

Abstract
Utilization of carbohydrate in the form of intramuscular glycogen stores and glucose delivered from plasma becomes an increasingly important energy substrate to the working muscle with increasing exercise intensity. This review gives an update on the molecular signals by which glucose transport is increased in the contracting muscle followed by a discussion of glycogen mobilization and synthesis by the action of glycogen phosphorylase and glycogen synthase, respectively. Finally, this review deals with the signalling relaying the well-described increased sensitivity of glucose transport to insulin in the post-exercise period which can result in an overshoot of intramuscular glycogen resynthesis post exercise (glycogen supercompensation).
Professor MD, DMSci, Erik A. Richter (E.A.R., left) and post doc Thomas E. Jensen (T.E.J., right) work in the Department of Exercise and Sport Sciences at the University of Copenhagen, investigating the signalling mechanisms that regulate skeletal muscle substrate metabolism in health and disease. E.A.R. has for three decades contributed exercise-metabolism research, receiving numerous awards and distinctions. T.E.J. did a PhD with E.A.R. in Copenhagen followed by post-doctoral training with Amira Klip in Toronto. His main research interest is regulation of glucose transport by contraction and insulin.
 |
Introduction
Carbohydrate in the form of glucose and intramuscular glycogen becomes an increasingly important energy substrate with rising exercise intensity (Holloszy & Kohrt, 1996). Carbohydrate oxidation accounts for 10–15% of total energy production during low intensity aerobic exercise (∼30%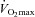 ), increasing progressively to roughly 70–80% of total energy during exercise of about 85%
), increasing progressively to roughly 70–80% of total energy during exercise of about 85%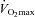 to about 100% of energy consumption at exercise intensities of 100% of
to about 100% of energy consumption at exercise intensities of 100% of 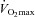 and above (Romijn et al. 1993; Holloszy & Kohrt, 1996). There are two sources of glucose molecules available to the working muscle; plasma glucose and muscle glycogen. While very little net glycogen breakdown is observed at low-intensity exercise, glycogen-breakdown becomes the predominant glucose source at higher intensities (Hargreaves & Richter, 1988). In terms of athletic performance, low muscle glycogen depots seem detrimental to both high and moderate intensity exercise performance (Hargreaves & Richter, 1988). This has resulted in the widespread practice of high-carbohydrate diet regimens to increase pre-exercise glycogen levels (carbohydrate loading) (Hargreaves & Richter, 1988). In this review we discuss the current thinking on the molecular signals that acutely control glucose uptake and glycogen use by the working muscle. Then we discuss the mechanisms by which skeletal muscle may accomplish an increase in glycogen stores above pre-exercise levels, focusing on the mechanisms enhancing insulin-stimulated glucose uptake post-exercise.
and above (Romijn et al. 1993; Holloszy & Kohrt, 1996). There are two sources of glucose molecules available to the working muscle; plasma glucose and muscle glycogen. While very little net glycogen breakdown is observed at low-intensity exercise, glycogen-breakdown becomes the predominant glucose source at higher intensities (Hargreaves & Richter, 1988). In terms of athletic performance, low muscle glycogen depots seem detrimental to both high and moderate intensity exercise performance (Hargreaves & Richter, 1988). This has resulted in the widespread practice of high-carbohydrate diet regimens to increase pre-exercise glycogen levels (carbohydrate loading) (Hargreaves & Richter, 1988). In this review we discuss the current thinking on the molecular signals that acutely control glucose uptake and glycogen use by the working muscle. Then we discuss the mechanisms by which skeletal muscle may accomplish an increase in glycogen stores above pre-exercise levels, focusing on the mechanisms enhancing insulin-stimulated glucose uptake post-exercise.
Glucose metabolism during exercise-regulation of glucose transport
Glucose delivery to the working muscle is increased by a marked increase in capillary perfusion during exercise as originally described by August Krogh in frog muscle and recently confirmed by real time contrast enhanced ultrasound in humans (Sjoberg et al. 2011). Another way to increase delivery is to increase plasma glucose concentrations by ingestion of carbohydrate rich meals or drinks. The magnitude of increase depends on the type and quantity of carbohydrates and the reader is referred to other reviews for discussion of how to optimize carbohydrate availability during exercise (Hawley et al. 2011). At the fibre level, it is still debated whether the rate-limiting step in vivo is GLUT4-dependent transport across the plasma membrane or intracellular phosphorylation by hexokinase II. However, increased recruitment of GLUT4 from intracellular vesicular structures to the cell surface during acute muscle contraction/exercise is a well-described acute adaptation in both rodents and humans (for refs see Jessen & Goodyear, 2005; Rose & Richter, 2005) and a necessary contributor to increased skeletal muscle glucose uptake in exercising muscle since in mouse muscles where GLUT4 has been genetically ablated, contraction-induced glucose uptake is abrogated (Zisman et al. 2000). In addition, a contribution from an increase in GLUT4 intrinsic activity, which is clearly dissociable from GLUT4 translocation in some studies, cannot be discounted (Klip, 2009) although effects of exercise on GLUT4 intrinsic activity have not been rigorously demonstrated.
Overall, the GLUT4-translocation response to contraction has been proposed to involve feed-forward activation by sarcoplasmic reticulum (SR) Ca2+ release with subsequent fine-tuning by changes secondary to contraction (e.g. mechanical stretch, metabolism, redox-state). The feed forward proposition is supported by ex vivo rat muscle studies where caffeine-stimulated Ca2+ release from the SR was sufficient to elicit an increase in glucose transport in the absence of measurable increases in force development, nucleotide-status or activation of the AMP/ATP and ADP/ATP-sensitive AMP-activated protein kinase (AMPK) (Wright et al. 2004). However, whereas the original studies did not find changes in energy status or AMPK activation by subcontraction threshold Ca2+ release, more recent studies have reported nucleotide-changes and AMPK activation using similar Ca2+ concentrations (Jensen et al. 2007; Raney & Turcotte, 2008; Egawa et al. 2009), questioning the usefulness of the caffeine-approach to isolate the Ca2+ response independently of energy turnover and other contraction-activated events. Furthermore, an old observation is that the glucose uptake response correlates excellently with the intensity of muscular work during both human exercise and in more reductionistic rodent muscle contraction models (Rose & Richter, 2005). Of particular interest, Ihlemann and coworkers, by adjusting the length of ex vivo stimulated rat muscles and as a consequence force production and metabolic stress, demonstrated that the glucose transport response correlates with the degree of tension development rather than stimulation frequency (Ihlemann et al. 2000). These studies were recently followed up by another approach where pharmacological inhibition of fast-twitch myosin II-dependent crossbridge cycling partially reduced electrically stimulated glucose transport in rat epitrochlearis muscle (Blair et al. 2009). Using a lower intensity tetanic stimulation protocol to minimize energy turnover by e.g. SERCA-dependent Ca2+ reuptake, we have data showing that the increase in glucose transport by electrical stimulation of mouse muscles ex vivo is fully prevented by myosin II inhibition despite normal Ca2+ activated phosphorylation events (T. E. Jensen, E. A. Richter, unpublished data). This suggests that, while some Ca2+ activated proteins provide necessary signals for contraction-stimulated glucose transport (Rose & Richter, 2005), Ca2+per se is probably not sufficient to increase muscle glucose transport.
Based on experiments using the AMP-mimetic aminoimidazole carboxamide ribonucleotide (AICAR), activation of AMPK appears sufficient to cause a partial increase in glucose transport in rodent fast-twitch muscle. In contrast, this response is lower in the mixed type I and II fibre mouse soleus and often absent in the type I fibre-dominated rat soleus despite activation of AMPK (Jørgensen et al. 2004; Wright et al. 2005). This does not seem to relate to differential expression of potential downstream mediators of GLUT4 translocation such as TBC1D1 and TBC1D4/AS160 in the rat (Castorena et al. 2011) but may relate to differential expression of AMPK β and γ subunits in different rodent muscles (Treebak et al. 2009). In humans, despite a lack of measurable changes in total AMPK phosphorylation early on during intense exercise, the α2β2γ3 containing subset of AMPK complexes are rapidly activated with exercise consistent with a role in promoting glucose transport (Birk & Wojtaszewski, 2006). A necessary role of AMPK for contraction-stimulated glucose transport is more controversial, with some studies reporting decreased glucose transport in AMPK deficient mouse models and others not, probably due to redundancy of signalling, differential contraction protocols, and transgenic manipulation strategies (Rose & Richter, 2005). Recently, conditional muscle-specific knockout of both β-AMPK regulatory subunits abolished AMPK activity and potently inhibited exercise-stimulated glucose uptake in vivo and contraction-stimulated glucose transport ex vivo (O'Neill et al. 2011). In parallel to AMPK, proposed to act through TBC1D1/4 (Cartee & Wojtaszewski, 2007; Cartee & Funai, 2009) and eNOS (Lee-Young et al. 2009), a number of other pathways have been proposed to signal to increase glucose transport and may include LKB1 signalling through the AMPK-related kinase SNARK (Koh et al. 2010), and stretch-activated p38 MAPK (Chambers et al. 2009). In relation to the latter, it is, however, worth mentioning that at least one of the p38 MAPK inhibitors used by Chambers and co-workers, SB203580, has been shown interact with and inhibit GLUT4 directly (Antonescu et al. 2005; Ribe et al. 2005). Teasing out how AMPK and other signals blend to elicit a given level of increase in glucose transport in different muscle fibre types during exercise remains a challenging subject for future study.
Regulation of glycogen breakdown
Glycogenolysis is regulated by glycogen phosphorylase (GP), acting on the terminal α-1,4-glycosidic linked glucose residues, and debranching enzyme, targeting the α-1,6-branchpoints in the glycogen molecule (Roach, 2002). Most studies have focused on the regulation of GP, the activity of which is increased by allosteric binding of AMP or IMP and competed by ATP or glucose-6-phosphate (G-6-P). In addition, since GP requires inorganic phosphate to produce glucose-1-phosphate from glycogen, inorganic phosphate from ATP and creatine phosphate (CrP)-turnover has been speculated to limit GP activity at the substrate level (for refs see Hargreaves & Richter, 1988). Finally, high initial muscle glycogen concentration clearly augments net glycogen breakdown during contractions likely due to activation of GP by glycogen (Hespel & Richter, 1992). Apart from its allosteric and presumably substrate-level regulation, phosphorylation of GP on Ser14 by phosphorylase kinase (PK) increases the activity of GP measured in vitro. Classically, PK is thought to integrate local and systemic signals to promote glycogen breakdown by being activated initially by Ca2+ binding to the PK δ-subunit (identical to calmodulin), and then plasma adrenaline acting though a β2-adrenergic receptor–adenylate cyclase–PKA cascade (Hargreaves & Richter, 1988). In the absence of adrenaline stimulation, GP activity measured in vitro (reflecting its phosphorylation state) increases rapidly at the onset of contraction and then reverts towards resting activity within a few minutes despite continued contraction and therefore presence of Ca2+ transients (Richter et al. 1982). This is probably a result of dephosphorylation at Ser14 following the initial activation by Ca2+ although, to our knowledge, the mechanism behind this has not been studied in detail. With regards to the adrenergic stimulation of glycogenolysis, it is also worth noting that adrenaline-stimulated glycogen breakdown in incubated rat muscles is potently inhibited by the sodium–potassium pump inhibitor ouabain, suggesting a link to adrenaline-stimulated sodium-potassium pump activity (James et al. 1999). Whether this connection relates to local changes in e.g. nucleotides or K+ is not clear. In humans, the evidence for adrenergic stimulation of glycogenolysis is not clear-cut, with some studies reporting increased glycogen use and GP activation with adrenaline infusion, while others do not (see e.g. Kjaer et al. 2000; Watt et al. 2001 and refs therein). As discussed by Watt and coworkers (2001), this may relate in part to the intensity of exercise, with allosteric regulation of GP playing a larger regulatory role with increasing intensity.
Within a given fibre, glycogen particles have been proposed to be present in at least three distinct subcellular locations, with ∼80% between the myofibrils in close vicinity to the SR and mitochondria and two smaller compartments located within the myofibrils and underneath the sarcolemma contributing ∼10% each (Nielsen et al. 2011; Prats et al. 2011). The detailed roles of these different glycogen compartments to muscle contraction–metabolism remain to be uncovered but the various glycogen pools are differentially depleted and supercompensated by different kinds of exercise and training (for refs and discussion, see Prats et al. 2009; Nielsen et al. 2011). In relation to fatigue, the emptying of intramyofibrillar glycogen correlates somewhat with lower SR Ca2+ release by 4-chloro-m-cresol in vitro (r2 = 0.23) (for ref see Nielsen et al. 2011), suggesting a potential contribution to the unexplained relation between fatigue and low glycogen. It would be interesting to examine if e.g. most of the ∼20% depletion of muscle glycogen with 30 s of all-out bicycle sprint exercise (Birk & Wojtaszewski, 2006) preferentially stems from intramyofibrillar glycogen. The regulation of glucose transport and glycogen turnover in working muscle is summarized in Fig. 1.
Figure 1. Glucose utilization in the working muscle is increased through increased delivery and uptake of plasma glucose and increased glycogenolysis.
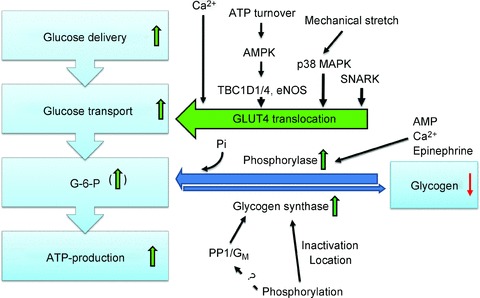
Transport of glucose across the sarcolemma and T-tubular membranes is determined by the amount of contraction- and insulin-responsive glucose transporter 4 (GLUT4) proteins in the outer membrane. This magnitude of glucose transport response with contraction correlates with work intensity with evidence suggesting the involvement of kinases like AMPK, p38 MAPK and SNARK whereas Ca2+ activated proteins are probably required but likely to be insufficient to stimulate glucose transport. Allosteric and covalent regulation increases both glycogen mobilization by glycogen phosphorylase (GP) and resynthesis by glycogen synthase (GS) simultaneously during exercise by altering enzyme activity and/or location. GP may also be regulated by the availability of its substrates glycogen and inorganic phosphate (Pi). Depending on the work intensity and duration, glucose-6-phosphate (G-6-P), an important allosteric inhibitor of GP and stimulator of GS, may increase.
Regulation of glycogen synthesis
Glycogen synthase (GS) catalyses the rate-limiting incorporation of UDP-glucose via α-1,4-glycosidic linkages into the growing glycogen polymer, with branching enzyme catalysing formation of α-1,6-branchpoints (Roach, 2002). Counterintuitively, this UTP-requiring anabolic glycogen synthase is not only stimulated by insulin but also by exercise although unchanged or inhibited GS activity at high intensity has been described (for refs see Nielsen & Wojtaszewski, 2004). In stark contrast to GP regulation, the regulation of GS by post-translational modifications is quite complex, with at least nine phosphorylation sites targeted by multiple kinases (Nielsen & Wojtaszewski, 2004; Jensen & Lai, 2009). The dephosphorylated state of GS, in particular at sites 2, 2a, 3a and 3b, increases GS activity in vitro. This dephoshorylation is catalysed by protein phosphatase 1 (PP1), which is also the phosphatase for GP and PK. At least one glycogen-binding protein, GM, targets PP1 to glycogen and has been shown in mice to be required for exercise-stimulated GS activation (Aschenbach et al. 2001). It is tempting to speculate that the co-localization between PP1–GM and glycogen regulatory enzymes like GS is linked to the well described inverse correlation between in vitro GS activity and glycogen content in muscle (Danforth, 1965; Nielsen et al. 2001). Worth mentioning, PP1 is also known to be regulated by phosphorylation of endogenous inhibitors of PP1 like inhibitor-1 and −2, both of which are expressed in skeletal muscle (for refs see Nicolaou et al. 2009). In addition, GS activity shows a partial resistance to phosphatase treatment in vitro, suggesting that other described covalent modifications of GS such as glycolysation (Parker et al. 2003) may contribute to GS regulation in skeletal muscle. In vivo, allosteric activation by G-6-P is probably an all-important point of regulation. This is evidenced by recent data in mice where muscle-specific replacement of wild-type GS with a G-6-P insensitive mutant GS protein potently reduced insulin and prevents AICAR-stimulated glycogen synthesis (Bouskila et al. 2010; Hunter et al. 2011), suggesting that sensing of G-6-P from transported glucose is required for most of the stimulation of glycogen synthesis. Interestingly, the stimulatory effect of AICAR on glycogen synthesis occurred despite the fact that direct AMPK-dependent phosphorylation of GS at sites 2+2a causes a moderate reduction of GS activity in vitro (Jorgensen et al. 2004), arguing that allosteric regulation can override covalent regulation, at least on sites 2+2a.
In relation to the recently re-emphasized distinct subcellular depots of muscle glycogen, GS seems located to different compartments depending on its phosphorylation state. Hence, GS phosphorylation on site 1b, presumably by adrenaline-activated PKA, is located intramyofibrillarly following roughly 2 h of exhaustive human knee-extension exercise, while GS phosphorylation on the AMPK sites 2+2a is located with subsarcolemmal and intermyofibrillar glycogen depots (Prats et al. 2009). The details of how GS re-localizes to these compartments are not clear but have been suggested to depend on the actin cytoskeleton in as much as GS and GP assemble with β-actin, but not γ-actin, into spherical structures after glycogen-depleting electrical rabbit tibialis anterior muscle stimulation (Prats et al. 2005).
Glycogen resynthesis post-exercise – the role of increased insulin-stimulated glucose uptake
Mechanistically, while increased microvascular recruitment may play a role in insulin sensitization in vivo by increasing glucose delivery (for refs see Wojtaszewski & Richter, 2006), prior contraction also sensitizes glucose transport and GLUT4 translocation ex vivo independently of the capillary network (Fisher et al. 2002; Geiger et al. 2005), suggesting that part of insulin sensitization by prior exercise stems from an effect on GLUT4-mediated glucose transport. It is worth noting, however, that if exercise involves muscle damaging eccentric components, then insulin sensitivity may in fact be decreased in the days after exercise due to decreased GLUT4 expression and impaired insulin signalling (for refs see Maarbjerg et al. 2011).
What might be the molecular mechanisms behind the exercise effect on insulin-stimulated glucose transport when no muscle damage is induced? One study has shown that AMPK activation in incubated rat epitrochlearis by AICAR or hypoxia, in conjunction with one or more unknown serum proteins >10 kDa (Gao et al. 1994), can increase submaximal insulin-stimulated glucose transport ex vivo 3.5 h after removal of the AICAR stimulus (Fisher et al. 2002). Importantly, this occurred without a measurable potentiation of proximal steps of insulin signalling like PI3K activity and Akt phosphorylation, consistent with previous observations in humans (Wojtaszewski & Richter, 2006). This effect could speculatively be relayed by downstream phosphorylation of TBC1D4, an emerging regulator of GLUT4 trafficking, which shows an increase lasting many hours post-exercise at certain residues including known AMPK sites in rats and humans (Sakamoto & Holman, 2008; Maarbjerg et al. 2011). Low muscle glycogen content correlates with high AMPK activity (Jorgensen et al. 2004) and glycogen has been shown to directly bind and inactivate AMPK through the carbohydrate-binding domain of the AMPK β-subunit (McBride et al. 2009). This makes it tempting to speculate about a connection between the release of AMPK from glycogen during exercise and the ensuing increase in insulin sensitivity. Supporting a regulatory role of glycogen is the finding that the increased post-exercise insulin sensitivity correlates significantly with the amount of glycogen broken down during the preceding exercise bout (r2 = 0.53; Richter et al. 2001). However, if a serum-factor is required for contraction to cause insulin sensitization ex vivo (Gao et al. 1994) but not for contraction-stimulated glycogen breakdown, then the relationship between contraction-stimulated glycogen use and insulin sensitivity is probably non-causal.
Both exercise and the protein synthesis inhibitor anisomycin acutely increased p38 MAPK activation in incubated rat soleus and epitrochlearis and increased submaximal insulin-stimulated glucose transport 3 h after cessation of either stimulus (Geiger et al. 2005). The effect of anisomycin was prevented by the selective p38 MAPK inhibitor SB202190, whereas the exercise-effect was not, suggesting that contraction could utilize redundant signalling pathways to increase insulin sensitivity after exercise. Interestingly, resting p38 MAPK phosphorylation 3 h post-exercise has been observed to remain 50% higher in previously exercised leg muscles compared to controls (Thong et al. 2003).
Studies in incubated rat muscles have indicated that any glucose transport-increasing stimulus including insulin itself may enhance insulin-stimulated glucose transport some hours after stimulation, possibly by re-location of GLUT4 to a more easily recruitable pool, thereby allowing a larger GLUT4 mobilization by an unaltered insulin signal (Geiger et al. 2006). Worth mentioning, this attractive hypothesis is not supported by a recent study showing no insulin-sensitizing effect of doing sequential insulin clamps in humans (Lucidi et al. 2010). However, muscle insulin sensitization of prior stimulation may have been overridden by the anti-hypoglycaemic hormonal and lipolytic responses that are activated in the period between the two clamps. A direct examination of the hypothesized altered location of insulin-responsive GLUT4 pools by insulin-sensitizing stimuli in rat and human muscle will be needed to directly test this hypothesis. The effects of prior exercise on insulin-stimulated glucose transport and glycogen resynthesis are recapitulated in Fig. 2.
Figure 2. Augmented glycogen resynthesis post-exercise is explained to a large part by sensitization of the insulin-stimulated glucose transport response and glycogen synthase activation.
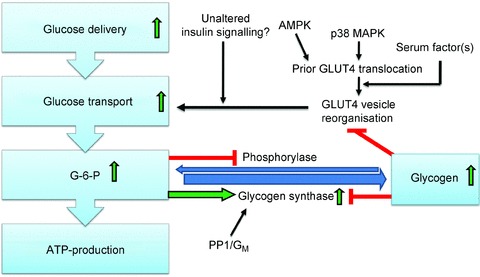
Although some signalling proteins display a prolonged increase in phosphorylation for many hours after exercise, perhaps contributing to insulin sensitization, many insulin-signalling endpoints seem unaffected by prior contraction. Mobilization of GLUT4 during contraction may cause a subsequent sorting into a more insulin-responsive pool or position. Insulin sensitization may require permissive input from one or more unidentified serum factors. Increased amounts of transported glucose are converted to glucose-6-phosphate (G-6-P) which allosterically increases glycogen synthase and inhibits phosphorylase activity, respectively. High glycogen shows a correlation with decreased insulin-stimulated glucose transport and glycogen synthase inactivation but whether this is a causal relationship remains unclear.
Conclusion
Carbohydrates in the form of plasma glucose and muscle glycogen are important fuels during exercise. The increase in muscle glucose uptake during exercise is dependent upon the delivery of glucose (capillary perfusion and plasma glucose concentration) and the permeability of the muscle membrane to glucose. The latter is regulated by a plethora of molecular signalling thought to include calcium, stretch and energy stress signalling and probably others. Muscle glycogen is utilized as a function of exercise intensity and duration and is controlled by the activity of the enzyme glycogen phosphorylase as well as the concentration of both of its substrates (glycogen and inorganic phosphate). In the post-exercise recovery period, muscle glucose uptake displays an increased sensitivity to insulin in this way increasing glucose uptake after a meal in the muscles that have performed the exercise and therefore are in need of rebuilding their glycogen stores. Whereas the molecular mechanisms involved in post-exercise increased insulin sensitivity are not fully understood, they could involve repackaging of the GLUT4 vesicles in more easily recruitable pools post-exercise. Furthermore, exercise-induced phosphorylation of proteins such as TBC1D4 and p38 MAPK, which remain phosphorylated for hours after exercise, may contribute to insulin-sensitization.
Acknowledgments
This work was supported by grants to E.A.R. by the Danish Medical Research Council, The Lundbeck Foundation, NovoNordisk Foundation and the research program of the UNIK: Food, Fitness & Pharma for Health and Disease (see http://www.foodfitnesspharma.ku.dk). The UNIK project is supported by the Danish Ministry of Science, Technology and Innovation. T.E.J. was supported by a postdoctoral fellowship from the Danish Medical Research Council.
References
- Antonescu CN, Huang C, Niu W, Liu Z, Eyers PA, Heidenreich KA, Bilan PJ, Klip A. Reduction of insulin-stimulated glucose uptake in L6 myotubes by the protein kinase inhibitor SB203580 is independent of p38MAPK activity. Endocrinology. 2005;146:3773–3781. doi: 10.1210/en.2005-0404. [DOI] [PubMed] [Google Scholar]
- Aschenbach WG, Suzuki Y, Breeden K, Prats C, Hirshman MF, Dufresne SD, Sakamoto K, Vilardo PG, Steele M, Kim JH, Jing SL, Goodyear LJ, DePaoli-Roach AA. The muscle-specific protein phosphatase PP1G/R(GL)(G(M))is essential for activation of glycogen synthase by exercise. J Biol Chem. 2001;276:39959–39967. doi: 10.1074/jbc.M105518200. [DOI] [PubMed] [Google Scholar]
- Birk JB, Wojtaszewski JF. Predominant α2/β2/γ3 AMPK activation during exercise in human skeletal muscle. J Physiol. 2006;577:1021–1032. doi: 10.1113/jphysiol.2006.120972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair DR, Funai K, Schweitzer GG, Cartee GD. A myosin II ATPase inhibitor reduces force production, glucose transport, and phosphorylation of AMPK and TBC1D1 in electrically stimulated rat skeletal muscle. Am J Physiol Endocrinol Metab. 2009;296:E993–E1002. doi: 10.1152/ajpendo.91003.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouskila M, Hunter RW, Ibrahim AF, Delattre L, Peggie M, Diepen vanJA, Voshol PJ, Jensen J, Sakamoto K. Allosteric regulation of glycogen synthase controls glycogen synthesis in muscle. Cell Metab. 2010;12:456–466. doi: 10.1016/j.cmet.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Cartee GD, Funai K. Exercise and insulin: Convergence or divergence at AS160 and TBC1D1? Exerc Sport Sci Rev. 2009;37:188–195. doi: 10.1097/JES.0b013e3181b7b7c5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartee GD, Wojtaszewski JF. Role of Akt substrate of 160 kDa in insulin-stimulated and contraction-stimulated glucose transport. Appl Physiol Nutr Metab. 2007;32:557–566. doi: 10.1139/H07-026. [DOI] [PubMed] [Google Scholar]
- Castorena CM, Mackrell JG, Bogan JS, Kanzaki M, Cartee GD. Clustering of GLUT4, TUG and RUVBL2 protein levels correlate with myosin heavy chain isoform pattern in skeletal muscles, but AS160 and TBC1D1 levels do not. J Appl Physiol. 2011;111:1106–1117. doi: 10.1152/japplphysiol.00631.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers MA, Moylan JS, Smith JD, Goodyear LJ, Reid MB. Stretch-stimulated glucose uptake in skeletal muscle is mediated by reactive oxygen species and p38 MAP-kinase. J Physiol. 2009;587:3363–3373. doi: 10.1113/jphysiol.2008.165639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danforth WH. Glycogen synthetase activity in skeletal muscle: interconversion of two forms and control of glycogen synthesis. J Biol Chem. 1965;240:588–593. [PubMed] [Google Scholar]
- Egawa T, Hamada T, Kameda N, Karaike K, Ma X, Masuda S, Iwanaka N, Hayashi T. Caffeine acutely activates 5′ adenosine monophosphate-activated protein kinase and increases insulin-independent glucose transport in rat skeletal muscles. Metabolism. 2009;58:1609–1617. doi: 10.1016/j.metabol.2009.05.013. [DOI] [PubMed] [Google Scholar]
- Fisher JS, Gao J, Han DH, Holloszy JO, Nolte LA. Activation of AMP kinase enhances sensitivity of muscle glucose transport to insulin. Am J Physiol Endocrinol Metab. 2002;282:E18–E23. doi: 10.1152/ajpendo.2002.282.1.E18. [DOI] [PubMed] [Google Scholar]
- Gao J, Gulve EA, Holloszy JO. Contraction-induced increase in muscle insulin sensitivity: requirement for a serum factor. Am J Physiol Endocrinol Metab. 1994;266:E186–E192. doi: 10.1152/ajpendo.1994.266.2.E186. [DOI] [PubMed] [Google Scholar]
- Geiger PC, Han DH, Wright DC, Holloszy JO. How muscle insulin sensitivity is regulated: testing of a hypothesis. Am J Physiol Endocrinol Metab. 2006;291:E1258–E1263. doi: 10.1152/ajpendo.00273.2006. [DOI] [PubMed] [Google Scholar]
- Geiger PC, Wright DC, Han DH, Holloszy JO. Activation of p38 MAP kinase enhances sensitivity of muscle glucose transport to insulin. Am J Physiol Endocrinol Metab. 2005;288:E782–E788. doi: 10.1152/ajpendo.00477.2004. [DOI] [PubMed] [Google Scholar]
- Hargreaves M, Richter EA. Regulation of skeletal muscle glycogenolysis during exercise. Can J Sport Sci. 1988;13:197–203. [PubMed] [Google Scholar]
- Hawley JA, Burke LM, Phillips SM, Spriet LL. Nutritional modulation of training-induced skeletal muscle adaptations. J Appl Physiol. 2011;110:834–845. doi: 10.1152/japplphysiol.00949.2010. [DOI] [PubMed] [Google Scholar]
- Hespel P, Richter EA. Mechanism linking glycogen concentration and glycogenolytic rate in perfused contracting rat skeletal muscle. Biochem J. 1992;284:777–780. doi: 10.1042/bj2840777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloszy JO, Kohrt WM. Regulation of carbohydrate and fat metabolism during and after exercise. Annu Rev Nutr. 1996;16:121–138. doi: 10.1146/annurev.nu.16.070196.001005. [DOI] [PubMed] [Google Scholar]
- Hunter RW, Treebak JT, Wojtaszewski JF, Sakamoto K. Molecular mechanism by which AMP-activated protein kinase activation promotes glycogen accumulation in muscle. Diabetes. 2011;60:766–774. doi: 10.2337/db10-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihlemann J, Ploug T, Hellsten Y, Galbo H. Effect of stimulation frequency on contraction-induced glucose transport in rat skeletal muscle. Am J Physiol Endocrinol Metab. 2000;279:E862–E867. doi: 10.1152/ajpendo.2000.279.4.E862. [DOI] [PubMed] [Google Scholar]
- James JH, Wagner KR, King JK, Leffler RE, Upputuri RK, Balasubramaniam A, Friend LA, Shelly DA, Paul RJ, Fischer JE. Stimulation of both aerobic glycolysis and Na+-K+-ATPase activity in skeletal muscle by epinephrine or amylin. Am J Physiol Endocrinol Metab. 1999;277:E176–E186. doi: 10.1152/ajpendo.1999.277.1.E176. [DOI] [PubMed] [Google Scholar]
- Jensen J, Lai YC. Regulation of muscle glycogen synthase phosphorylation and kinetic properties by insulin, exercise, adrenaline and role in insulin resistance. Arch Physiol Biochem. 2009;115:13–21. doi: 10.1080/13813450902778171. [DOI] [PubMed] [Google Scholar]
- Jensen TE, Rose AJ, Hellsten Y, Wojtaszewski JF, Richter EA. Caffeine-induced Ca2+ release increases AMPK-dependent glucose uptake in rodent soleus muscle. Am J Physiol Endocrinol Metab. 2007;293:E286–E292. doi: 10.1152/ajpendo.00693.2006. [DOI] [PubMed] [Google Scholar]
- Jessen N, Goodyear LJ. Contraction signaling to glucose transport in skeletal muscle. J Appl Physiol. 2005;99:330–337. doi: 10.1152/japplphysiol.00175.2005. [DOI] [PubMed] [Google Scholar]
- Jorgensen SB, Nielsen JN, Birk JB, Olsen GS, Viollet B, Andreelli F, Schjerling P, Vaulont S, Hardie DG, Hansen BF, Richter EA, Wojtaszewski JF. The α2–5′AMP-activated protein kinase is a site 2 glycogen synthase kinase in skeletal muscle and is responsive to glucose loading. Diabetes. 2004;53:3074–3081. doi: 10.2337/diabetes.53.12.3074. [DOI] [PubMed] [Google Scholar]
- Jørgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA, Wojtaszewski JFP. Knockout of the α2 but not α1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside but not contraction-induced glucose uptake in skeletal muscle. J Biol Chem. 2004;279:1070–1079. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- Kjaer M, Howlett K, Langfort J, Zimmerman-Belsing T, Lorentsen J, Bulow J, Ihlemann J, Feldt-Rasmussen U, Galbo H. Adrenaline and glycogenolysis in skeletal muscle during exercise: a study in adrenalectomised humans. J Physiol. 2000;528:371–378. doi: 10.1111/j.1469-7793.2000.00371.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klip A. The many ways to regulate glucose transporter 4. Appl Physiol Nutr Metab. 2009;34:481–487. doi: 10.1139/H09-047. [DOI] [PubMed] [Google Scholar]
- Koh HJ, Toyoda T, Fujii N, Jung MM, Rathod A, Middelbeek RJ, Lessard SJ, Treebak JT, Tsuchihara K, Esumi H, Richter EA, Wojtaszewski JF, Hirshman MF, Goodyear LJ. Sucrose nonfermenting AMPK-related kinase (SNARK) mediates contraction-stimulated glucose transport in mouse skeletal muscle. Proc Natl Acad Sci U S A. 2010;107:15541–15546. doi: 10.1073/pnas.1008131107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Young RS, Griffee SR, Lynes SE, Bracy DP, Ayala JE, McGuinness OP, Wasserman DH. Skeletal muscle AMP-activated protein kinase is essential for the metabolic response to exercise in vivo. J Biol Chem. 2009;284:23925–23934. doi: 10.1074/jbc.M109.021048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucidi P, Rossetti P, Porcellati F, Pampanelli S, Candeloro P, Andreoli AM, Perriello G, Bolli GB, Fanelli CG. Mechanisms of insulin resistance after insulin-induced hypoglycemia in humans: the role of lipolysis. Diabetes. 2010;59:1349–1357. doi: 10.2337/db09-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maarbjerg SJ, Sylow L, Richter EA. Current understanding of increased insulin sensitivity after exercise – emerging candidates. Acta Physiol (Oxf) 2011;202:323–335. doi: 10.1111/j.1748-1716.2011.02267.x. [DOI] [PubMed] [Google Scholar]
- McBride A, Ghilagaber S, Nikolaev A, Hardie DG. The glycogen-binding domain on the AMPK beta subunit allows the kinase to act as a glycogen sensor. Cell Metab. 2009;9:23–34. doi: 10.1016/j.cmet.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou P, Hajjar RJ, Kranias EG. Role of protein phosphatase-1 inhibitor-1 in cardiac physiology and pathophysiology. J Mol Cell Cardiol. 2009;47:365–371. doi: 10.1016/j.yjmcc.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J, Holmberg HC, Schroder HD, Saltin B, Ortenblad N. Human skeletal muscle glycogen utilization in exhaustive exercise: role of subcellular localization and fibre type. J Physiol. 2011;589:2871–2885. doi: 10.1113/jphysiol.2010.204487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen JN, Derave W, Kristiansen S, Ralston E, Ploug T, Richter EA. Glycogen synthase localization and activity in rat skeletal muscle is strongly dependent on glycogen content. J Physiol. 2001;531:757–769. doi: 10.1111/j.1469-7793.2001.0757h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen JN, Wojtaszewski JF. Regulation of glycogen synthase activity and phosphorylation by exercise. Proc Nutr Soc. 2004;63:233–237. doi: 10.1079/PNS2004348. [DOI] [PubMed] [Google Scholar]
- O'Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jorgensen SB, Schertzer JD, Shyroka O, Kiens B, Denderen vanBJ, Tarnopolsky MA, Kemp BE, Richter EA, Steinberg GR. AMP-activated protein kinase (AMPK) β1β2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci U S A. 2011;108:16092–16097. doi: 10.1073/pnas.1105062108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker GJ, Lund KC, Taylor RP, McClain DA. Insulin resistance of glycogen synthase mediated by o-linked N-acetylglucosamine. J Biol Chem. 2003;278:10022–10027. doi: 10.1074/jbc.M207787200. [DOI] [PubMed] [Google Scholar]
- Prats C, Cadefau JA, Cusso R, Qvortrup K, Nielsen JN, Wojtaszewski JF, Hardie DG, Stewart G, Hansen BF, Ploug T. Phosphorylation-dependent translocation of glycogen synthase to a novel structure during glycogen resynthesis. J Biol Chem. 2005;280:23165–23172. doi: 10.1074/jbc.M502713200. [DOI] [PubMed] [Google Scholar]
- Prats C, Gomez-Cabello A, Hansen AV. Intracellular compartmentalization of skeletal muscle glycogen metabolism and insulin signalling. Exp Physiol. 2011;96:385–390. doi: 10.1113/expphysiol.2010.052860. [DOI] [PubMed] [Google Scholar]
- Prats C, Helge JW, Nordby P, Qvortrup K, Ploug T, Dela F, Wojtaszewski JF. Dual regulation of muscle glycogen synthase during exercise by activation and compartmentalization. J Biol Chem. 2009;284:15692–15700. doi: 10.1074/jbc.M900845200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raney MA, Turcotte LP. Evidence for the involvement of CaMKII and AMPK in Ca2+-dependent signaling pathways regulating FA uptake and oxidation in contracting rodent muscle. J Appl Physiol. 2008;104:1366–1373. doi: 10.1152/japplphysiol.01282.2007. [DOI] [PubMed] [Google Scholar]
- Ribe D, Yang J, Patel S, Koumanov F, Cushman SW, Holman GD. Endofacial competitive inhibition of glucose transporter-4 intrinsic activity by the mitogen-activated protein kinase inhibitor SB203580. Endocrinology. 2005;146:1713–1717. doi: 10.1210/en.2004-1294. [DOI] [PubMed] [Google Scholar]
- Richter EA, Derave W, Wojtaszewski JF. Glucose, exercise and insulin: emerging concepts. J Physiol. 2001;535:313–322. doi: 10.1111/j.1469-7793.2001.t01-2-00313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter EA, Ruderman NB, Gavras H, Belur ER, Galbo H. Muscle glycogenolysis during exercise: dual control by epinephrine and contractions. Am J Physiol Endocrinol Metab. 1982;242:E25–E32. doi: 10.1152/ajpendo.1982.242.1.E25. [DOI] [PubMed] [Google Scholar]
- Roach PJ. Glycogen and its metabolism. Curr Mol Med. 2002;2:101–120. doi: 10.2174/1566524024605761. [DOI] [PubMed] [Google Scholar]
- Romijn JA, Coyle EF, Sidossis LS, Gastaldelli A, Horowitz JF, Endert E, Wolfe RR. Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am J Physiol Endocrinol Metab. 1993;265:E380–E391. doi: 10.1152/ajpendo.1993.265.3.E380. [DOI] [PubMed] [Google Scholar]
- Rose AJ, Richter EA. Skeletal muscle glucose uptake during exercise: how is it regulated? Physiology (Bethesda) 2005;20:260–270. doi: 10.1152/physiol.00012.2005. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Holman GD. Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am J Physiol Endocrinol Metab. 2008;295:E29–E37. doi: 10.1152/ajpendo.90331.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoberg KA, Rattigan S, Hiscock N, Richter EA, Kiens B. A new method to study changes in microvascular blood volume in muscle and adipose tissue: real-time imaging in humans and rat. Am J Physiol Heart Circ Physiol. 2011;301:H450–H458. doi: 10.1152/ajpheart.01174.2010. [DOI] [PubMed] [Google Scholar]
- Thong FS, Derave W, Urso B, Kiens B, Richter EA. Prior exercise increases basal and insulin-induced p38 mitogen-activated protein kinase phosphorylation in human skeletal muscle. J Appl Physiol. 2003;94:2337–2341. doi: 10.1152/japplphysiol.00036.2003. [DOI] [PubMed] [Google Scholar]
- Treebak JT, Birk JB, Hansen BF, Olsen GS, Wojtaszewski JF. A-769662 activates AMPK beta1-containing complexes but induces glucose uptake through a PI3-kinase-dependent pathway in mouse skeletal muscle. Am J Physiol Cell Physiol. 2009;297:C1041–C1052. doi: 10.1152/ajpcell.00051.2009. [DOI] [PubMed] [Google Scholar]
- Watt MJ, Howlett KF, Febbraio MA, Spriet LL, Hargreaves M. Adrenaline increases skeletal muscle glycogenolysis, pyruvate dehydrogenase activation and carbohydrate oxidation during moderate exercise in humans. J Physiol. 2001;534:269–278. doi: 10.1111/j.1469-7793.2001.t01-1-00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszewski JF, Richter EA. Effects of acute exercise and training on insulin action and sensitivity: focus on molecular mechanisms in muscle. Essays Biochem. 2006;42:31–46. doi: 10.1042/bse0420031. [DOI] [PubMed] [Google Scholar]
- Wright DC, Geiger PC, Holloszy JO, Han DH. Contraction- and hypoxia-stimulated glucose transport is mediated by a Ca2+-dependent mechanism in slow-twitch rat soleus muscle. Am J Physiol Endocrinol Metab. 2005;288:E1062–E1066. doi: 10.1152/ajpendo.00561.2004. [DOI] [PubMed] [Google Scholar]
- Wright DC, Hucker KA, Holloszy JO, Han DH. Ca2+ and AMPK both mediate stimulation of glucose transport by muscle contractions. Diabetes. 2004;53:330–335. doi: 10.2337/diabetes.53.2.330. [DOI] [PubMed] [Google Scholar]
- Zisman A, Peroni OD, Abel ED, Michael MD, Mauvais-Jarvis F, Lowell BB, Wojtaszewski JF, Hirshman MF, Virkamaki A, Goodyear LJ, Kahn CR, Kahn BB. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat Med. 2000;6:924–928. doi: 10.1038/78693. [DOI] [PubMed] [Google Scholar]