tkachenko E, Botoshansky M, Gross Z. J Am Chem Soc. 2002;124:7411–7420. doi: 10.1021/ja025851g. [DOI] [PubMed] [Google Scholar]
- 12.Stefanelli M, Mastroianni M, Nardis S, Licoccia S, Fronczek FR, Smith KM, Zhu W, Ou Z, Kadish KM, Paolesse R. Inorg Chem. 2007;46:10791–10799. doi: 10.1021/ic7014572. [DOI] [PubMed] [Google Scholar]
- 13.Pomarico G, Fronczek FR, Nardis S, Smith KM, Paolesse R. J Porphyrins Phthalocyanines. 2011;15:1086–1092. doi: 10.1142/S1088424611004038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stefanelli M, Nardis S, Tortora L, Fronczek FR, Smith KM, Licoccia S, Paolesse R. Chem Commun. 2011;47:4255–4257. doi: 10.1039/c0cc05491g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stefanelli M, Shen J, Zhu W, Mastroianni M, Mandoj F, Nardis S, Ou Z, Kadish KM, Fronczek FR, Smith KM, Paolesse R. Inorg Chem. 2009;48:6879–6887. doi: 10.1021/ic900859a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Mandoj F, Nardis S, Pomarico G, Paolesse R. J Porphyrins Phthalocyanines. 2008;12:19–26. doi: 10.1142/S1088424610002513. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ngo TH, Van Rossom W, Dehaen W, Maes W. Org Biomol Chem. 2009;7:439–443. doi: 10.1039/b819185a. [DOI] [PubMed] [Google Scholar]; (c) Capar C, Thomas KE, Ghosh A. J Porphyrins Phthalocyanines. 2008;12:964–967. [Google Scholar]
- 17.Sheldrick GM. Acta Crystallogr, Sect A. 2008;64:112. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 18.APEX2, SAINT and SADABS. Bruker AXS Inc.; Madison, Wisconsin, USA: 2007. [Google Scholar]
- 19.Flack HD. Acta Cryst. 1983;A39:876–881. [Google Scholar]
- 20.Spek AL. Acta Crystallogr Sect D-Biol Crystallogr. 2009;65:148. doi: 10.1107/S090744490804362X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin XQ, Kadish KM. Anal Chem. 1985;57:1498–1501. doi: 10.1021/ac00284a079. [DOI] [PubMed] [Google Scholar]
- 22.(a) TURBOMOLE V6.3 2011, a development of University of Karlsruhe and Forschungzentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007. available from http://www.turbomole.com.; (b) Ahlrichs R, Bär M, Häser M, Horn H, Kölmel C. Chem Phys Lett. 1989;162:165. [Google Scholar]
- 23.(a) Becke A. Phys Rev A. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]; (b) Perdew JP. Phys Rev B. 1986;33:8822. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 24.(a) Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]; (b) Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 25.Weigend F, Ahlrichs R. Phys Chem Chem Phys. 2005;7:3297. doi: 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- 26.Eichkorn K, Treutler Chem Phys Lett. 1995;240:283. [Google Scholar]
- 27.Weigend F. Phys Chem Chem Phys. 2006;8:1057–1065. doi: 10.1039/b515623h. [DOI] [PubMed] [Google Scholar]
- 28.(a) Klamt A, Schürmann G. J Chem Soc Perkin Trans. 1993;2:799. [Google Scholar]; (b) Klamt A, Jonas V. J Chem Phys. 1996;105:9972. [Google Scholar]
- 29.(a) Gross EUK, Dobson JF, Petersilka M. In: Density Functional Theory, Springer Series “Topics in Current Chemistry”. Nalewajski RF, editor. Springer; Heidelberg: 1996. [Google Scholar]; (b) Casida ME. In: Recent Advances in Density Functional Methods. Chong DP, editor. Vol. 1. World Scientific; Singapore: 1995. p. 155. [Google Scholar]; (c) Parr RG, Yang W. Density Functional Theory of Atoms and Molecules. Oxford University Press; New York: 1989. [Google Scholar]; (d) Drew A, Head-Gordon M. Chem Rev. 2005;105:4009. doi: 10.1021/cr0505627. [DOI] [PubMed] [Google Scholar]
- 30.(a) Klamt A. J Phys Chem. 1996;100:3349–3353. [Google Scholar]; (b) Scalmani G, Frisch MJ, Mennucci B, Tomasi J, Cammi R, Barone V. J Chem Phys. 2006;124:094107–094121. doi: 10.1063/1.2173258. [DOI] [PubMed] [Google Scholar]
- 31.Tortora L, Nardis S, Fronczek FR, Smith KM, Paolesse R. Chem Commun. 2011;47:4243–4245. doi: 10.1039/c0cc05837h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nardis S, Pomarico G, Fronczek FR, Vicente MGH, Paolesse R. Tetrahedron Lett. 2007;48:8643–8646. [Google Scholar]
- 33.Nardis S, Pomarico G, Mandoj F, Fronczek FR, Smith KM, Paolesse R. J Porphyrins Phthalocyanines. 2010;14:753–757. doi: 10.1142/S1088424610002513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Setsune J, Tsukajima A, Watanabe J. Tetrahedron Lett. 2006;47:1817–1820. [Google Scholar]
- 35.Flint DL, Fowler RL, LeSaulnier TD, Long AC, O'Brien AY, Geier GR., III J Org Chem. 2010;75:553–563. doi: 10.1021/jo902452c. [DOI] [PubMed] [Google Scholar]
- 36.(a) Ou Z, Shen J, Shao J, E W, Galezowski M, Gryko DT, Kadish KM. Inorg Chem. 2007;46:2775–2786. doi: 10.1021/ic0617893. [DOI] [PubMed] [Google Scholar]; (b) Shen J, Shao J, Ou Z, E W, Koszarna B, Gryko DT, Kadish KM. Inorg Chem. 2006;45:2251–2265. doi: 10.1021/ic051729h. [DOI] [PubMed] [Google Scholar]
- 37.Nardis S, Stefanelli M, Mohite P, Pomarico G, Tortora L, Manowong M, Chen P, Kadish KM, Fronczek FR, McCandless GT, Smith KM, Paolesse R. Inorg Chem. 2012;51:3910–3920. doi: 10.1021/ic3002459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.(a) Kadish KM, Chen P, Enakieva YY, Nefedov SE, Gorbunova YG, Tsivadze AY, Lemeune A, Stern C, Guilard R. J Electroanal Chem. 2011;656:61–71. [Google Scholar]; (b) Inisan C, Saillard JY, Guilard R, Tabardc A, Mest YL. New J Chem. 1998:823–830. [Google Scholar]
- 39.(a) Gouterman M, Rentzepis PM, Straub KD, editors. Porphyrins: excited states and dynamics. Vol. 321 American Chemical Society; New York: 1987. [Google Scholar]; (b) Storm CB, Teklu Y. J Am Chem Soc. 1972;94:1745–1747. doi: 10.1021/ja00760a056. [DOI] [PubMed] [Google Scholar]; (c) Ding T, Harvey JD, Ziegler CJ. J Porphyrins Phthalocyanines. 2005;9:22–27. [Google Scholar]
- 40.(a) Gross Z, Galili N, Simkhovich L, Saltsman I, Botoshansky M, Bläser D, Boese R, Goldberg I. Organic Letters. 1999;1:599. [Google Scholar]; (b) Simkhovich L, Goldberg I, Gross Z. J Inorg Biochem. 2000;80:235–238. doi: 10.1016/s0162-0134(00)00077-5. [DOI] [PubMed] [Google Scholar]; (c) Paolesse R, Nardis S, Venanzi M, Mastroianni M, Russo M, Fronczek FR, Vicente MGH. Chem Eur J. 2003;9:1192–1197. doi: 10.1002/chem.200390136. [DOI] [PubMed] [Google Scholar]
- 41.Palummo M, Hogan C, Sottile F, Bagala P, Rubio A. J Chem Phys. 2009;131:084102–084107. doi: 10.1063/1.3204938. [DOI] [PubMed] [Google Scholar]
- 42.De Luca G, Romeo A, Monsù Scolaro L, Ricciardi G, Rosa A. Inorg Chem. 2009;48:8493. doi: 10.1021/ic9012153. [DOI] [PubMed] [Google Scholar]
- 43.Kelly CP, Cramer CJ, Truhlar DG. J Phys Chem B. 2006;110:16066–16081. doi: 10.1021/jp063552y. [DOI] [PubMed] [Google Scholar]
- 44.Bard AJ, Faulkner LR. Electrochemical Methods: Fundamentals and Applications. Wiley; New York: 2001. [Google Scholar]
- 45.(a) Krishtalik LI, Alpatova NM, Ovsyannikova EV. J Electroanal Chem. 1992;329:1–8. [Google Scholar]; (b) Tsierkezos N. J Sol Chem. 2008;37:1437–1448. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.