

Abstract
Activation of transient receptor potential (TRP) canonical TRPC3/C6/C7 channels by diacylglycerol (DAG) upon stimulation of phospholipase C (PLC)-coupled receptors results in the breakdown of phosphoinositides (PIPs). The critical importance of PIPs to various ion-transporting molecules is well documented, but their function in relation to TRPC3/C6/C7 channels remains controversial. By using an ectopic voltage-sensing PIP phosphatase (DrVSP), we found that dephosphorylation of PIPs robustly inhibits currents induced by carbachol (CCh), 1-oleolyl-2-acetyl-sn-glycerol (OAG) or RHC80267 in TRPC3, TRPC6 and TRPC7 channels, though the strength of the DrVSP-mediated inhibition (VMI) varied among the channels with a rank order of C7 > C6 > C3. Pharmacological and molecular interventions suggest that depletion of phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) is most likely the critical event for VMI in all three channels. When the PLC catalytic signal was vigorously activated through overexpression of the muscarinic type-I receptor (M1R), the inactivation of macroscopic TRPC currents was greatly accelerated in the same rank order as the VMI, and VMI of these currents was attenuated or lost. VMI was also rarely detected in vasopressin-induced TRPC6-like currents in A7r5 vascular smooth muscle cells, indicating that the inactivation by PI(4,5)P2 depletion underlies the physiological condition. Simultaneous fluorescence resonance energy transfer (FRET)-based measurement of PI(4,5)P2 levels and TRPC6 currents confirmed that VMI magnitude reflects the degree of PI(4,5)P2 depletion. These results demonstrate that TRPC3/C6/C7 channels are differentially regulated by depletion of PI(4,5)P2, and that the bimodal signal produced by PLC activation controls these channels in a self-limiting manner.
Key points
From brain to digestive tract, electro-chemical signals are broadly utilized to control the activity of the organs; however, the formation of such signals is very varied in each cell and still unknown in many cells.
In this study, we found a novel mechanism for forming an electrical signal, produced by channels of the transient receptor potential canonical (TRPC) family of channels, which allow the permeation of ions such as sodium and calcium and are opened by the actions of hormones such as adrenaline and noradrenaline.
Such hormones can activate an enzyme (phospholipase C) by which PI(4,5)P2, a member of the membrane lipid ‘phosphoinositide’, is degraded: the degradation of PI(4,5)P2 to produce an agonist (diacylglycerol) involved in the opening of TRPC channels, while the degradation itself is surprisingly critical to the closing of these channels.
As a result of such a self-limiting effect via membrane lipid degradation, TRPC channels can produce a unique electro-chemical signal which is tightly bound to the arrangement of membrane lipid and hormones.
Differential sensitivity for PIP2 of TRPC
Introduction
TRPC3/C6/C7 channels are mammalian homologues of light-sensitive Drosophila TRP channels and are widely distributed in the central nervous system, kidney, digestive tract and blood vessels, as well as in lymphocytes and uterine smooth muscle cells (Ramsey et al. 2006). These channels are categorized as non-selective cation channels, which in most cases show severalfold higher permeability to Ca2+ than other cations. Their activation has a critical impact on cardiovascular physiology, inducing vasoconstriction and contributing to pathological remodelling such as atherosclerosis and cardiac hypertrophy (Inoue et al. 2006; Eder & Groschner, 2008; Dietrich et al. 2010). They have also been implicated in a variety of other functions, including synaptic transmission, neuronal dendritic growth, respiratory rhythm and the progression of Alzheimer's disease (Li et al. 1999; Strubing et al. 2003; Zhou et al. 2008; Becker et al. 2009; Ben-Mabrouk & Tryba, 2010; Li et al. 2010). Their roles in pathophysiological processes highlight the importance of the contribution made by TRPC3/C6/C7 channels to the biophysical properties of cells and their regulatory mechanisms.
Diacylglycerol (DAG) is a critical activator of TRPC3/C6/C7 channels; thus application of a membrane-permeant DAG analogue (e.g. OAG), the DAG-lipase inhibitor RHC80267 (RHC), a Gαq/11-protein-coupled receptor (GqPCR) agonist or a receptor tyrosine kinase engaged with phospholipase C (PLC) induces cationic currents in cells expressing these channels (Hofmann et al. 1999; Li et al. 1999; Okada et al. 1999). In native systems, TRPC3/C6/C7 channel agonists include various neurohormones, noradrenaline (norepinephrine), endothelin, acetylcholine and brain-derived neurotrophic factor (Moran et al. 2011). These agonists all stimulate PLC to produce DAG, IP3 and protons from membrane phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) (Huang et al. 2009).
In contrast to the triggering effect of DAG for the activation of TRPC3/C6/C7 channels, the functional role of PIPs, especially PI(4,5)P2, remains less clear and controversial (Hardie, 2007; Voets & Nilius, 2007). Lemonnier et al. (2008) reported that intracellular application of PI(4,5)P2 induces the opening of expressed TRPC6 and TRPC7 channels, but not TRPC3 channels. In another study, however, depletion of PI(4,5)P2 from rabbit mesenteric artery smooth muscle cells increased endogenous TRPC6-like channel activity (Albert et al. 2008). In both of these studies, isolated membrane patches were utilized to assess the effects of PIPs under steady-state conditions. However, given the dynamic nature of PIP turnover during DAG production, the results from steady-state experiments may be misleading. In that regard, new tools, such as VSPs (voltage-sensing phosphatases, a membrane-resident PI-5 phosphatase) and rapamycin-inducible PIP 5-phosphatase are proving to be effective for investigation of PIPs function, as they enable rapid depletion of PIPs content (Suh et al. 2006; Okamura et al. 2009).
In the present study, therefore, we used Danio rerio VSP (DrVSP) to investigate the dynamic influence of PIP depletion on TRPC3/C6/C7 channel gating at various levels of PLC-coupled GqPCR activation. Our results show that DrVSP-mediated depletion of PI(4,5)P2 inhibits TRPC3/C6/C7 channel activity in an isoform-specific manner. Moreover, the degree of DrVSP inhibition correlates well with the inactivation kinetics of the macroscopic currents.
Methods
Plasmids and cells
The pCDNA3 expression vector harbouring DNA encoding human TRPC6 (accession number: NM_004621) was provided by Thomas Hofmann (Institut für Pharmakologie und Toxikologie, Marburg, Germany); murine TRPC3 (NM_019510) and TRPC7 (NM_012035) in pCI-neo expression vector were provided by Dr Y. Mori (Kyoto University); DrVSP and its derivative mutants in pIRES-eGFP vector (Invitrogen) are identical to those described in a previous report (Hossain et al. 2008); human M1R in pEF-BOS expression vector was provided by Dr T. Haga (Gakushuin University) (distributed by Dr M. Nishida, Kyushu University). eCFP-PH and eYFP-PH domain (fusion proteins of pleckstrin homology domain from PLC delta1 and eCFP or eYFP) in pCDNA3 were provided by K. Jalink (The Netherland Cancer Institutes).
Human embryonic kidney 293 (HEK293) cells obtained from ATCC were maintained in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (GIBCO) and antibiotics (Pen/Strep, GIBCO) at 37°C (5% CO2). For transfection, the cells were reseeded onto poly-l-lysine-coated cover glass (Matsunami) in 35 mm culture dishes. They were then transfected with a mixture of plasmid vector-incorporated DNAs using the transfection reagent SuperFect (Qiagen). Electrophysiological measurements were made within 24–72 h after transfection. A7r5 cells, a rat thoracic aortic smooth muscle cell line (Brandt et al. 1976), were obtained from ATCC and maintained in medium identical to that used for HEK293 cells and were passaged every 5–7 days. The transfection protocol was essentially the same as used with HEK cells. Non-transfected or DrVSP-transfected A7r5 cells were reseeded onto poly-l-lysine-coated glass coverslips, incubated at 37°C (5% CO2) for 15 min and used within 2 h.
Solutions and drugs
The pipette solution for whole-cell recording contained (mm): 120 CsOH, 120 aspartate, 20 CsCl, 2 MgCl2, 5 EGTA, 1.5 CaCl2, 10 Hepes, 2 ATP-Na2, 0.1 GTP, 10 glucose (pH 7.2, adjusted with Tris base; 290–295 mosmol l−1, adjusted with glucose). The resulting free calcium concentration was calculated to be ∼100 nm based on a previous report (Shi et al. 2004). The standard external solution contained (mm): 140 NaCl, 5 KCl, 1 CaCl2, 1.2 MgCl2, 10 Hepes and 10 glucose (pH 7.4, adjusted with Tris base; 300 mosmol l−1, adjusted with glucose). Carbachol (CCh; Sigma-Aldrich) was diluted in the standard external solution from its stock (100 mm) in H2O. To confirm monovalent cationic currents, NMDG solution (150 N-methyl-d-glucamine chloride, 10 Hepes, 1 CaCl2, pH 7.4 adjusted with HCl) was applied at the end of every stimulus. In addition, 100 μm DIDS (4,4-diisothiocyanostilbene-2,2-disulphonic acid, 2 Na, purchased from Calbiochem) was applied in the external solutions when needed. Stock solutions of wortmannin (Sigma-Aldrich), LY-294002 (Sigma-Aldrich), RHC80267 (RHC, Calbiochem) and 1-oleoyl-2-acetyl-sn-glycerol (OAG, Cayman Chemical) were dissolved in dimethyl sulfoxide (DMSO, Wako, Japan) at concentrations that were more than 1000 times higher than needed for experimental use. Consequently, the highest concentration of DMSO used never exceeded 0.1%. OAG dissolved in external solution was sonicated for 10 min before use. RHC dissolved in external solution was filtered through a 0.8 μm pore filter (Millipore) to remove small particles that often appeared. Stock solutions (10 mm) of diC8-PI(4,5)P2, diC8-PI(3,4,5)P3 and diC8-PI(4)P (Wako) were prepared in distilled H2O. Stock solutions of Arg8-vasopressin (AVP; MP Biomedical) (100 μm) and nifedipine (Calbiochem) (10 mm) were prepared in H2O and DMSO, respectively. AVP and nifedipine diluted in the standard external solution to final concentrations of 50 nm and 5 μm were used for stimulation of A7r5 cells. During experiments, HEK or A7r5 cells were continuously perfused with gravity-fed external solution at a flow rate of 0.25 ml min−1. The perfusion could be turned on and off using electromagnetic solenoid microvalves (The Lee Co., USA), which enabled us to exchange the external solutions within 1 s. Adenylyl-imidodiphosphate, tetralithium salt (AMP-PNP; illustrated in Supplemental Fig. S3, available online only) purchased from Calbiochem was added to the pipette solution to obtain a final concentration of 2 mm.
Electrophysiology and data analysis
The whole-cell patch-clamp technique was employed in the present study. Patch electrodes with resistances of 4–6 MΩ (when filled with internal solution) were made from 1.5 mm borosilicate glass capillaries using an automated electrode puller (both obtained from Sutter Instruments) and heat polished. Series-resistance errors were compensated more than 70%. Voltage generation and current signal acquisition were accomplished using a high-impedance low-noise patch clamp amplifier (AxoPatch 200B, Axon Instruments) in conjunction with an A/D, D/A-converter (Digidata 1200, Axon Instruments). Sampled data were low-pass filtered at 1 kHz, digitized at 5 kHz and analysed using Clampfit v. 9.2 (Axon Instruments) and custom-written software (MATLAB). The currents were recorded at a holding potential of −50 mV. All experiments were performed at room temperature (22–25°C). To activate DrVSP, a depolarizing pulse (+100 mV, 500 ms duration) was repetitively delivered every 10 or 20 s. The ratio of the current before and after DrVSP activation (r) was used to quantify the level of inhibition. Prior to calculation, leak components were subtracted based on the minimum current in NMDG solution. All data were acquired from the time of the first stimulus currents elicited by any agonist, except in Fig. 9D.
Figure 7. VMI in A7r5 aortic smooth muscle cells.
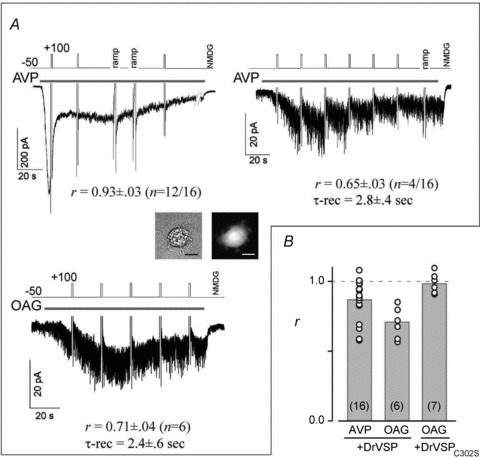
A, depolarization had almost no effect on TRPC6-like currents elicited in A7r5 cells by 50 nm AVP (left top, 12 of 16 cells). Co-transfection of the cells with DrVSP was confirmed by GFP expression (inset: bright-field (left) and GFP fluorescence (right)). A few cells exhibited clear VMI (right, 4 of 16 cells). OAG-induced currents also exhibited substantial VMI (left bottom). B, summary of VMI of AVP- and OAG-induced currents. VMI measured in each A7r5 cell (open circles); grey bars indicate the means of the VMI. Cells co-transfected with DrVSPC302S exhibited no VMI of OAG-induced currents.
Simultaneous imaging of [Ca2+]i and PIP2
In recent reports, measurement of [Ca2+]i in individual cells using Fura-2 simultaneously with CFP/YFP-based FRET was accomplished using cross-talk correction protocols described previously (Mori et al. 2011). We applied this imaging and calculation algorithm to detect Ca2+ and PI(4,5)P2 dynamics elicited by CCh. In brief, HEK293 cells were transiently transfected with equal amounts of TRPC channels, eYFP-PH and eCFP-PH, with or without M1R. Co-transfected cells on coverslips were incubated with 5 μm of the acetoxy methyl ester form of Fura-2, Fura-2/AM (Dojindo Laboratories, Japan) for 30 min then washed and bathed in standard external solution (see ‘Solutions and drugs’) for at least 5 min before stimulation with CCh. The coverslips were then placed in a perfusion chamber mounted on the stage of an inverted fluorescence microscope (DMI 6000 B Leica, Switzerland) equipped with filter cubes for Fura-2 and C/Y mounted on a rotating turret that enabled five successive images (F340, F360, FCFP, FYFP, and FFRET: subscript is the name of the filter setting) to be obtained in 3.5 s every 5 s. All imaging data were acquired using Slidebook v.4.2 software (Intelligent Imaging Innovations, Denver, CO, USA) and analysed using a custom-written MATLAB program. Free [Ca2+]i were estimated using a magnesium-containing calcium calibration kit supplied by Molecular Probes (Eugene, OR, USA). FRET ratios (FRs) were calculated using the ‘3-cube’ method described previously (Erickson et al. 2003).
Simultaneous detection of TRPC currents and PIP2
HEK293 cells on coverslips were co-transfected with equal amounts of TRPC channels, eYFP-PH and eCFP-PH, without or with M1R, and placed in a perfusion chamber. FRET detection from single living cells was accomplished using a Nikon TE300 Eclipse microscope (×60, 0.9 NA objective) equipped with beam-splitter (Dual-View2, Roper Scientific, USA) and CCD camera (Evolve512, Roper Scientific). Excitation light filtered at 427 nm/10 nm was introduced via an optical fibre from a lamp house (150 W xenon lamp, OSP-EXA; Olympus). The epifluorescence passing into the beam-splitter was separated using a multiband dichroic mirror (449–483 and 530–569 nm) and filtered at 464 nm/23 nm (short wavelength) or 542 nm/27 nm (long wavelength). Optical filters were obtained from Semrock. Images were digitized as 512 × 512 pixels by 16-bit arrays. For sampling, the illumination shutter in the lamp house was opened for 100 ms every 300 ms. Averaged intensities from ROIs (regions of interest, typically 10 × 10 to 25 × 25 square pixels) that include the cell membrane and cytosol were used for FRET calculations, which were taken as the ratio of the long-wavelength to the short-wavelength emission (L/S) during 427 nm illumination after correcting for background fluorescence. Imaging data were acquired using Micro-Manager 1.3 (http://www.micro-manager.org) and analysed using a custom-written MATLAB program. To synchronize the FRET and electrophysiology data, triggers from the A/D,D/A converter were linked to the excitation light shutter.
Statistics
All data are expressed as mean ± SEM, paired Student's t test (denoted by asterisks (*)) and one-way ANOVA (denoted by daggers (†)) were used for single- and multiple-comparison statistical analyses, respectively.
Results
Activation of DrVSP leads to robust and transient inhibition of ITRPC
HEK293 cells endogenously express muscarinic GqPCRs (muscarinic type I receptors; M1Rs) (Mundell et al. 2002); consequently, application of the muscarinic agonist carbachol (CCh) induced sustained inward currents (ITRPC6) when TRPC6 protein was transiently expressed. ITRPC6 were not noticeably affected by brief repetitive depolarizations (+100 mV, 500 ms) applied with an interval of 10 s (data not shown), but they underwent robust and transient inhibition when DrVSP was co-expressed with TRPC6 (Fig. 1A, left panel). The DrVSP-mediated inhibition (VMI) of ITRPC6 occurred repetitively and disappeared quickly after each depolarization. To evaluate the extent of this inhibition, we calculated the ratio (r) of the current amplitudes before and after depolarization (right panel in Fig. 1A; r = Ipost/Ipre) and plotted it against the number of stimuli. This calculation showed the VMI index r to be nearly constant over the entire time course of ITRPC6 (filled circles in Fig. 1C; r = 0.50 ± 0.06, n = 11). Likewise, the rate of recovery from VMI also remained nearly constant (Fig. 1D; time constant of recovery: τ-recovery = 1.6 ± 0.2 s).
Figure 1. DrVSP-mediated inhibition (VMI) of TRPC6 currents.
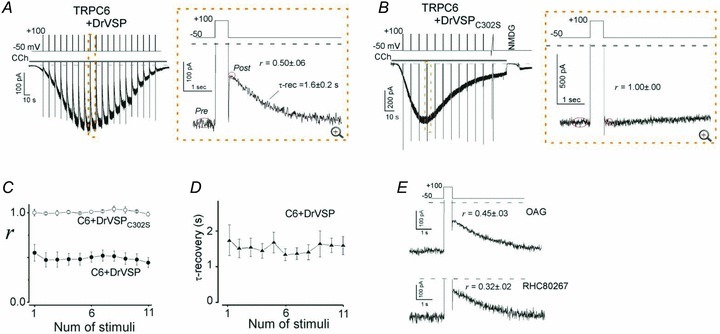
A, current evoked by carbachol (CCh; 100 μm) recorded in the whole-cell mode from cells transfected with TRPC6 and DrVSP (left). Depolarization (+100 mV, 500 ms) was applied every 10 s (protocol displayed above). The area enclosed by the dashed box is enlarged in the right panel. Tail and outward currents are clipped to the frame here and throughout. B, data from cells transfected with TRPC6 and the enzyme-defective mutant DrVSPC302S. The conditions are the same as in A.C, averaged VMI elicited by repetitive depolarization in the presence of DrVSP (filled circles; n = 11) and DrVSPC302S (open circles; n = 5). D, averaged time constants of a single-exponential fit to the recovery from VMI. E, VMI of OAG (100 μm)- and RHC (100 μm)-induced TRPC6 currents (upper and lower panel, respectively). All VMI data are summarized in Fig. 2G and Table 1.
By contrast, when a VSP mutant carrying a loss-of-function mutation in its phosphatase domain (DrVSPC302S) was co-expressed with TRPC6, the VMI of ITRPC6 was totally abolished (Fig. 1B and C open circles). This suggests the inhibition reflects the dephosphorylation of PIPs and excludes the possibility that voltage-dependent activation of M1Rs (via its altered agonist affinity) or regulation by RGS (regulator of G-protein signalling) (Kurachi & Ishii, 2004; Ben-Chaim et al. 2006) contributes to the VMI of ITRPC6.
The VMI phenotype was reproduced when ITRPC6 were elicited using the membrane-permeant DAG analogue OAG or the DAG lipase inhibitor RHC, which bypass the M1R (top and bottom panels in Fig. 1E, respectively). Notably, ITRPC6 induced by RHC were more susceptible to VMI (i.e. more profoundly inhibited) than those activated by CCh (r = 0.32 ± 0.02; †P < 0.05, vs. 1 μm to 1 mm CCh, in Fig. 2G left). Moreover, both OAG- and RHC-induced ITRPC6 showed a significantly slower recovery from VMI than CCh-induced ITRPC6 (OAG: τ-rec = 2.4 ± 0.5 s, n = 9; RHC: τ-rec = 2.5 ± 0.4 s, n = 12).
Figure 2. VMI of TRPC7 and TRPC3 currents.
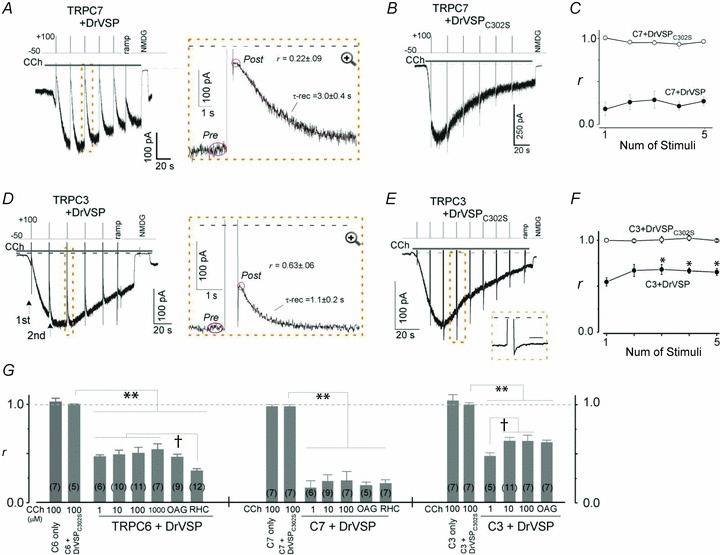
A–C and D–F, the conditions are largely the same as in Fig. 1A–C, except that for TRPC7 and TRPC3 the intervals between depolarizations are 20 s. G, summary of DrVSP effects on TRPC6 (left), C7 (middle) and C3 (right). The inhibition ratio (r) in the bar graph is averaged from the median r value for each cell, here and throughout. The numbers in parentheses indicate the numbers of cells averaged. Numbers at the bottom indicate the CCh concentration (μm). Asterisks indicate statistical significance (**P < 0.001 vs. Cx + DrVSPC302S). The daggers indicate statistically significant differences among the significant inhibition group (†P < 0.05).
Essentially the same type of VMI was observed when TRPC7, another TRPC channel isoform, was co-expressed with DrVSP. However, as shown in Fig. 2A, CCh-induced currents in TRPC7-expressing cells (ITRPC7) exhibited more robust VMI (r = 0.22 ± 0.09 for 100 μm CCh; Fig. 2G middle) than ITRPC6, as did ITRPC7 induced with 100 μm OAG or 100 μm RHC (r = 0.17 ± 0.05 and 0.19 ± 0.05, respectively; middle panel in Fig. 2G). The recovery of ITRPC7 from VMI was proportionally much slower (CCh: τ-rec = 3.0 ± 0.4 s, n = 7; OAG: 5.3 ± 1.8 s, n = 5; RHC: 3.1 ± 0.4 s, n = 7; each 100 μm) than that of ITRPC6. This may imply that TRPC7 has a lower affinity for PIPs than TRPC6. The VMI of ITRPC7 was also abolished by co-expression of the non-functional DrVSPC302S mutant, again suggesting that the inhibition depends on the dephosphorylation of PIPs (Fig. 2B and C open circles).
CCh-induced currents in cells expressing TRPC3 (ITRPC3) with DrVSP exhibited the least inhibition in response to depolarization and the fastest recovery (r = 0.63 ± 0.06 and τ-rec = 1.1 ± 0.2 s for 100 μm CCh; Fig. 2D and right bar in Fig. 2G). Although RHC failed to elicit ITRPC3, OAG-induced ITRPC3 also exhibited significantly weaker VMI (r = 0.54 ± 0.03, right panel in Fig. 2G) with a faster recovery (τ-rec = 1.5 ± 0.3 s) than OAG-induced ITRPC6 or ITRPC7. Interestingly, the VMI of CCh-induced ITRPC3 was progressively attenuated as the current grew larger (Fig. 2D). After the third depolarization (approximately 45 s after the onset of CCh application), the magnitude of the VMI was significantly smaller than the first VMI (the first r = 0.54 ± 0.04 vs. the third r = 0.68 ± 0.06, n = 7, *P < 0.05; Fig. 2F). Conversely, VMI was more pronounced at lower concentrations of CCh, when the amplitude of ITRPC3 was smaller (†P < 0.05, Fig. 2G right panel). These data suggest that the extent of concurrent receptor-mediated hydrolysis of PIPs may time-dependently affect the ability of DrVSP to dephosphorylate PIPs.
To more accurately quantify the differential VMI of TRPC3/C6/C7 channels, we next examined the time- and voltage-dependence of the inhibition by varying the duration and strength of the depolarizing pulses. The top panel of Fig. 3A shows a representative example of the VMI of RHC-induced ITRPC6 elicited by depolarizations of various duration (100–800 ms, +100 mV). It can be clearly seen that prolongation of the pulse resulted in more pronounced inhibition of the current. The lower panel of Fig. 3A summarizes the relationships between depolarization duration and VMI (r) for ITRPC3, ITRPC6 and ITRPC7. For the sake of convenience, the data points for the respective TRPC channels were empirically fitted with a decaying sigmoidal function to determine the half-maximal duration (Khalf), at which 50% inhibition of agonist-induced current occurs. Using a similar approach with depolarizing pulses of various amplitude (0 to +140 mV, 500 ms duration), we also determined the half-maximal voltage (Vhalf), causing 50% inhibition of ITRPC3, ITRPC6 or ITRPC7 (Fig. 3B). Notably, both the Khalf and Vhalf values indicate that the susceptibility to depolarization-induced DrVSP activation is in the order: TRPC3 (384 ± 54 ms and 78 ± 5 mV) < TRPC6 (178 ± 54 ms and 62 ± 4 mV) < TRPC7 (103 ± 12 ms and 50 ± 3 mV). Although this is in good agreement with the results described above, the differential values of Khalf and Vhalf more convincingly demonstrate the distinctive PIP sensitivities of the respective TRPC isoforms (VMI data are also summarized in Table 1).
Figure 3. Pulse-duration and voltage-step protocols reproduce the differential VMI of TRPC3/C6/C7 channels.
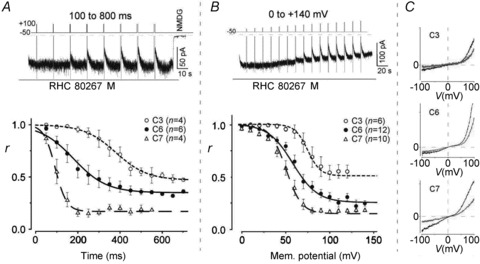
A, top; typical example of time-dependent VMI of a RHC80267-induced TRPC6 current. Bottom; time dependence of the VMI (r) of TRPC3/C6/C7-derived currents. TRPC6 or C7 and C3 were evoked by external application of RHC80267 (100 μm) and OAG (100 μm), respectively. B, top; example of voltage-dependent VMI of a RHC-induced TRPC6 current. Bottom; voltage-dependence of VMI (r) of TRPC3/C6/C7-derived currents. The agonists were the same as in A for the respective currents. C, typical I–V relationships for TRPC3/C6/C7 currents generated using a ramp protocol before (black) and just after VMI (grey).
Table 1.
| Channel and VSP | Agonist (conc. μm) | r (Ipost/Ipre) | τ-rec (s) | Khalf (ms) | Vhalf (mV) | n |
|---|---|---|---|---|---|---|
| TRPC3 + DrVSP | CCh (1) | 0.49 ± 0.04 | 1.3 ± 0.3 | n.d. | n.d. | 5 |
| CCh (10) | 0.64 ± 0.03 | 1.0 ± 0.1 | n.d. | n.d. | 11 | |
| CCh (100) | 0.63 ± 0.06 | 1.1 ± 0.2 | n.d. | n.d. | 7 | |
| OAG (100) | 0.54 ± 0.03 | 1.5 ± 0.3 | 384 ± 54 | 78 ± 5 | 7 | |
| TRPC3 + DrVSPC302S | CCh (100) | 1.03 ± 0.06 | — | — | — | 7 |
| TRPC6 + DrVSP | CCh (1) | 0.45 ± 0.02 | 1.8 ± 0.2 | n.d. | n.d. | 6 |
| CCh (10) | 0.48 ± 0.05 | 1.7 ± 0.6 | n.d. | n.d. | 10 | |
| CCh (100) | 0.50 ± 0.06 | 1.6 ± 0.2 | n.d. | n.d. | 11 | |
| CCh (1000) | 0.54 ± 0.06 | 1.8 ± 0.2 | n.d. | n.d. | 7 | |
| OAG (100) | 0.46 ± 0.03 | 2.4 ± 0.5 | n.d. | n.d. | 9 | |
| RHC (100) | 0.32 ± 0.02 | 2.5 ± 0.4 | 178 ± 54 | 62 ± 4 | 12 | |
| TRPC6 + DrVSPC302S | CCh (100) | 1.00 ± 0.00 | — | — | — | 5 |
| TRPC7 + DrVSP | CCh (1) | 0.15 ± 0.07 | 3.4 ± 0.8 | n.d. | n.d. | 6 |
| CCh (10) | 0.21 ± 0.07 | 2.6 ± 0.2 | n.d. | n.d. | 9 | |
| CCh (100) | 0.22 ± 0.09 | 3.0 ± 0.4 | n.d. | n.d. | 7 | |
| OAG (100) | 0.17 ± 0.05 | 5.3 ± 1.8 | n.d. | n.d. | 5 | |
| RHC (100) | 0.19 ± 0.05 | 3.1 ± 0.4 | 103 ± 12 | 50 ± 3 | 7 | |
| TRPC7 + DrVSPC302S | CCh (100) | 0.98 ± 0.02 | — | — | — | 7 |
| TRPC6-like (A7r5) + DrVSP | AVP (0.05) | 0.86 ± 0.03 | 2.8 ± 0.4* | n.d. | n.d. | 16 |
| OAG (50) | 0.71 ± 0.04 | 2.4 ± 0.6 | n.d. | n.d. | 6 | |
| TRPC6-like (A7r5) + DrVSPC302S | OAG (50) | 0.97 ± 0.03 | — | — | — | 7 |
Values represent means ± SEM. A median value of repetitive stimulation from each cell is utilized for calculation of the inhibition ratio (r) and the recovery time constant (τ-rec). The asterisk in A7r5 row indicates cells exhibiting a significant VMI (n = 4). n.d., not determined.
The current–voltage (I–V) relationships of ITRPC3, ITRPC6 and ITRPC7 before and after VMI are compared in Fig. 3C. With all three TRPC channels, both the inward and outward portions of the I–V relationships were nearly the same before (black traces) and after VMI (grey traces). This suggests the effects of PIPs dephosphorylation by DrVSP on TRPC3/C6/C7 channels gating does not depend on the transmembrane potential gradient; instead, the site of PIP–TRPC channel interaction may be closer to the inner leaflet of the cell membrane (Balla et al. 2009).
Identification of the causal PIP for VMI
DrVSP preferentially removes the 5-phosphate from PI(3,4,5)P3 and PI(4,5)P2, but it can also catalyse dephosphorylation of other PIPs, such as PI(3,4)P2 and PI(3,5)P2 (Iwasaki et al. 2008). Because, like its homologue PTEN (phosphatase and tensin homologue deleted on chromosome 10), DrVSP has the highest affinity for PI(3,4,5)P3, we examined the contribution to VMI made by PI(3,4,5)P3 and related PIPs using DrVSPG306A, which has activity toward PI(3,4,5)P3 but not PI(4,5)P2 (Iwasaki et al. 2008). As shown in Fig. 4A, DrVSPG306A failed to induce VMI of any ITRPC (Fig. 4A and B), and VMI was not restored by application of excess diC8-PI(3,4,5)P3 (100 μm) through the patch pipette (for TRPC6; left second column in Fig. 4B). Moreover, neither a selective inhibitor of phosphoinositide (PI)-3 kinase (LY-294002; 50 μm), nor a low concentration of wortmannin (WN; 5 μm) had any effect on the VMI of ITRPC3, ITRPC6 or ITRPC7 (Fig. 4B). These results preclude the involvement of PIPs in which the inositol was 3-phosphorylated (i.e. PI(3,4)P2, PI(3,5)P2 and PI(3,4,5)P3), as well as the secondary involvement of monophosphate released upon dephosphorylation of PI(3,4,5)P3. In addition, they suggest PI(4,5)P2 plays a pivotal role in the VMI of ITRPC3, ITRPC6 and ITRPC7.
Figure 4. Identification of PI(4,5)P2 as the cause of VMI.
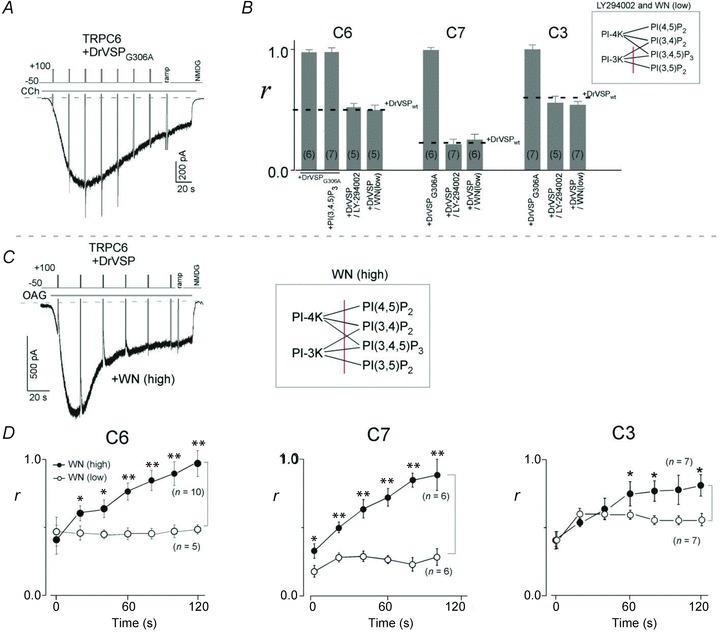
A, co-transfected PTEN-like DrVSPG306A exhibited no ability to dephosphorylate PI(4,5)P2 and failed to inhibit TRPC6 currents. B, summary of the effects of DrVSPG306A and LY-294002. No inhibition of any TRPC channel was detected by the activation of DrVSPG306A, even with additional PI(3,4,5)P3 in the patch pipette. Pretreatment (for 0.5 h) with LY-294002 (50 μm) or a low concentration of wortmannin (WN) had no effect on the VMI of the respective currents. C, loss of VMI of TRPC6, C7 and C3 currents after pretreatment for several minutes with a high concentration of WN (50 μm). Currents were evoked by external application of OAG (30 μm). D, time dependence of the attenuation of VMI (r) by a high concentration of WN (filled circles). The time after the first depolarization was set to zero. **P < 0.001 and *P < 0.05 vs. the identical time in low WN (5 μm; open circles).
High concentrations of WN reportedly retard replenishment of PI(4,5)P2 by inhibiting PI-4 kinase (Nakanishi et al. 1995), which could affect the extent of VMI during the course of GqPCR stimulation. Consistent with that report, after pretreatment with a high concentration of WN (50 μm), VMI gradually disappeared from OAG-induced ITRPC6, ITRPC7 and ITRPC3 (Fig. 4C and filled circles in Fig. 4D). The time required for the disappearance of the VMI was around 120 and 100 s for ITRPC6 and ITRPC7, respectively (left and middle panels in Fig. 4D), whereas some inhibition of ITRPC3 continued to be detected, even after 120 s (right panel in Fig. 4D). These observations strongly suggest that VMI is not the result of reduced DAG production caused by decreased availability of substrate for PLC (i.e. PI(4,5)P2), but may more directly correlate with local levels of PIP2 in the vicinity of the TRPC3/C6/C7 channels.
An alternative explanation is that VMI is induced by PI(4)P, a product of the DrVSP-catalysed dephosphorylation of PI(4,5)P2. To test its involvement, soluble PI(4)P (diC8-PI(4)P) was directly added to cells from the pipette solution. The results show that PI(4)P had no effect on CCh-induced current densities with any of the channels, which excludes PI(4)P from participating in VMI (see Supplemental Fig. S1).
In summary, our findings strongly support the idea that depletion of PI(4,5)P2 (generally known as PIP2) per se is the most probable cause of the VMI of TRPC3/C6/C7 currents, and that effect is also variably modulated by concurrent activation of PLC-coupled GqPCRs, another pathway linked to PIP2 metabolism.
Vigorous stimulation of M1R attenuates VMI
Given the results summarized above, we reasoned that overexpression of PLC-coupled M1Rs would greatly enhance the degradation of PIP2 through robust stimulation of PLC, thereby neutralizing VMI. As shown in Fig. 5A (red traces), overexpression of M1R sharply accelerated the activation and inactivation processes of CCh-induced ITRPC6, ITRPC7 and ITRPC3. The activation times (Δact10-90%; defined as the time required for 10–90% activation) for ITRPC6, ITRPC7 and ITRPC3 in cells overexpressing M1Rs were 4.0 ± 0.4, 1.0 ± 0.1 and 1.6 ± 0.4 s, respectively (red bars in Fig. 5B), which are much faster than those obtained with cells expressing only endogenous M1R (16.2 ± 3.2, 14.1 ± 3.8 and 19.0 ± 6.1 s for ITRPC6, ITRPC7 and ITRPC3, respectively; grey bars in Fig. 5B). For inactivation, the times required for 90–50% reduction from the peak amplitude (Δinac90-50%) were also severalfold shorter than in cells overexpressing M1R, as compared to endogenous expression, i.e. Δinac90-50% for ITRPC6, ITRPC7 and ITRPC3 were 7.1 ± 1.6, 2.3 ± 0.9 and 25.7 ± 3.4 s, respectively, in cells overexpressing MIR and 28.3 ± 4.9, 34.5 ± 6.3 and 55.6 ± 9.3 s in cells only expressing the endogenous receptor (M1R vs. endo; Fig. 5C, see also Table 2). Thus, the activation and inactivation kinetics of CCh-induced ITRPC6, ITRPC7 and ITRPC3 appear to be greatly accelerated by M1R overexpression, with ITRPC7 being the most affected (Fig. 5B and C†P < 0.05, ‡P < 0.001).
Figure 5. Effect of vigorous PLC stimulation in cells overexpressing M1R on VMI.
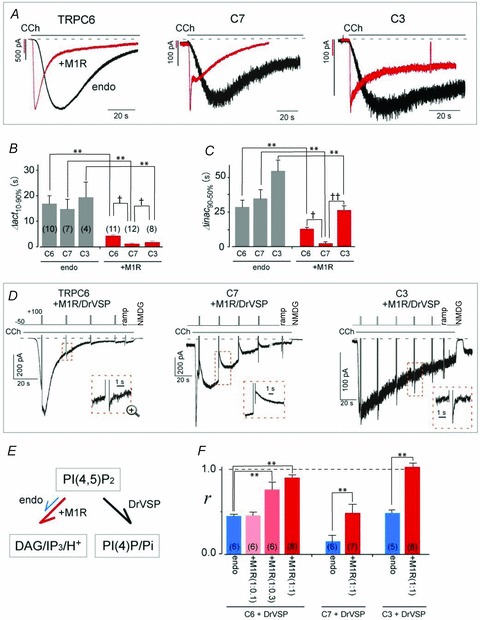
A, typical example of currents elicited by CCh (10 μm) in HEK293 cells transfected with TRPC6 (left), C7 (middle) or C3 (right) channels, with (red trace) and without (black trace) muscarinic receptor type-I (M1R). Membrane potentials were held at −50 mV. Comparison of the activation (B) and inactivation (C) kinetics obtained from A (†P < 0.05 **P < 0.001 by t test). D, co-transfection of M1R eliminates VMI of TRPC6 (left) and C3 (right) currents and reduces C7 currents (middle). E, PI(4,5)P2 catalytic schemes for GqPCR (red arrow: M1R co-transfection, blue arrow: no exogenous GqPCR) and DrVSP (black arrow), which lead to production of DAG/IP3/H+ and PI(4)P/Pi, respectively. F, summary of the mean VMI (r) from Fig. 5D. Blue bars represent the transient inhibition of CCh-evoked (1 μm) currents in HEK293 cells expressing only endogenous M1R. The ratio of the expression vectors (TRPC:M1R) is shown below ‘+M1R’, in parentheses.
Table 2.
Summary of the macroscopic current kinetics
| Channel isoform | Agonist (conc. μm) | Condition (PIP2 conc. μm) | Δact10-90% (s) | Δinac90-50% (s) | n |
|---|---|---|---|---|---|
| TRPC3 | CCh (10) | endo | 19.0 ± 6.1 | 55.6 ± 9.3 | 4 |
| CCh (10) | +M1R | 1.6 ± 0.4 | 25.7 ± 3.4 | 8 | |
| CCh (10) | +M1R + PIP2 (100) | 3.7 ± 0.6 | 38.2 ± 3.0 | 4 | |
| CCh (10) | +M1R + PIP2 (250) | 3.5 ± 1.6 | 48.3 ± 6.3 | 7 | |
| TRPC6 | CCh (10) | endo | 16.2 ± 3.1 | 28.3 ± 4.9 | 10 |
| CCh (10) | +PIP2 (100) | 28.3 ± 4.4 | 60.2 ± 13.5 | 7 | |
| CCh (1) | +M1R | 4.0 ± 0.4 | 12.9 ± 1.3 | 11 | |
| CCh (10) | +M1R | 2.3 ± 0.6 | 7.1 ± 1.6 | 13 | |
| CCh (1) | +M1R + PIP2 (100) | 4.3 ± 0.8 | 16.4 ± 1.7 | 6 | |
| CCh (1) | +M1R + PIP2 (250) | 4.9 ± 0.8 | 29.2 ± 2.1 | 6 | |
| CCh (1) | +M1R + PIP2 (500) | 6.5 ± 1.5 | 32.2 ± 4.0 | 7 | |
| TRPC7 | CCh (10) | endo | 14.1 ± 3.8 | 34.5 ± 6.3 | 7 |
| CCh (10) | +M1R | 1.0 ± 0.1 | 2.3 ± 0.9 | 12 | |
| CCh (10) | +M1R + PIP2 (100) | 1.4 ± 0.5 | 13.8 ± 2.2 | 8 | |
| CCh (10) | +M1R + PIP2 (250) | 1.2 ± 0.2 | 14.5 ± 4.3 | 6 | |
| CCh (10) | +M1R + PIP2 (500) | 1.7 ± 0.4 | 17.0 ± 4.8 | 5 | |
| TRPC6-like (A7r5 cells) | AVP (0.05) | n.t. | 3.4 ± 0.5 | 7.1 ± 2.2 | 8 |
| AVP (0.05) | n.t. + PIP2 (100) | 3.6 ± 0.5 | 13.9 ± 1.5 | 6 | |
| AVP (0.05) | n.t. + PIP2 (500) | 8.8 ± 1.6 | 20.8 ± 5.1 | 7 |
Values represent means ± SEM. n.t., no transfection.
Intensive stimulation of PLC via overexpressed M1Rs also markedly affected the VMI of ITRPC6, ITRPC7 and ITRPC3. Both ITRPC6 and ITRPC3 no longer exhibited VMI when the transfection ratio of the expression vectors encoding M1R and the TRPC channel was 1:1 (left and right panels in Fig. 5D; r = ∼1.0; red bars in Fig. 5F). However, as the amount of plasmid harbouring the M1R DNA was reduced, VMI recovered such that when the TRPC6:M1R transfection ratio was 1:0.3 and 1:0.1, r = 0.77 ± 0.10 and 0.45 ± 0.04, respectively (pink and light red bars in Fig. 5F left). In addition, the VMI of ITRPC7 persisted, even when the TRPC6:M1R transfection ratio was 1:1 (middle panel in Fig. 5D), though the magnitude of the inhibition was less than in cells expressing only the endogenous receptor (r = 0.49 ± 0.1, **P < 0.001; middle bars in Fig. 5F). That M1R-overexpressing cells show greatly enhanced CCh reactivity (more than 10-fold, data not shown) suggests that the loss or attenuation of VMI reflects the vigorous depletion of PIP2.
Confirmation of PIP2 depletion by overexpression of M1R
To confirm depletion of PIP2 in the cells overexpressing M1R, we used our bleed-through correction approach (Mori et al. 2011) to simultaneously record CCh-induced [Ca2+]i transients and FRET signals reflecting the interaction of PIP2 with its sensor protein carrying the PH domain of PLC (van der Wal et al. 2001; Falkenburger et al. 2009; Suh et al. 2010). We found that application of CCh to the M1R transfectants markedly reduced the FRET ratio (FR) (i.e. it depleted PIP2), and the time course of that response was well matched by the concomitant [Ca2+]i transients (red traces in Fig. 6A). In addition, the response was comparatively modest in cells expressing only endogenous muscarinic receptors (black traces in Fig. 6A). Interestingly, the extent of the reduction was smaller in cells co-expressing M1R with TRPC7 than in those expressing TRPC6 (†P < 0.05) or TRPC3 (Fig. 6C). These data indicate that depletion of PIP2 in cells co-expressing M1Rs and TRPC may partly explain why VMI persisted only for TRPC7 channels (Fig. 5D, middle panel).
Figure 6. Measurement of CCh-induced [Ca2+]i and PIP2 dynamics.
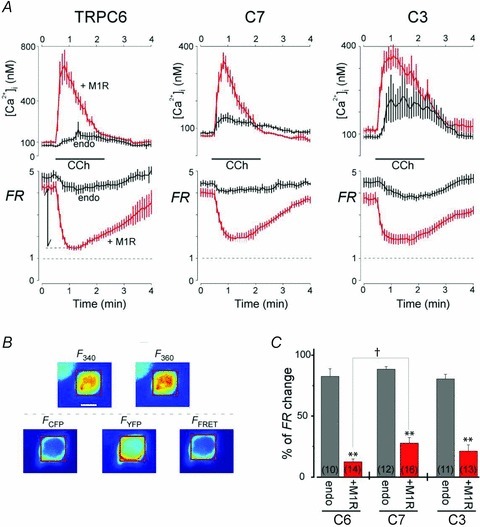
A, simultaneous cell imaging of CCh (100 μm)-induced changes in [Ca2+]i (upper) using Fura-2 and PI(4,5)P2 using FRET (FR, lower) of eCFP- and eYFP-PH domain proteins for TRPC6 (left), C7 (middle) and C3 (right). HEK293 cells were co-transfected with the indicated TRPC channels and PIP2-sensing proteins, with or without M1R (red and black traces, respectively). FR is the fractional increase of YFP emission due to FRET. Ca2+-dependent Fura-2 bleed-through was subtracted from the FRET signals (see Methods). B, representative images used for Fura-2 (upper panels; F340 and F360) and FRET measurements (bottom panels; FCFP, FYFP and FFRET). The scale bar in the F340 panel is 10 μm. Fura-2 and FR were determined using the same ROI (red square). C, summary of fold changes in FR from initial baseline to the minimum elicited by CCh stimulation. **P < 0.001 vs. no co-transfection of M1R with unpaired t test, †P < 0.05).
Detection of VMI was rare in vascular smooth muscle cells
To explore the effects of PIP2 depletion in a more physiological setting, we transfected a bicistronic IRES/GFP vector harbouring DrVSP into cells of the A7r5 aortic smooth muscle line. These cells are not contractile, but other phenotypic features of native vascular smooth muscle, such as vasopressin (AVP) receptors and L-type Ca2+ and TRPC6 channels, are preserved (Jung et al. 2002; Soboloff et al. 2005; Inoue et al. 2009). Interestingly, the majority of A7r5 cells co-expressing GFP and DrVSP did not exhibit VMI of AVP-evoked currents (12 of 16 cells, Fig. 7A left panel). However, currents elicited in 4 of the 16 cells exhibited VMI with slightly larger r (0.65 ± 0.03) and τ-recovery (2.8 ± 0.4 s, Fig. 7A right panel) values than were seen with ITRPC6 elicited by stimulation in HEK cells expressing only endogenous M1R. We therefore speculate that in A7r5 cells, the amount of PIP2 is inherently small and/or the coupling of AVP receptors to the TRPC6 channels is tight or similar to that in HEK cells overexpressing M1R. Consistent with this idea, the VMI of OAG-induced currents in A7r5 cells (r = 0.71 ± 0.04 and τ-rec = 2.4 ± 0.6 s) was often substantially larger than the VMI of AVP-induced currents (Fig. 7A bottom left and Fig. 7B), indicating PIP2 is well preserved in the plasma membrane.
Supplying diC8-PIP2 through the patch pipette slows TRPC3/C6/C7 current decay
The inhibitory effects of PIP2 depletion on macroscopic TRPC3/C6/C7 currents can also be tested by supplying PIP2 exogenously. As shown in Fig. 8A, application of diC8-PIP2 through the patch pipette had little impact on the activation phase of CCh-induced ITRPC6, ITRPC7 or ITRPC3 (summarized in Fig. 8B). On the other hand, the inactivation of all three TRPC currents was significantly slowed (Fig. 8A, +PIP2; black traces), and the effect was concentration dependent (summarized in Fig. 8C). Notably, ITRPC7, which exhibit the fastest kinetics upon receptor stimulation and are the most susceptible to VMI, were the most affected by diC8-PIP2. The slowing of ITRPC7 inactivation was particularly prominent in cells overexpressing M1R, with Δinac90-50% values of 2.3 ± 0.9, 13.8 ± 2.2 and 17.0 ± 4.8 s for 0 (control), 100 and 500 μm diC8-PIP2, respectively (middle left columns in Fig. 8C; **P < 0.001). The inactivation of ITRPC6 was somewhat less sensitive to diC8-PIP2, requiring a higher concentration (500 μm) to elicit overt effects (leftmost columns in Fig. 8C, Δinac90-50% = 32.2 ± 4.0 s, **P < 0.001). The inactivation of ITRPC3 was the least sensitive to diC8-PIP2 (middle right columns in Fig. 8C), which is analogous to its VMI showing the least sensitivity to PIP2. Similarly, the effects of diC8-PIP2 on the inactivation of AVP-induced TRPC6-like currents in A7r5 cells were also concentration dependent with graded prolongation of Δinac90-50% (rightmost columns in Fig. 8C and see also Table 2).
Figure 8. Effect of PIP2 in patch pipettes in M1R transfectants and A7r5 cells.
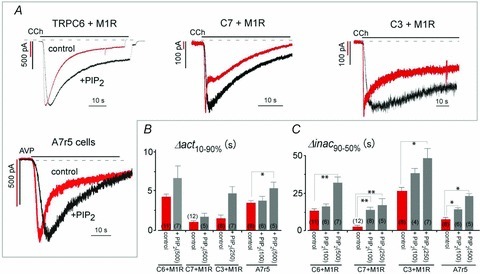
A, black traces (+PIP2) are typical examples recorded in M1R-expressing HEK293 cells with water-soluble PI(4,5)P2 in the patch pipettes (500 μm diC8-PIP2 for TRPC6, 250 μm for C3 and 100 μm for C7). All of the currents exhibited slower decay than currents recorded without diC8-PIP2 (red traces). Currents elicited by AVP (50 nm) in A7r5 cells exhibited slower activation and inactivation with 100 μm diC8-PIP2 in the pipette (left bottom). Membrane potentials were held at −50 mV. B and C, effect of diC8-PIP2 on channel activation (B) and inactivation (C) time courses. **P < 0.001, *P < 0.05 vs. control (i.e. no diC8-PIP2 in the patch pipette).
We also examined the sensitivity of VMI to diC8-PIP2 in cells co-transfected with TRPC3/C6/C7 channels and M1R (Supplemental Fig. S2). Intracellular application of diC8-PIP2 concentration-dependently restored the VMI of CCh-induced ITRPC6 and ITRPC7 (Supplemental Fig. S2A and B). At 500 μm diC8-PIP2, the magnitude of the VMI was close to that observed with stimulation of endogenous M1R (dotted lines in Supplemental Fig. S2B), and the time course of recovery was also comparable (τ-recovery of 2.5 ± 0.2 and 2.8 ± 0.6 s for ITRPC6 and ITRPC7, respectively). By contrast, the VMI of ITRPC3 in M1R-overexpressing cells was partially rescued by diC8-PIP2 (r = 0.85 ± 0.05). In this case, however, the time course of recovery from VMI (τ-rec of 0.86 ± 0.07 s) was still similar to that seen with endogenous muscarinic receptor stimulation (Supplemental Fig. S2B).
Overall, the functional effects of vigorous stimulation of PIP2–PLC–DAG signalling clearly demonstrate the importance of PIP2 in TRPC3/C6/C7 inactivation and established the existence of a sharing mechanism for the regulation of PIP2 for DAG-triggered activation of a TRPC channel subfamily.
Acute local depletion of PIP2 by CCh attenuates VMI
Finally, to understand the relationship between the macroscopic current inactivation and VMI elicited by depletion of PIP2, we simultaneously monitored ITRPC and PIP2 content using the same FRET-based sensor mentioned above. Figure 9 shows the synchronous detection of ITRPC6 and PIP2 in cells co-transfected with a PIP2-FRET sensor set (eCFP-PH and eYFP-PH), TRPC6 and DrVSP, with (panel A) or without (endo, panel B) M1R. Prior to CCh stimulation, depolarizing pulses from the holding potential to +60 to +140 mV (in +40 mV steps) elicited substantial FRET (L/S) reductions, confirming the functionality of DrVSP (Fig. 9A and B, lower panels). Moreover, the FRET reduction was observed throughout the cells, demonstrating the global functionality of DrVSP (Fig. 9C right panel). Soon after the application of CCh (its onset is set to zero; marked as a vertical dashed line in Fig. 9A and B), M1R-overexpressing cells rapidly developed a large inward current (Fig. 9A upper panel), and there was a corresponding gradual reduction in FRET (lower panel). With prolonged CCh stimulation, depolarizing pulses produced no obvious VMI and almost no further FRET reduction. Intriguingly, only a 10–30% reduction in PIP2 from baseline was necessary to attenuate VMI (i.e. the first or second depolarization after CCh stimulation in Fig. 9A). These observations suggest that vigorous PLC activation via potentiated M1R stimulation may facilitate local hydrolysis of PIP2 in the vicinity of the TRPC channels. By contrast, stimulation of endogenous muscarinic receptors caused only a slight reduction in basal PIP2 (a green dashed line in Fig. 9B lower panel), while the transient PIP2 reductions induced by DrVSP activation were clearly visible upon repetitive depolarization. These data could well account for the stability of the VMI observed here and in Figs 1A and 2A and D.
Figure 9. Simultaneous detection of TRPC6 currents (ITRPC6) and PIP2.
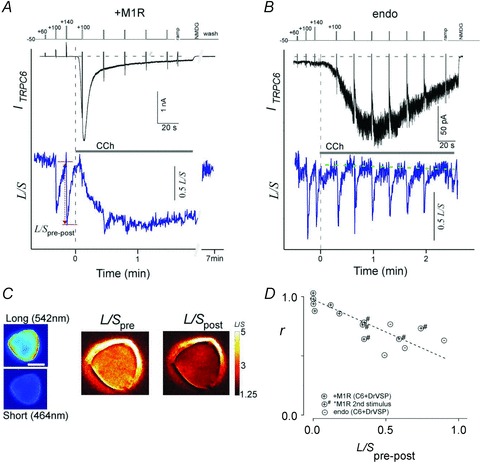
HEK293 cells were co-transfected with TRPC6 channels, PIP2-sensing eCFP/eYFP-PH proteins and DrVSP, with (A) and without (B) M1R. ITRPC6 (upper panel) and FRET between eCFP-PH and eYFP-PH domain (lower panel) signals were simultaneously collected. Depolarization pulses from the holding potential to +60 to +140 mV (40 mV steps) were delivered prior to agonist stimulation (1 μm CCh in A and 10 μm CCh in B). Robust VMI and FRET changes elicited by repetitive depolarizations were observed in cells expressing only endogenous M1R (B) but not in cells co-transfected with M1R (A). C, representative images of the long- and short-wavelength emission (left). FRET ratio (L/S) images were obtained using Flong and Fshort before (L/Spre) and after the depolarization (L/Spost) (right). Data correspond to changes in A (red arrow). D, VMI of ITRPC6 (r) plotted against FRET changes (L/Spre-post) elicited in the presence of CCh stimulation in cells, with (circles with +) and without (endo; circles with –) co-transfection of M1R. # indicates data from secondary CCh-evoked currents from M1R transfectants.
Figure 9D shows the correlation between the changes in FRET induced by DrVSP activation (L/Spre-post) and the concomitant VMI (r). The plotted data were collected during the first and second CCh stimulations of M1R-overexpressing cells and the first CCh stimulation of non-transfected HEK cells. Despite the differences in the experimental conditions, a good linear correlation was found between these two parameters. This correlation strongly supports the idea that reduction of PIP2 occurs in response to either PLC or DrVSP activation, which means the observed disappearance of VMI in M1R-overexpressing cells probably arises from the vigorous depletion of local PIP2 around the TRPC channels.
Discussion
By using DrVSP to rapidly dephosphorylate PI(4,5)P2 (PIP2), but not PI(3,4,5)P3, as well as GqPCR/PLC-mediated hydrolysis of PIP2, we were able to obtain compelling evidence that depletion of PIP2 differentially inhibits the DAG-evoked activation of TRPC3/C6/C7 channels. This conclusion was reinforced by the simultaneous monitoring of membrane PIP2 content and current using a FRET-based PIP2 sensor and the patch clamp technique. The results also suggest a unique regulatory system operating for the TRPC3/C6/C7 subfamily, whereby DAG, the major PLC product generated from PIP2, acts as the essential activator of these channels, and the catalytic depletion in membrane PIP2 through PLC stimulation exerts an opposing effect on channel activation.
Comparison with earlier studies on PIPs-mediated regulation of TRPC3/C6/C7 channels
Phosphoinositides (PIPs) function as spatially restricted membrane signalling molecules that regulate diverse cellular processes, including various types of ion channels (Hilgemann, 2007). In that regard, Kwon et al. demonstrated that the binding of several types of PIP, including PI(3,4,5)P3, to the C-terminal domain of the TRPC6 channel is coupled to the channel opening (Kwon et al. 2007). To date, no significant effect of VSP activation on PI(3,4,5)P3 has been detected; instead, we found critical regulation by PIP2 of a subfamily of TRPC channels. Lemonnier et al. 2008 reported that application of PIP2 to excised patch membranes caused robust activation of single TRPC6 and TRPC7 channels (order of responsiveness: TRPC7 > TRPC6), but not TRPC3 channels. This result is essentially consistent with our present findings. On the other hand, we observed no obvious direct inhibitory effects of PIP2 itself on the exogenous expression system or A7r5 cells in the whole-cell clamp mode. Application of PIP2 to excised patch membranes from rabbit mesenteric arterial myocytes reduced the activity of single TRPC6-like channels, whereas depletion of PIP2 using an antibody enhanced the channel's activity (Albert et al. 2008). These results suggest PIP2 acts to negatively regulate TRPC6 channel activity, which seemingly contradicts our finding that AVP stimulation depletes PIP2 in A7r5 cells. There has not yet been a quantitative analysis of basal PIP2 levels in native smooth muscle cells, but such an analysis will be needed to resolve this contradiction. In addition, many TRPC channel family members, including TRPC3/C6/C7 channels, contain an ankyrin repeat domain within their N-terminal regions. Given that this domain often acts to tether a protein to the cytoskeletal matrix or to compartmentalize it within a plasma membrane domain (Bennett & Baines, 2001), it is unclear whether TRPC channels in isolated membrane patches retain their native gating properties. In this regard, our present results may provide new physiological information essential for understanding the in situ role of PIP2 in the regulation of TRPC3/C6/C7 channel activity.
Our finding that DrVSP has broad specificity and acts toward a number of phosphoinositides, particularly PI(3,4,5)P3, is consistent with earlier reports on VSPs (Iwasaki et al. 2008). Likewise, there are no reported findings that are at variance with our observations of DrVSP-evoked depletion of PI(4,5)P2, its outcome (inhibition) or the rank order of TRPC channel susceptibility (C7 > C6 > C3).
Mechanistic considerations of PIP2 sensitivity
To gain deeper insight into the differential sensitivities of TRPC3/C6/C7 channels to PIP2 depletion, we estimated the relative equilibrium binding constants for the interaction of PIP2 with the TRPC3/C6/C7 channels, based on the following enzymatic scheme:
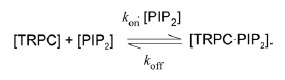 |
Here the 1/Khalf and 1/τ-rec values obtained for VMI of RHC- and OAG-induced TRPC currents are attributed to the off (koff) and on (kon×[PIP2]) rate constants, respectively. The equilibrium dissociation constant (Kd) for PIP2-TRPC3/C6/C7 binding (denoted as arrows) can then be calculated using the relationship: Kd = koff/kon×[PIP2]. The concentration of PIP2 is not known, but it seems reasonable to assume that the local concentration of PIP2 is comparable among TRPC3/C6/C7 channels, because replenishment of PIP2 by PI-5 kinase is independent of channel species (Suh et al. 2010). The relative Kd values calculated in this way suggest that the binding affinity of PIP2 for TRPC7 is 3 and 10 fold weaker than for TRPC6 and TRPC3, respectively. This order of PIP2-binding affinity could well explain the observed differential sensitivities of these channels to PIP2 depletion. For example, the binding and unbinding of PIP2 to TRPC3 appear to occur on comparable time scales (about 1.5 and 0.4 s, respectively). Thus, depletion of PIP2 by DrVSP would not be sufficient to detach all bound PIP2 from TRPC3. With respect to the VMI of TRPC7, by contrast, the unbinding of PIP2 is almost 30 times faster than the binding, so that local depletion of PIP2 could easily facilitate the dissociation of PIP2 from the TRPC7 channel. In apparent accordance with these considerations, the application of the ATP analogue AMP-PNP from the patch pipette, which would inhibit repletion of PIP2, slowed the recovery from VMI and increased its magnitude (r) to a greater degree in TRPC7 channels (by about 50%) than in TRPC3 or TRPC6 channels (by about 25%) (Supplemental Fig. S3).
As graphically illustrated in Fig. 10A, the more robust VMI of TRPC7 channel activity reflects the faster decay of the macroscopic currents induced by M1R stimulation. By contrast, the weaker VMI of TRPC3 channel activity clearly corresponds to the inactivation of its macroscopic current, as its decay time is 2 and 4 times longer than that of TRPC6 and C7, respectively. With regard to the depletion of PIP2, a clear relation between VMI and the macroscopic inactivation of TRPC channels is surprisingly demonstrated. Therefore, our observation that overexpression of M1R enormously accelerates their macroscopic inactivation kinetics attests to the importance of the relation between VMI and the macroscopic currents kinetics.
Figure 10. Schematic diagram for autonomic regulation.
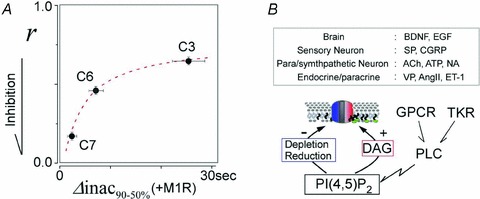
A, VMI in the currents recorded from the endogenous stimulation of HEK293 cells (r, Y-axis) plotted against the fast inactivation in the M1R-overexpressing HEK293 cells (Δinac90-50, X-axis) of the respective TRPC channels. Data obtained at 10 μm CCh-induced currents were used. A curvature relation was observed between VMI and the current decay (dotted line, curve fitting by double exponential formula). B, schematic diagram of the autonomic regulation of TRPC3/C6/C7 channel activity by PI(4,5)P2. Agonists released from various organs are listed in the upper box. Receptors for these agonists (GqPCR or TKR (tyrosine kinase receptor)) activate PLC, leading to DAG production and reduction or depletion of PI(4,5)P2. As PLC activity increases, bimodal signals from PI(4,5)P2 accelerates the channel opening and closing.
However, other factors have been demonstrated in the TRPC channel inactivation process such as changes in [Ca2+]i, phosphorylation by PKC or CaMKII, and association with IP3 (Venkatachalam et al. 2003; Shi et al. 2004; Bousquet et al. 2010; Ju et al. 2010). It would be interesting to see how those factors affect TRPC channel regulation by PIP2.
Physiological impact of variation in PIP2 sensitivity
Recent studies have suggested that TRPC3/C6/C7 channels may be involved in the pathophysiology of hypertensive disorders in human and animal models. For example, TRPC3 expression is up-regulated in spontaneously hypertensive rats (SHRs), and TRPC6-deficient mice exhibit unexpectedly high blood pressure (Dietrich et al. 2005; Liu et al. 2005; Nishida & Kurose, 2008; Thilo et al. 2008). Perhaps the relatively modest inhibitory effect of PIP2 depletion on TRPC3 activity accounts for the induction of vascular hypersensitivity to neurotransmitters released from sympathetic nerves. On the other hand, while there is no clear evidence yet, up-regulation of TRPC7 could protect against hypertension through its strong negative regulation by PIP2 and/or its physical or functional coupling with TRPC3 or TRPC6 channels. In fact, functional hetero-oligomeric assembly TRPC7 with TRPC3 and TRPC6 has been identified in various cell types, including vascular smooth muscle cells (Hofmann et al. 2002; Strubing et al. 2003; Wu et al. 2004; Maruyama et al. 2006; Poteser et al. 2006; Liao et al. 2008). It is also noteworthy that some mutations found in focal segmental glomerulosclerosis patients correspond to the PIP2 binding site in the PH-like domain of the TRPC6 channel (Winn et al. 2005; Dryer & Reiser, 2010), though the TRP box (proximal to transmembrane) and distal portion of the C-terminal domain have also been proposed as PIP association sites in TRPC channels (Rohacs et al. 2005; van Rossum et al. 2005; Kwon et al. 2007). We have not yet identified the PIP2 binding site on the TRPC channel that would enable it to sense the presence or depletion of PIP2. The identification of that site may clarify the pathophysiological significance of PIP2 depletion to TRPC channel activity.
Autonomic regulation by PI(4,5)P2–PLC–DAG signalling
In voltage-gated channels, membrane voltage often controls both the activation and inactivation kinetics. A typical manifestation of this simple regulatory system is that voltage-gated sodium channels are able to produce ultratransient ionic flows, which are physiologically critical for generating the action potential and its refractory period (Hodgkin & Huxley, 1952; Bezanilla & Armstrong, 1977). The negative feedback regulation of Ca2+-permeable channels by the entering Ca2+ is also essential for preventing excessive Ca2+ influx through voltage-gated Ca2+ channels (Brehm & Eckert, 1978) other than TRPC channels (Scott et al. 1997; Minke & Parnas, 2006). In addition, acute desensitization caused by the agonist is often observed in ligand-gated ion channels, such as nicotinic acetylcholine receptors (Katz & Thesleff, 1957), other cys-loop proteins (Swope et al. 1995) and TRPV1 channels (Tominaga & Tominaga, 2005). Those regulatory mechanisms are simple but robust and result in self-limiting systems able to mediate transient electrical signals in a highly efficient manner.
TRPC3/C6/C7 channels are activated by a variety of neurohormonal factors (Fig. 10B). For that reason, these channels can be categorized as ligand (DAG)-triggered; however, their negative regulation by PIP2 depletion differs from desensitization in that it is agonist independent and more quickly reversible. The depletion of PLC substrate (PIP2) needed to produce the activator (DAG) would not only slow the rate of activator production, but the depletion itself would vitally inhibit channel opening in a more direct fashion. Thus PIP2–PLC–DAG signalling generates bimodal effects on TRPC3/C6/C7 channels such that the activation and inactivation of the channels are closely related to prevent excessive ionic influx through these channels. Factors including the agonist concentration, quantity of PIP2, and microstructure of the lipid domain and signal proteins would have a dynamic impact on TRPC channel opening. Furthermore, the different sensitivities to PIP2 and rates of recovery from PIP2 depletion of the TRPC isoforms may be of considerable physiological significance, as they would enable diversification of Ca2+ influx through variation in the cellular distribution of the channel isoforms.
Acknowledgments
We thank all those who provided generous gifts of DNA constructs (Methods). We thank Dr David T. Yue and members of our laboratory for critical comments (J. Ichikawa, H. Lin, Y. Duan, Y. Hu). This work was supported by a Grant-in-aid for Young Scientists (M.X.M.) from the Japan Society for the Promotion of Sciences and from the Naito Foundation (M.X.M.). The authors have no conflicts of interest to disclose.
Glossary
Abbreviations
- CCh
carbachol
- CFP
cyan fluorescent protein
- DrVSP
Danio rerio voltage-sensing phosphatase
- DAG
diacylglycerol
- FR
FRET ratios
- FRET
fluorescence resonance energy transfer
- M1R
muscarinic type I receptor
- OAG
1-oleolyl-2-acetyl-sn-glycerol
- PIPs
phosphoinositides
- PI(4,5)P2 or PIP2
phosphatidylinositol 4,5-bisphosphate
- PLC
phospholipase C
- RHC
- TRPC
transient receptor potential canonical
- VMI
DrVSP-mediated inhibition
- YFP
yellow fluorescent protein
Author contributions
Y.I. and M.X.M. conceived the study. Y.I., K.I. and M.X.M. performed the experiments and analysed the data. Y.O. and R.I. provided guidance and support throughout. All authors contributed to writing the paper.
References
- Albert AP, Saleh SN, Large WA. Inhibition of native TRPC6 channel activity by phosphatidylinositol 4,5-bisphosphate in mesenteric artery myocytes. J Physiol. 2008;586:3087–3095. doi: 10.1113/jphysiol.2008.153676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla T, Szentpetery Z, Kim YJ. Phosphoinositide signaling: new tools and insights. Physiology (Bethesda) 2009;24:231–244. doi: 10.1152/physiol.00014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker EB, Oliver PL, Glitsch MD, Banks GT, Achilli F, Hardy A, Nolan PM, Fisher EM, Davies KE. A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc Natl Acad Sci U S A. 2009;106:6706–6711. doi: 10.1073/pnas.0810599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Chaim Y, Chanda B, Dascal N, Bezanilla F, Parnas I, Parnas H. Movement of ‘gating charge’ is coupled to ligand binding in a G-protein-coupled receptor. Nature. 2006;444:106–109. doi: 10.1038/nature05259. [DOI] [PubMed] [Google Scholar]
- Ben-Mabrouk F, Tryba AK. Substance P modulation of TRPC3/7 channels improves respiratory rhythm regularity and ICAN-dependent pacemaker activity. Eur J Neurosci. 2010;31:1219–1232. doi: 10.1111/j.1460-9568.2010.07156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett V, Baines AJ. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev. 2001;81:1353–1392. doi: 10.1152/physrev.2001.81.3.1353. [DOI] [PubMed] [Google Scholar]
- Bezanilla F, Armstrong CM. Inactivation of the sodium channel. I. Sodium current experiments. J Gen Physiol. 1977;70:549–566. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousquet SM, Monet M, Boulay G. Protein kinase C-dependent phosphorylation of transient receptor potential canonical 6 (TRPC6) on serine 448 causes channel inhibition. J Biol Chem. 2010;285:40534–40543. doi: 10.1074/jbc.M110.160051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt BL, Kimes BW, Klier FG. Development of a clonal myogenic cell line with unusual biochemical properties. J Cell Physiol. 1976;88:255–275. doi: 10.1002/jcp.1040880302. [DOI] [PubMed] [Google Scholar]
- Brehm P, Eckert R. Calcium entry leads to inactivation of calcium channel in Paramecium. Science. 1978;202:1203–1206. doi: 10.1126/science.103199. [DOI] [PubMed] [Google Scholar]
- Dietrich A, Kalwa H, Gudermann T. TRPC channels in vascular cell function. Thromb Haemost. 2010;103:262–270. doi: 10.1160/TH09-08-0517. [DOI] [PubMed] [Google Scholar]
- Dietrich AMederos, Schnitzler M, Gollasch M, Gross V, Storch U, Dubrovska G, Obst M, Yildirim E, Salanova B, Kalwa H, Essin K, Pinkenburg O, Luft FC, Gudermann T, Birnbaumer L. Increased vascular smooth muscle contractility in TRPC6−/− mice. Mol Cell Biol. 2005;25:6980–6989. doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryer SE, Reiser J. TRPC6 channels and their binding partners in podocytes: role in glomerular filtration and pathophysiology. Am J Physiol Renal Physiol. 2010;299:F689–F701. doi: 10.1152/ajprenal.00298.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder P, Groschner K. TRPC3/6/7: Topical aspects of biophysics and pathophysiology. Channels (Austin) 2008;2:94–99. doi: 10.4161/chan.2.2.6015. [DOI] [PubMed] [Google Scholar]
- Erickson MG, Liang H, Mori MX, Yue DT. FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron. 2003;39:97–107. doi: 10.1016/s0896-6273(03)00395-7. [DOI] [PubMed] [Google Scholar]
- Falkenburger BH, Jensen JB, Hille B. Kinetics of PIP2 metabolism and KCNQ2/3 channel regulation studied with a voltage-sensitive phosphatase in living cells. J Gen Physiol. 2009;135:99–114. doi: 10.1085/jgp.200910345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie RC. TRP channels and lipids: from Drosophila to mammalian physiology. J Physiol. 2007;578:9–24. doi: 10.1113/jphysiol.2006.118372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann DW. Local PIP2 signals: when, where, and how? Pflugers Arch. 2007;455:55–67. doi: 10.1007/s00424-007-0280-9. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. The dual effect of membrane potential on sodium conductance in the giant axon of Loligo. J Physiol. 1952;116:497–506. doi: 10.1113/jphysiol.1952.sp004719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci U S A. 2002;99:7461–7466. doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain MI, Iwasaki H, Okochi Y, Chahine M, Higashijima S, Nagayama K, Okamura Y. Enzyme domain affects the movement of the voltage sensor in ascidian and zebrafish voltage-sensing phosphatases. J Biol Chem. 2008;283:18248–18259. doi: 10.1074/jbc.M706184200. [DOI] [PubMed] [Google Scholar]
- Huang J, Liu CH, Hughes SA, Postma M, Schwiening CJ, Hardie RC. Activation of TRP channels by protons and phosphoinositide depletion in Drosophila photoreceptors. Curr Biol. 2009;20:189–197. doi: 10.1016/j.cub.2009.12.019. [DOI] [PubMed] [Google Scholar]
- Inoue R, Jensen LJ, Jian Z, Shi J, Hai L, Lurie AI, Henriksen FH, Salomonsson M, Morita H, Kawarabayashi Y, Mori M, Mori Y, Ito Y. Synergistic activation of vascular TRPC6 channel by receptor and mechanical stimulation via phospholipase C/diacylglycerol and phospholipase A2/ω-hydroxylase/20-HETE pathways. Circ Res. 2009;104:1399–1409. doi: 10.1161/CIRCRESAHA.108.193227. [DOI] [PubMed] [Google Scholar]
- Inoue R, Jensen LJ, Shi J, Morita H, Nishida M, Honda A, Ito Y. Transient receptor potential channels in cardiovascular function and disease. Circ Res. 2006;99:119–131. doi: 10.1161/01.RES.0000233356.10630.8a. [DOI] [PubMed] [Google Scholar]
- Iwasaki H, Murata Y, Kim Y, Hossain MI, Worby CA, Dixon JE, McCormack T, Sasaki T, Okamura Y. A voltage-sensing phosphatase, Ci-VSP, which shares sequence identity with PTEN, dephosphorylates phosphatidylinositol 4,5-bisphosphate. Proc Natl Acad Sci U S A. 2008;105:7970–7975. doi: 10.1073/pnas.0803936105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju M, Shi J, Saleh SN, Albert AP, Large WA. Ins(1,4,5)P3 interacts with PIP2 to regulate activation of TRPC6/C7 channels by diacylglycerol in native vascular myocytes. J Physiol. 2010;588:1419–1433. doi: 10.1113/jphysiol.2009.185256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Strotmann R, Schultz G, Plant TD. TRPC6 is a candidate channel involved in receptor-stimulated cation currents in A7r5 smooth muscle cells. Am J Physiol Cell Physiol. 2002;282:C347–C359. doi: 10.1152/ajpcell.00283.2001. [DOI] [PubMed] [Google Scholar]
- Katz B, Thesleffs A study of the desensitization produced by acetylcholine at the motor end-plate. J Physiol. 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurachi Y, Ishii M. Cell signal control of the G protein-gated potassium channel and its subcellular localization. J Physiol. 2004;554:285–294. doi: 10.1113/jphysiol.2003.048439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y, Hofmann T, Montell C. Integration of phosphoinositide- and calmodulin-mediated regulation of TRPC6. Mol Cell. 2007;25:491–503. doi: 10.1016/j.molcel.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemonnier L, Trebak M, Putney JW., Jr Complex regulation of the TRPC3, 6 and 7 channel subfamily by diacylglycerol and phosphatidylinositol-4,5-bisphosphate. Cell Calcium. 2008;43:506–514. doi: 10.1016/j.ceca.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HS, Xu XZ, Montell C. Activation of a TRPC3-dependent cation current through the neurotrophin BDNF. Neuron. 1999;24:261–273. doi: 10.1016/s0896-6273(00)80838-7. [DOI] [PubMed] [Google Scholar]
- Li Y, Calfa G, Inoue T, Amaral MD, Pozzo-Miller L. Activity-dependent release of endogenous BDNF from mossy fibers evokes a TRPC3 current and Ca2+ elevations in CA3 pyramidal neurons. J Neurophysiol. 2010;103:2846–2856. doi: 10.1152/jn.01140.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Erxleben C, Abramowitz J, Flockerzi V, Zhu MX, Armstrong DL, Birnbaumer L. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc Natl Acad Sci U S A. 2008;105:2895–2900. doi: 10.1073/pnas.0712288105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Scholze A, Zhu Z, Kreutz R, Wehland-von-Trebra M, Zidek W, Tepel M. Increased transient receptor potential channel TRPC3 expression in spontaneously hypertensive rats. Am J Hypertens. 2005;18:1503–1507. doi: 10.1016/j.amjhyper.2005.05.033. [DOI] [PubMed] [Google Scholar]
- Maruyama Y, Nakanishi Y, Walsh EJ, Wilson DP, Welsh DG, Cole WC. Heteromultimeric TRPC6-TRPC7 channels contribute to arginine vasopressin-induced cation current of A7r5 vascular smooth muscle cells. Circ Res. 2006;98:1520–1527. doi: 10.1161/01.RES.0000226495.34949.28. [DOI] [PubMed] [Google Scholar]
- Minke B, Parnas M. Insights on TRP channels from in vivo studies in Drosophila. Annu Rev Physiol. 2006;68:649–684. doi: 10.1146/annurev.physiol.68.040204.100939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran MM, McAlexander MA, Biro T, Szallasi A. Transient receptor potential channels as therapeutic targets. Nat Rev Drug Discov. 2011;10:601–620. doi: 10.1038/nrd3456. [DOI] [PubMed] [Google Scholar]
- Mori MX, Imai Y, Itsuki K, Inoue R. Quantitative measurement of Ca2+-dependent calmodulin-target binding by Fura-2 and CFP and YFP FRET imaging in living cells. Biochemistry. 2011;50:4685–4696. doi: 10.1021/bi200287x. [DOI] [PubMed] [Google Scholar]
- Mundell SJ, Matharu AL, Pula G, Holman D, Roberts PJ, Kelly E. Metabotropic glutamate receptor 1 internalization induced by muscarinic acetylcholine receptor activation: differential dependency of internalization of splice variants on nonvisual arrestins. Mol Pharmacol. 2002;61:1114–1123. doi: 10.1124/mol.61.5.1114. [DOI] [PubMed] [Google Scholar]
- Nakanishi S, Catt KJ, Balla T. A wortmannin-sensitive phosphatidylinositol 4-kinase that regulates hormone-sensitive pools of inositolphospholipids. Proc Natl Acad Sci U S A. 1995;92:5317–5321. doi: 10.1073/pnas.92.12.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida M, Kurose H. Roles of TRP channels in the development of cardiac hypertrophy. Naunyn Schmiedebergs Arch Pharmacol. 2008;378:395–406. doi: 10.1007/s00210-008-0321-8. [DOI] [PubMed] [Google Scholar]
- Okada T, Inoue R, Yamazaki K, Maeda A, Kurosaki T, Yamakuni T, Tanaka I, Shimizu S, Ikenaka K, Imoto K, Mori Y. Molecular and functional characterization of a novel mouse transient receptor potential protein homologue TRP7. Ca2+-permeable cation channel that is constitutively activated and enhanced by stimulation of G protein-coupled receptor. J Biol Chem. 1999;274:27359–27370. doi: 10.1074/jbc.274.39.27359. [DOI] [PubMed] [Google Scholar]
- Okamura Y, Murata Y, Iwasaki H. Voltage-sensing phosphatase: actions and potentials. J Physiol. 2009;587:513–520. doi: 10.1113/jphysiol.2008.163097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poteser M, Graziani A, Rosker C, Eder P, Derler I, Kahr H, Zhu MX, Romanin C, Groschner K. TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J Biol Chem. 2006;281:13588–13595. doi: 10.1074/jbc.M512205200. [DOI] [PubMed] [Google Scholar]
- Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- Rohacs T, Lopes CM, Michailidis I, Logothetis DE. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat Neurosci. 2005;8:626–634. doi: 10.1038/nn1451. [DOI] [PubMed] [Google Scholar]
- Scott K, Sun Y, Beckingham K, Zuker CS. Calmodulin regulation of Drosophila light-activated channels and receptor function mediates termination of the light response in vivo. Cell. 1997;91:375–383. doi: 10.1016/s0092-8674(00)80421-3. [DOI] [PubMed] [Google Scholar]
- Shi J, Mori E, Mori Y, Mori M, Li J, Ito Y, Inoue R. Multiple regulation by calcium of murine homologues of transient receptor potential proteins TRPC6 and TRPC7 expressed in HEK293 cells. J Physiol. 2004;561:415–432. doi: 10.1113/jphysiol.2004.075051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboloff J, Spassova M, Xu W, He LP, Cuesta N, Gill DL. Role of endogenous TRPC6 channels in Ca2+ signal generation in A7r5 smooth muscle cells. J Biol Chem. 2005;280:39786–39794. doi: 10.1074/jbc.M506064200. [DOI] [PubMed] [Google Scholar]
- Strubing C, Krapivinsky G, Krapivinsky L, Clapham DE. Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J Biol Chem. 2003;278:39014–39019. doi: 10.1074/jbc.M306705200. [DOI] [PubMed] [Google Scholar]
- Suh BC, Inoue T, Meyer T, Hille B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science. 2006;314:1454–1457. doi: 10.1126/science.1131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Leal K, Hille B. Modulation of high-voltage activated Ca2+ channels by membrane phosphatidylinositol 4,5-bisphosphate. Neuron. 2010;67:224–238. doi: 10.1016/j.neuron.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swope SL, Qu Z, Huganir RL. Phosphorylation of the nicotinic acetylcholine receptor by protein tyrosine kinases. Ann N Y Acad Sci. 1995;757:197–214. doi: 10.1111/j.1749-6632.1995.tb17476.x. [DOI] [PubMed] [Google Scholar]
- Thilo F, Scholze A, Liu DY, Zidek W, Tepel M. Association of transient receptor potential canonical type 3 (TRPC3) channel transcripts with proinflammatory cytokines. Arch Biochem Biophys. 2008;471:57–62. doi: 10.1016/j.abb.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Tominaga T. Structure and function of TRPV1. Pflugers Arch. 2005;451:143–150. doi: 10.1007/s00424-005-1457-8. [DOI] [PubMed] [Google Scholar]
- Wal vanderJ, Habets R, Varnai P, Balla T, Jalink K. Monitoring agonist-induced phospholipase C activation in live cells by fluorescence resonance energy transfer. J Biol Chem. 2001;276:15337–15344. doi: 10.1074/jbc.M007194200. [DOI] [PubMed] [Google Scholar]
- Rossum vanDB, Patterson RL, Sharma S, Barrow RK, Kornberg M, Gill DL, Snyder SH. Phospholipase Cγ1 controls surface expression of TRPC3 through an intermolecular PH domain. Nature. 2005;434:99–104. doi: 10.1038/nature03340. [DOI] [PubMed] [Google Scholar]
- Venkatachalam K, Zheng F, Gill DL. Regulation of canonical transient receptor potential (TRPC) channel function by diacylglycerol and protein kinase C. J Biol Chem. 2003;278:29031–29040. doi: 10.1074/jbc.M302751200. [DOI] [PubMed] [Google Scholar]
- Voets T, Nilius B. Modulation of TRPs by PIPs. J Physiol. 2007;582:939–944. doi: 10.1113/jphysiol.2007.132522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- Wu X, Zagranichnaya TK, Gurda GT, Eves EM, Villereal ML. A TRPC1/TRPC3-mediated increase in store-operated calcium entry is required for differentiation of H19-7 hippocampal neuronal cells. J Biol Chem. 2004;279:43392–43402. doi: 10.1074/jbc.M408959200. [DOI] [PubMed] [Google Scholar]
- Zhou J, Du W, Zhou K, Tai Y, Yao H, Jia Y, Ding Y, Wang Y. Critical role of TRPC6 channels in the formation of excitatory synapses. Nat Neurosci. 2008;11:741–743. doi: 10.1038/nn.2127. [DOI] [PubMed] [Google Scholar]