

Abstract
Intermedin (IMD) protects rodent heart and vasculature from oxidative stress and ischaemia. Less is known about distribution of IMD and its receptors and the potential for similar protection in man. Expression of IMD and receptor components were studied in human aortic endothelium cells (HAECs), smooth muscle cells (HASMCs), cardiac microvascular endothelium cells (HMVECs) and fibroblasts (v-HCFs). Receptor subtype involvement in protection by IMD against injury by hydrogen peroxide (H2O2, 1 mmol l−1) and simulated ischaemia and reperfusion were investigated using receptor component-specific siRNAs. IMD and CRLR, RAMP1, RAMP2 and RAMP3 were expressed in all cell types. When cells were treated with 1 nmol l−1 IMD during exposure to 1 mmol l−1 H2O2 for 4 h, viability was greater vs. H2O2 alone (P < 0.05 for all cell types). Viabilities under 6 h simulated ischaemia differed (P < 0.05) in the absence and presence of 1 nmol l−1 IMD: HAECs 63% and 85%; HMVECs 51% and 68%; v-HCFs 42% and 96%. IMD 1 nmol l−1 present throughout ischaemia (3 h) and reperfusion (1 h) attenuated injury (P < 0.05): viabilities were 95%, 74% and 82% for HAECs, HMVECs and v-HCFs, respectively, relative to those in the absence of IMD (62%, 35%, 32%, respectively). When IMD 1 nmol l−1 was present during reperfusion only, protection was still evident (P < 0.05, 79%, 55%, 48%, respectively). Cytoskeletal disruption and protein carbonyl formation followed similar patterns. Pre-treatment (4 days) of HAECs with CRLR or RAMP2, but not RAMP1 or RAMP3, siRNAs abolished protection by IMD (1 nmol l−1) against ischaemia–reperfusion injury. IMD protects human vascular and cardiac non-vascular cells from oxidative stress and ischaemia–reperfusion, predominantly via AM1 receptors.
Key points
Coronary artery disease occurs when fatty deposits cause obstruction to blood flow in the coronary arteries, reducing the supply of blood to the heart. This can damage the heart muscle (heart attack).
In this study, a small protein named intermedin is shown to be present in cells from the human heart and blood vessels.
Intermedin, acting on a specific type of receptor protein shown to be present on the surface of these cells, is found to protect against damage occurring during experiments conducted in human cardiac and vascular cells in culture under conditions designed to simulate initial obstruction and subsequent restoration of blood flow, respectively.
These results suggest that administration of intermedin might provide a novel therapeutic strategy to minimise damage to heart muscle following a heart attack.
Introduction
Intermedin (adrenomedullin-2, IMD), a member of the calcitonin/calcitonin gene-related peptide (CGRP) family, is an emerging counter-regulatory peptide in the cardiovascular and renal systems (Bell & McDermott, 2008). Cleavage sites demarcated by paired basic amino acids at various positions within the mammalian prepro-IMD precursor yield a series of peptides of varying length, namely IMD1-53, IMD1-47 and IMD8-47 (Roh et al. 2004; Yang et al. 2005). IMD has similar but distinct vasodilator and hypotensive actions to adrenomedullin (AM) and CGRP (Takei et al. 2004; Fujisawa et al. 2007; Bell & McDermott, 2008; Jolly et al. 2009). IMD augments cardiac contractility (Dong et al. 2006), prevents calcification of vascular smooth muscle (Cai et al. 2010), inhibits collagen synthesis, attenuates proliferation of cardiac fibroblasts (Yang et al. 2009), and attenuates cardiomyocyte hypertrophy (Pan et al. 2005; Zhao et al. 2006; Bell & McDermott, 2008; Yang et al. 2010). IMD exerts its physiological effects mainly through the common calcitonin receptor-like receptor (CRLR)–receptor activity-modifying protein (RAMP) receptor system shared with CGRP and AM, which gives rise to CGRP1, AM1 and AM2 receptor subtypes at which IMD interacts non-selectively (Bell and McDermott, 2008), although the existence of additional receptors specific for IMD has been suggested (Taylor et al. 2006; Owji et al. 2008; Zeng et al. 2009).
Surplus generation of reactive oxygen species (ROS) such as superoxide and hydrogen peroxide, termed oxidative stress, has been implicated in the pathophysiology of hypertension, atherosclerosis, myocardial ischaemia–reperfusion injury and cardiac remodelling (Cai, 2005; Pearson et al. 2009). IMD is protective in vitro and in vivo against endothelial damage induced by oxidative stress (Chen et al. 2006; Hagiwara et al. 2008; Song et al. 2009). Adenoviral vector-mediated delivery of the IMD gene promotes angiogenesis and improved blood flow in a rodent model of chronic hindlimb ischaemia (Smith et al. 2009). IMD attenuates myocardial injury in a rodent model of β-adrenergic drive (Jia et al. 2006); this could be attributed to an indirect coronary vasodilator effect of the peptide (Pan et al. 2005). Similarly IMD reduces ischaemia–reperfusion injury acutely in the isolated perfused rat heart (Yang et al. 2005). Receptor subtype involvement in the actions of IMD was not determined in these studies. IMD is expressed less abundantly and in a more restricted fashion in the rodent cardiovascular system than AM (Bell & McDermott, 2008). Although upregulation of myocardial expression of IMD and each of its receptor components was demonstrated in the model of chronic β-adrenergic drive (Jia et al. 2006) and in a model of long term nitric oxide deficiency (Zhao et al. 2006; Bell et al. 2007, 2008), it is not clear that such upregulation would occur in acute ischaemic insult and there is lack of consensus regarding the magnitude of increase for each receptor component. IMD is protective during reperfusion in a mouse model of ischaemia induced by left anterior descending coronary artery ligation (mechanical intimal-to-medial injury); receptor subtype involvement was not determined, however. RAMP3 expression was upregulated earlier during reperfusion that that of RAMP2 while RAMP1 expression remained unchanged (Zhang et al. 2009).
In humans, CRLR is expressed in endothelium of blood vessels including large and small arteries, veins and capillaries and in myocardium and endocardium. Differential RAMP expression has been detected in endothelium of human coronary and cerebral arteries, coronary artery vascular smooth muscle and myocardial trabeculae (Kamitani et al. 1999; Frayon et al. 2000; Saetrum et al. 2000; Sams et al. 2000; Hagner et al. 2002; Oliver et al. 2002; Jansen-Olesen et al. 2003; Nagoshi et al. 2004; Nikitenko et al. 2006). In contrast, information on IMD expression in human heart and vasculature is limited. IMD is found in vascular smooth muscle of human skin (Kindt et al. 2007), and, in patients without evident cardiac disease post mortem, in left ventricular cardiomyocytes, pericardial adipocytes, endothelial cells of pericardial veins and vascular smooth muscle cells of coronary arteries (Morimoto et al. 2007), and is present in plasma of healthy volunteers (Morimoto et al. 2007), albeit less abundantly than AM (Bell & McDermott, 2008), suggestive of a more restricted distribution within the cardiovascular system.
It is not clear how ischaemic studies in rodents translate to humans since the methods used in rodent models could not be employed in human studies and the extracellular environment and temporal aspect differ somewhat from the clinical manifestation of acute coronary syndrome in humans; differences in upregulation of the receptor components may be species dependent, may be related to duration of insult, and may also vary by cell type within the tissue beds investigated. IMD is expressed, less abundantly than AM, in endothelial cells from human aorta and umbilical vein, but in contrast to AM it is up-regulated markedly due to metabolic stressors, including hydrogen peroxide, and is protective of aortic endothelial cells against oxidative injury and apoptosis (Pearson et al. 2009). Further research in isolated human cells is required to provide the justification for clinical trials. The response to ischaemic and/or reperfusion injury and the potential for protection by IMD has not been investigated in human cells.
The aim of the present study was first to examine the expression of IMD at both the mRNA and the protein level relative to that of AM and components of the CRLR/RAMP receptor system in a range of adult human cells in culture; specifically to compare expression patterns of these peptides and their receptor system in macrovascular (human aorta) and microvascular (coronary microvasculature) endothelial cells and in aortic vascular smooth muscle cells and non-vascular cells from myocardium, namely ventricular cardiac fibroblasts. Secondly, the direct cardioprotective effects of exogenous IMD were examined in response to oxidative stress, induced in cultured cells by (1) administration of hydrogen peroxide; (2) simulated ischaemia–reperfusion injury using a protocol designed to simulate the ischaemic environment, namely impaired supply of oxygen and glucose, markedly acidic pH and increased extracellular K+ ion (Van den Hoek et al. 2000; Townsend et al. 2004; McDermott et al. 2007). In the latter, the effects of IMD administration given throughout the ischaemia and reperfusion period were compared with those of giving the peptide during the reperfusion phase only (investigating a potential benefit for IMD as a post-myocardial infarction adjunct therapy). Finally, evidence for involvement of CRLR and the various RAMPs in the protective action of IMD was investigated using silencing RNA targeting specific CRLR/RAMP components.
Methods
Cell culture
Adult human cardiac microvascular endothelial cells (HMVECs) and human aortic endothelium cells (HAECs) were obtained from PromoCell, Heidelberg, Germany; adult human aortic smooth muscle cells (HASMCs) were, as previously described, primary explant-derived from transplant donors with written consent and ethical approval (Campbell et al. 2008); and human ventricular cardiac fibroblast (v-HCFs) (ScienCell Research Laboratories, San Diego CA, USA/TCS Cellworks, Buckingham, UK) were a gift from University College, Dublin. Cells were cultured in cell type-specific media supplemented with 200 μmol l−1 glutamine dimer, 2 μg ml−1 amphotericin-B, 60 μg ml−1 penicillin-G and 100 μg ml−1 streptomycin sulphate (Invitrogen, UK). Cells were grown in T25 flasks (Iwaki, Japan), or on sterile glass coverslips placed in 12-well plates, at 37°C in 5% CO2 in a humidified incubator; medium was replaced every 3–4 days. Cells were subcultured every 10–14 days (at ∼80–90% confluence). Experiments were conducted at passage 4–8, at which time cells demonstrated good growth rates without loss of phenotype.
Hydrogen peroxide-induced oxidative stress
Cells were incubated in cell type-specific media as above in 5% CO2 at 37°C in the presence of hydrogen peroxide (≤10 mmol l−1, Sigma-Aldrich, UK) with or without IMD1-47 (≤100 nmol l−1, Bachem, St Helens, UK) for 4 h.
Ischaemia and ischaemia–reperfusion protocols
The medium used to simulate ischaemia was prepared freshly for each experiment and consisted of M199 plus Hepes (free acid) 12.5 mmol l−1, KCl 4 mmol l−1, 2-deoxyglucose 20 mmol l−1 (Fluka Biochemica, Switzerland; hinders glycolysis) and penicillin/streptomycin (2% v/v), gassed for 2 h with 100% N2 and maintained at pH 5.8. (Nitrogen was gassed through a humidified sealed container inside an adapted incubator and emptied through a water-seal trap into the incubator.) Normal medium consisted of M199, Hepes (free acid; 12.5 mmol l−1), and penicillin/streptomycin (2%, v/v), placed in an air incubator and maintained at pH 7.4. To examine the concentration dependence of the protective effect of IMD against ischaemic injury, cells were incubated in ischaemic medium in 100% N2 at 37°C for 4 h with or without IMD (≤100 nmol l−1); cells incubated in normal medium in an air incubator with or without IMD (≤100 nmol l−1) served as time-matched controls. To investigate the protective effects of IMD in simulated ischaemia–reperfusion, cells were incubated for 3 h in ischaemic medium in 100% N2 at 37°C; cells were then returned to normoxic conditions (normal medium in air incubator) for 1 h; cells which were maintained under ischaemic conditions or normoxic conditions throughout for 4 h served for comparison. The influence of IMD (1 nmol l−1), present throughout 3 h ischaemia and 1 h reperfusion, or during 1 h reperfusion only, was compared with that in cells incubated under identical conditions in the absence of IMD.
siRNA protocol
For each transfection, cells were incubated with siRNA duplex for the receptor (Sc-43705) or RAMP1–3 (Sc-40894, Sc-36378 and Sc-40896 respectively) together with transfection reagent in transfection medium (all Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 6 h. Cells then received normal growth medium containing twice as much serum and antibiotics as usual (Campbell et al. 2008). Cells were incubated for a further 18–24 h before changing to normal 1× growth medium. After 72 h under these conditions, cells were used in ischaemia–reperfusion protocols as described above.
Cell viability
Following trypsinisation, neutralisation with fetal bovine serum (20% v/v in normal medium) and centrifugation (200 g) of the resultant suspension, cells were re-suspended in phosphate-buffered saline (PBS, 100 μl) containing 0.2% Trypan blue (Sigma) by gentle pipetting and introduced at the edge of a haemocytometer chamber. Cells were counted, in triplicate aliquots, over four corner squares of the haemocytometer grid.
Real-time PCR
Total RNA was extracted from cells using an RNeasy Mini-Kit (Qiagen, UK). Reported sequences for each gene (Table 1) were used to design on Primer Express software (PE, Applied Biosystems) human-specific primers for each peptide and receptor component, adapted to RT-PCR conditions, which were synthesised by Invitrogen. Quantitative RT-PCR was performed as described previously (Zhao et al. 2006), using SYBR Green detection (Abgene Biotechnologies, UK) and the housekeeping genes, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and β-actin, as internal standards.
Table 1.
Primers sequences
| Forward | Backward | |
|---|---|---|
| IMD | TCCAGTGAGAATCCCCCTACC | GAGATTGATGGCGTTGGAGG |
| AM | AAACTGATTTCTCACGGCGTG | AATAGTGAGGCTTGCGCCC |
| CRLR | CATTCAACAAGCAGAAGGCG | CAGAGCCATCCATCCCAGG |
| RAMP1 | CTGGCCCATCACCTCTTCAT | CACCGTAGTTAGCCTCCTGGC |
| RAMP2 | CGAAAAGGATTGGTGCGACT | CAGGGTGCTATAAGGCCTGC |
| RAMP3 | GAGGTTCTCATCCCGCTGATC | CATGGCGACAGTCAGAACGA |
| GAPDH | ATCGATGACAACTTTGGTATCGTG | GGCATGGACTGTGGTCATGAG |
| β-Actin | GACTCCGGTGACGGGGTCACC | CACGATGGAGGGGCCGGACTC |
Accession numbers are taken from the European Molecular Biology Laboratory (EMBL) database, which is part of the International Nucleotide Sequence Database Collaboration.
Protein extraction
Medium was removed and cell monolayers washed twice with PBS prior to incubation in 300 μl RIPA buffer (1% v/v Triton X-100, 0.1% v/v SDS and protease inhibitors in PBS) for 20 min at 4°C. Cells were scraped from the flask surface, transferred to a micro-centrifuge tube and incubated for 1 h at 4°C before centrifugation (10,000 g for 10 min at 4°C). Where membrane isolation was undertaken, cell fractionation was carried out as described (Campbell et al. 2008). The supernatant was stored at −80°C pending analysis.
Oxidation of cellular proteins/detection of protein carbonylation
Protein concentration was determined by the method of Lowry. The carbonyl groups in the membrane proteins (5 μg protein per lane) were denatured by addition of 12% SDS and derivatised to 2,4-dinitrophenylhydrazone by reaction with 2,4-dinitrophenylhydrazine (DNPH) for 15 min prior to separation by 12% SDS-PAGE and transfer to PVDF membrane (0.45 μm, Millipore, UK). Non-derivatised protein samples served as negative control. The PVDF membrane was washed with PBS containing 0.05% v/v Tween-20 (Sigma) and blocked for 1 h in PBS/0.05% v/v Tween-20 solution containing 1% w/v bovine serum albumin (BSA). Immunoblotting was performed using a primary antibody directed specifically against the dinitrophenyl moiety (90451 Oxyblot-anti-DNP, raised in rabbit, Chemicon International, CA, USA) used at a dilution of 1:150. Immunocomplexes were detected using secondary antibodies conjugated to horseradish peroxidase (goat anti-rabbit 90452 Oxyblot, Chemicon) used at a dilution of 1:300, and ECL plus (Amersham, UK) as substrate, and quantified by densitometry (Analytical Imaging System, Pittsburgh, PA, USA).
Immunoprecipitation and Western blot
Immunoprecipitation
Primary antibody (2 μl) for CRLR or RAMP2 was added to membrane lysate containing 200 μg protein and incubated on a rotary mixer at 4°C for 2 h. Protein A-G Plus agarose beads (20 μl, Santa Cruz) were added and incubation continued at 4°C overnight. After centrifugation at 10,000 g for 5 min, the recovered pellet was washed four times with ice-cold RIPA buffer. After the final wash, the pellet was re-suspended in 40 μl of Laemmli buffer (Bio-Rad).
Western blotting
Either 20 μg of whole cell-lysate protein reduced in equal volume of Laemmli buffer or 20 μl of immunoprecipitate was denatured (95°C for 5 min) and resolved in 4–12% Bis-Tris gels (Invitrogen) and transferred to nitrocellulose membrane (Amersham). Membranes were blocked with TBS–4% BSA–0.1% Tween-20 at 4°C for 1 h. Primary antibody to CRLR (goat) or RAMPs (rabbit, all Santa Cruz) was used at a dilution of 1:200 at 4°C overnight. Detection was with horseradish peroxidase (HRP)-conjugated donkey secondary antibody (Abcam, UK) at 1:5000 and ECL plus substrate. Analysis was carried out using an AutoChemi system (UVP, Upland, CA, USA).
Staining of cells for AM/IMD, their receptor components and actin distribution
Medium was aspirated from the wells of 12-well plates and coverslips, containing adherent cells, then after two ice-cold PBS washes paraformaldehyde (4%) was added, 750 μl per well, and left for 20 min at room temperature to fix the cells. Coverslips were washed ×5 with PBS, for at least 10 min each time. Before staining, cells were permeabilised with PBS containing 0.5% Triton X-100 (20 min). Coverslips were washed ×3 with PBS, then incubated for 30 min with PBS containing 1% w/v BSA and 0.5% v/v Triton X-100 (PBS-BSA-T) to decrease non-specific binding. Primary anti-human antibodies for the various peptides and their receptor components (Phoenix Pharmaceuticals, Burlingame, CA, USA) were diluted in PBS-BSA-T at a ratio of 1:200 and 400 μl of this solution was applied per well. The coverslips were incubated for 1 h in a humid chamber, then washed ×3 in PBS before counterstaining for actin with anti-rabbit fluoroscein isothiocyanate and Alexafluor 535 nm diluted in PBS-BSA-T at a ratio of 1:500 and 1:50, respectively. The coverslips were incubated in darkness for 1 h in a humid chamber. Coverslips were washed ×3 in PBS, mounted on slides with 5 μl Vectashield (Vector Laboratories, UK) and sealed with clear nail varnish. The slides were kept in the dark at 4°C until examination by fluorescence microscopy (7.3 Three Shot Colour, Diagnostic Instruments Inc., Sterling Heights, MI, USA). Spot (version 4.1; Diagnostic Instruments) software was used for picture processing and semi-quantitative analysis (using standard settings – gain and time exposure). Semi-quantitative analysis measured the ratio of fluorescence signal emitted from the secondary antibody under investigation to the equivalent from that for actin, subtracting the size-matched background fluorescence signal for each peptide or receptor component/actin signal following indirect immunofluorescence staining.
Statistical analysis
Data were analysed using GraphPad Prism v. 5 for Windows (GraphPad Software inc., La Jolla, CA, USA). Cell counts were expressed as a percentage ratio of viable compared to normoxic control in the absence of IMD to standardise results across repeats. Comparison between groups was assessed by two-way analysis of variance (ANOVA) followed by Tukey's multiple comparison post hoc test. Comparison within groups was assessed by ANOVA followed by a post hoc Dunnett's multiple comparison test. For all results P < 0.05 was considered statistically significant.
Results
Cellular distribution of AM/IMD and their receptor components
IMD and AM mRNAs were detected in all cell types; IMD mRNA was more abundantly expressed than AM, particularly in endothelial cells (Fig. 1: results normalised to GAPDH (Fig. 1A) or β-actin (Fig. 1B)). IMD protein was also present in each cell type, with prominent perinuclear staining evident (Fig. 2; Supplementary Fig. A). Similarly, CRLR and RAMPs1–3 were expressed at the mRNA (Fig. 3) and protein (Fig. 4) level in each cell type studied. In endothelial cells, all three RAMPs were expressed more abundantly than CRLR at the mRNA level, while in HASMCs and v-HCFs expression levels were comparable relative to those of CRLR. CRLR and each of the RAMP proteins were widely distributed within all cell types studied although the intensity of staining tended to dissipate from the central perinuclear region towards the cell periphery (Fig. 4A). RAMP2 was the most abundant RAMP at the protein level, relative to the other RAMPs and to CRLR, and was especially abundant in non-endothelial cells (Fig. 4B–C).
Figure 1. Relative IMD (filled column) and AM (open column) mRNA expression normalised to GAPDH (A) or β-actin (B) in human aortic endothelial cells (HAECs) and smooth muscle cells (HASMCs), human coronary artery microvascular endothelial cells (HMVECs) and ventricular-human cardiac fibroblasts (v-HCFs) in basal conditions.
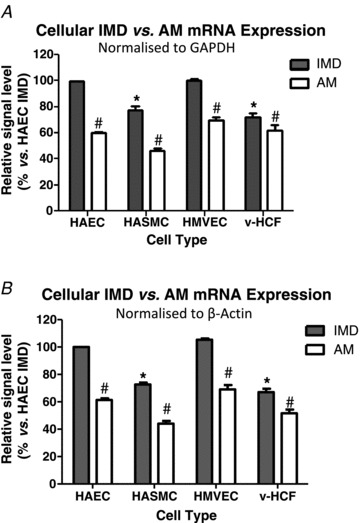
Results (means ± SEM) from n = 3 independent cell sources (or n = 3 individual cultures for v-HCFs) with each source (or culture) run in quadruplicate are presented as percentage expression compared to IMD expression in HAECs. Analysis (2-way ANOVA): *P < 0.05 vs. IMD in HAEC and #P < 0.05 vs. IMD in same cell type.
Figure 2. Cellular IMD vs. AM protein distribution.
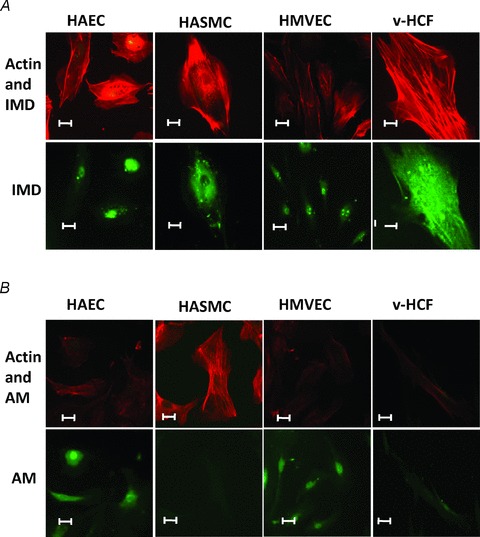
A, indirect immunofluorescence staining to detect the presence and location of IMD protein (green: signal enhanced in IMD alone micrographs) in the cell-types investigated in Fig. 1. Actin counter-staining (red) of the same cells facilitates normal cellular structure identification. B, for comparison AM (green) is shown. Scale bar: 10 μm.
Figure 3. Relative mRNA expression of the calcitonin receptor-like receptor (CRLR) receptor components in the cell-types investigated in Fig. 1 under basal conditions.
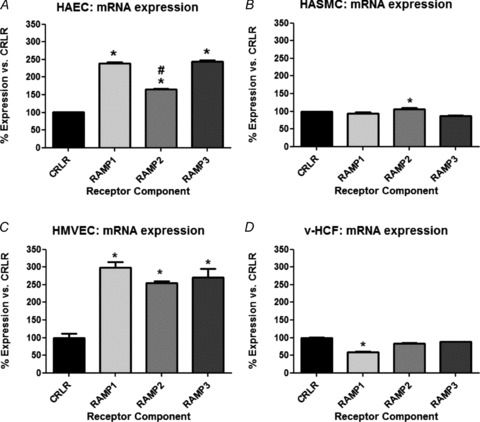
Results, normalised against GAPDH, are presented as relative to CRLR for each RAMP. n = 3 cell sources (or individual cultures for v-HCFs) run in triplicate: *P < 0.05 vs. CRLR and #P < 0.05 vs. other RAMPs. In data not shown similar results were found when β-actin was employed as the house-keeping gene.
Figure 4.
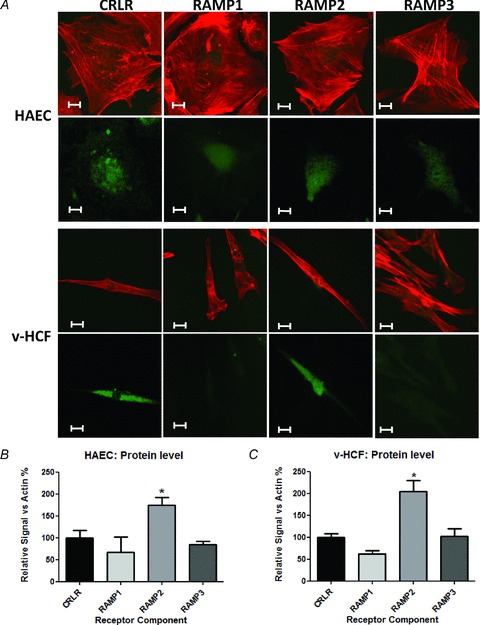
A, indirect immunofluorescence staining to detect the presence and location of CRLR receptor components (green) and actin (red) protein in HAECs and v-HCFs. RAMP1 and RAMP3 signals have been intensified to allow distribution to be seen in micrographs. Scale bar: 10 μm. B and C, indirect immunofluorescence staining images (using fixed fluorescence and image capture settings and no image intensification) were analysed for n≥ 30 cells (obtained from n≥ 3 microscopic fields) for n = 3–4 independent cultures for each condition, normalised for actin and presented (means ± SEM) as relative to CRLR for each RAMP. *P < 0.05 vs. CRLR.
Protection against the deleterious effects of hydrogen peroxide
Treatment for 4 h with 1 mmol l−1 H2O2 (see Supplementary Fig. B for choice of H2O2 concentration) reduced the number of viable cells in all cell types (P < 0.01) with associated membrane blebbing and thin cytoplasmic extensions present on phase contrast microscopy (Fig. 5A). Percentage reductions in viable cell numbers (Fig. 5B) were greater for smooth muscle (89%) and fibroblasts (72%) than for endothelial cells (56%). At 10 mmol l−1 H2O2 (4 h) viability was <10% for all cell types. IMD ≤1 nmol l−1 did not exert detectable cytotoxic effects in any cell type. When cells were treated with 1 nmol l−1 IMD during exposure to 1 mmol l−1 H2O2 for 4 h, viability was greater vs. H2O2 alone (P < 0.01 for all cell types). Disruption of the actin cytoskeleton (Fig. 5C) and protein carbonyl formation (Fig. 5D) followed similar patterns with increased damage due to 1 mmol l−1 H2O2 exposure (4 h), which was attenuated by the presence of 1 nmol l−1 IMD.
Figure 5.

A, phase contrast images showing the effect of 4 h exposure to 1 mmol l−1 H2O2 and/or 1 nmol l−1 IMD to endothelial, smooth muscle and fibroblast cells. Scale bar 100 μm. B, counts of viable cells for the cell-types and treatment conditions in Fig. 5A. n = 4 cell sources (or individual cultures for v-HCFs) run in quadruplicate. *P < 0.01 vs. untreated control and #P < 0.01 vs. H2O2 alone for that cell-type. C, indirect immunofluorescence staining of cells exposed to 1 mmol l−1 H2O2 (actin, red). With IMD addition a degree of normal cellular structure is maintained, represented by HAECs in the larger micrograph. Scale bar: 10 μm. D, protein carbonyl formation (an indicator of oxidative stress) in HAECs in response to the treatment conditions employed in Fig. 5A. n = 3 cell sources *P < 0.05 vs. control and #P < 0.05 vs. H2O2 alone. Representative blot shows, in order, both derivatised and non-derivatised signal for each treatment condition.
Protection against the deleterious effects of ischaemic injury
Preliminary experiments designed to optimise temporal dependence and pH of ischaemic conditions indicated that significant (P < 0.05) but submaximal reduction (30–80%) in cell viability occurred when cells were incubated for 4–6 h in ischaemic medium, pH 5.8 (see Supplementary Fig. C for v-HCF results; other data not shown). HASMCs and v-HCFs were more susceptible to ischaemic injury than HAECs or HMVECs. IMD (≤1 nmol l−1) exerted a concentration-dependent protective effect against ischaemic injury which was more apparent in those cells more susceptible to injury, namely v-HCFs and HASMCs, than in HMVECs and HAECs (comparable data for each cell type are shown in Fig. 6). Cell viabilities at 6 h under ischaemic conditions differed (P < 0.05, n = 3) in the absence and presence of 1 nmol l−1 IMD: HAECs 63% and 85%; HMVECs 51% and 68%; v-HCFs 42% and 96%; HASMCs 28% and 75%, respectively. IMD 100 nmol l−1 exerted a deleterious effect on cell viability (P < 0.05) – data not shown; 1 nmol l−1 IMD was therefore chosen for subsequent experiments.
Figure 6.
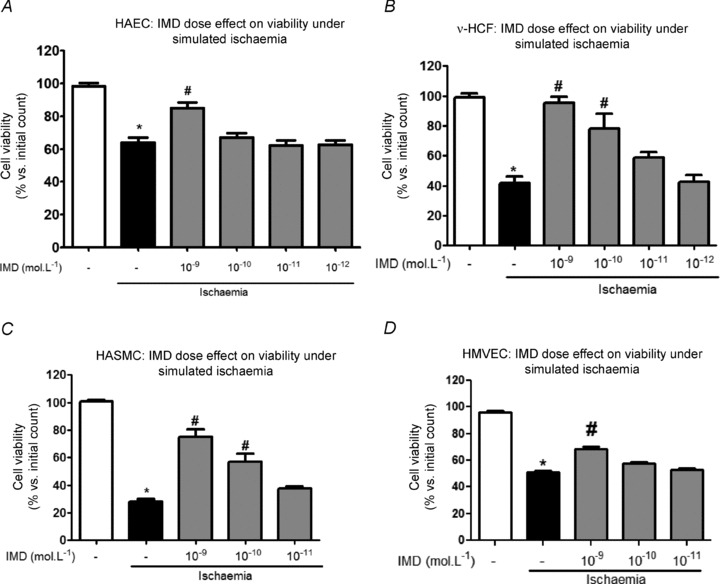
Dose-dependent effect of IMD (at concentrations ≤10−9 mol l−1; grey columns) on cell viability under simulated ischaemia (4 h) for those cell types investigated in Fig. 1. n = 3 cell sources (or individual cultures for v-HCFs) run in duplicate. *P < 0.05 vs. normoxia alone (open column) and #P < 0.05 vs. Ischaemia without IMD (black filled column).
Protection against the deleterious effects of ischaemia–reperfusion injury: receptor subtype involvement
Following 3 h of ischaemia, a further 1 h of reperfusion, simulated by replacing ischaemic medium with normoxic medium, resulted in further damage in all cell types: cell viability was lower (P < 0.05) following incubation under 3 h ischaemia plus 1 h reperfusion than under 4 h ischaemia alone. IMD (1 nmol l−1) present throughout the entire ischaemia and reperfusion period attenuated the extent of ischaemia–reperfusion injury (P < 0.01): cell viabilities were 95%, 74% and 82% for HAECs, HMVECs and v-HCFs, respectively, relative to those in the absence of IMD which were 62%, 35% and 32%, respectively (Fig. 7A). Attenuation of the disruption of the actin cytoskeleton (Fig. 7B) and protein carbonyl formation (Fig. 7C) followed similar patterns – representative data from HAECs experiments are shown. Furthermore, when IMD (1 nmol l−1) was present only during the reperfusion period, but not during the initial ischaemia phase, a protective effect was still evident (P < 0.01): cell viabilities were 79%, 55% and 48% for HAECs, HMVECs and v-HCFs, respectively.
Figure 7.
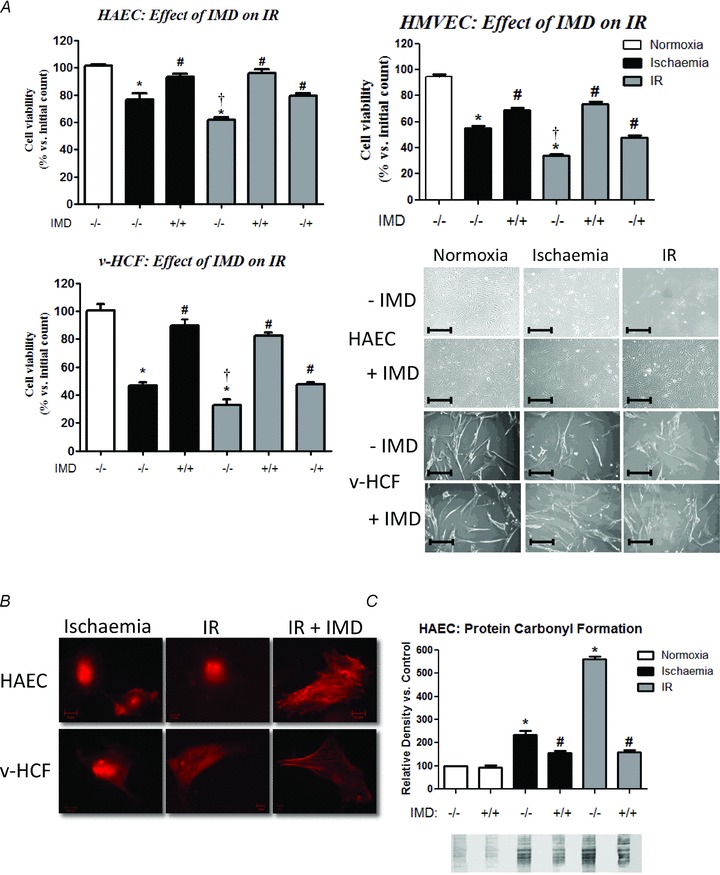
A, viable cell counts for cells exposed to normoxia (4 h), ischaemia (4 h) or ischaemia (3 h)–reperfusion (1 h; IR) without/with co-incubation with 10−9 mol l−1 IMD throughout the 4 h period or only during the 1 h of reperfusion. n = 4 cell sources (or individual cultures for v-HCFs) run in quadruplicate. *P < 0.01 vs. untreated control, †P < 0.05 vs. ischaemia alone, and #P < 0.01 vs. non-IMD counterpart. Representative phase contrast images showing the effect of exposure of endothelial cells and fibroblasts to IR and IMD are also included. Scale bar 100 μm. B, indirect immunofluorescence staining of cells exposed to ischaemia or IR (actin, red). With IMD addition during reperfusion alone a degree of normal cellular structure is maintained. Scale bar: 10 μm. C, protein carbonyl formation in HAECs in response to normoxia, ischaemia and IR without/with co-incubation with 10−9 mol l−1 IMD. n = 3 individual cultures *P < 0.05 vs. control and #P < 0.05 vs. non-IMD treated counterpart. Representative blot showing in order both derivatised and non-derivatised signal for each treatment condition.
IMD mRNA expression (Fig. 8A) and that for AM (Supplementary Fig. D) remained relatively unchanged during the experimental period. However intracellular protein level increased over 2-fold (Fig. 8B). Pre-treatment (4 days) of cells with each of the CRLR components’ siRNAs was sufficient to quench protein translation (Supplementary Fig. E). Pre-treatment (4 days) of HAECs with CRLR siRNA abolished the protective effect of IMD (1 nmol l−1) against ischaemia and ischaemia–reperfusion injury (Fig. 8C). Similarly pre-treatment with RAMP2 siRNA abolished the protective effect of IMD (Fig. 8D); in contrast, pre-treatment with RAMP1 (Fig. 8E) or RAMP3 (Fig. 8F) siRNA caused a small, but significant attenuation of the protective effect of IMD, present during simulated reperfusion only. siRNA pre-treatment alone had no detectable influence in the absence of IMD on cell viability under normoxic conditions or ischaemia–reperfusion (data not shown). CRLR and RAMP2 proteins isolated from the cell membrane were found to co-localise (Fig. 8G).
Figure 8.
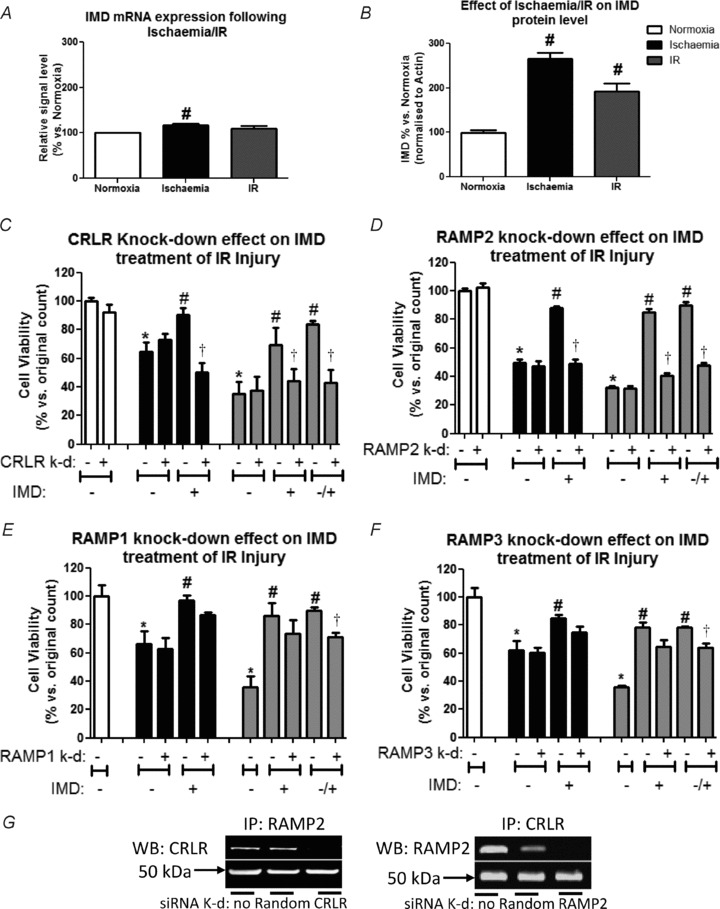
A and B, relative mRNA expression of IMD normalised against GAPDH (A; similar results were found using β-actin as a housekeeping gene – not shown) or protein level normalised against β-actin (B) are presented as relative to normoxia. n = 3 HMVECs sources run in triplicate: #P < 0.05 vs. normoxia. C–F, viable cell counts for cells which have had the CRLR receptor components knocked-down before exposure to normoxia, ischaemia and ischaemia–reperfusion without/with co-incubation with 10−9 mol l−1 IMD. n = 3 HAEC sources run in triplicate. *P < 0.05 vs. untreated control, †P < 0.05 vs. non knock-down counterpart, and #P < 0.05 vs. non-IMD counter part. G, blots obtained when membrane protein lysates were immunoprecipitated (IP) with RAMP2 and western blotted with CRLR antibody (and vice versa) to confirm co-precipitation of molecules. The heavy chain of the IP antibody also transfers to the blot and is employed as a loading control. Treatment conditions included no pre-treatment or pre-treatment with either random (non-targeting siRNA) or siRNA directed against CRLR or RAMP2.
Discussion
This study has demonstrated for the first time AM1 receptor-dependent protection by IMD against simulated ischaemia–reperfusion injury directly (independent of enhanced blood flow) in human macrovascular, microvascular and cardiac non-vascular cells. The augmented protein carbonyl formation during reperfusion indicates that further oxidative damage occurs as normoxia returns to ischaemic cells. Importantly, IMD was still found to be protective even when administration was restricted to the reperfusion phase only, raising the possibility of injection of exogenous IMD as an attractive treatment option post-myocardial infarction.
There is only one other previous report demonstrating protection of HAECs by IMD against biochemical stressors including oxidative stress, induced by hydrogen peroxide administration (Pearson et al. 2009), but neither ischaemia nor reperfusion was investigated. Protective effects of IMD have been demonstrated before, however, in a range of experimental rodent models in vitro namely: cultured rat cerebral endothelial cells subjected to hydrogen peroxide (Chen et al. 2006); rat neonatal cardiomyocytes subjected to hypoxia–reoxygenation (Song et al. 2009); Langendorff-perfused rat hearts subjected to 45 min global ischaemia and 30 min reperfusion (Yang et al. 2005). IMD has also be shown to be protective in vivo in mouse hearts subjected to myocardial ischaemia–reperfusion injury induced by coronary artery ligation (Zhang et al. 2009) and rat hearts subjected to isoprenaline-induced ischaemic insult (Jia et al. 2006). The current study therefore is the first to specifically address the direct protective effects of IMD against ischaemia–reperfusion injury in human cells, both vascular and cardiac non-vascular, using a protocol designed to simulate realistically the extracellular environment during ischaemia, namely impaired supply of oxygen and glucose, markedly acidic pH and increased extracellular K+ ion (Van den Hoek et al. 2000; Townsend et al. 2004; McDermott et al. 2007).
Furthermore, the present model faithfully reproduces the temporal characteristics associated with the onset of symptoms and cellular injury during an acute coronary syndrome in humans and typical clinical delay experienced before reperfusion. A variety of endpoints have been assessed previously to determine injury arising in vitro or in vivo as a result of oxidative stress and/or ischaemic insult (Yang et al. 2005; Chen et al. 2006; Jia et al. 2006; Pearson et al. 2009; Song et al. 2009; Zhang et al. 2009), including cell viability assays, leakage of enzymes including creatine kinase and lactate dehydrogenase, cellular lipid peroxidation and protein carbonylation, disruption to the cytoskeleton, altered membrane potential and haemodynamic parameters. Although the present study focused primarily on cell viability as an end-point, supporting evidence for the protective effects of IMD was provided by assessment of protein carbonyl formation and disruption of the actin cytoskeleton.
The cellular protective effects of IMD were observed in the concentration range 10 pmol l−1–10 nmol l−1; higher concentrations of IMD were not protective and potentially detrimental. These concentrations are at least an order of magnitude lower than those associated with functional effects in rodents in published reports (Yang et al. 2009; Song et al. 2009). There are only two published reports relating to IMD concentrations in human plasma; both found IMD to be present at <30 pmol l−1 in plasma of healthy subjects (Morimoto et al. 2007; Bell & McDermott, 2008), which is also at least an order of magnitude lower than in rodents (reviewed in Bell & McDermott, 2008). Plasma IMD is elevated significantly in humans in chronic heart failure and in acute coronary syndrome (D. Bell and M. Harbinson, unpublished observation) but still does not exceed 100 pmol l−1.
In the present study, IMD was detected at the mRNA and protein levels in all human cell types investigated. This is in agreement with two other reports regarding the presence of IMD in HAECs (Pearson et al. 2009) and at autopsy in cardiac tissue obtained from humans devoid of cardiovascular disease (Morimoto et al. 2007); in the latter, IMD was expressed more abundantly in cardiac non-vascular cells but was also present in the coronary vasculature, notably endothelial cells of pericardial veins, and smooth muscle cells of coronary arteries. IMD is also expressed, though sparsely, in adult rodent heart and cardiomyocytes (Pan et al. 2005); such expression is upregulated markedly in hypertrophying cardiomyocytes (Pan et al. 2005), in response to chronic oxidative stress and ischaemic insult (Zhao et al. 2006; Bell et al. 2007, 2008) and in the post-infarct failing heart (Hirose et al. 2008; Zhang et al. 2009). AM and IMD expression are differentially influenced by oxidative stress both in cardiomyocytes from chronic NO-deficient rats (Zhao et al. 2006; Bell et al. 2007) and HAECs subjected to acute metabolic stressors (Pearson et al. 2009), IMD showing much greater up-regulation, indicative of a more prominent pathophysiological role in response to oxidative stress.
However, in contrast to the observation of Pearson's group that AM was the more abundant of the two peptides in cultured HAECs maintained under optimal conditions (Pearson et al. 2009), we observed IMD to be the more abundantly expressed peptide in all human cell types under basal conditions. This discrepancy might be attributed to differences in the origin of the human cells or differences in the composition of media and conditions under which cells were routinely cultured.
It is probable that local levels of IMD would be much greater than those circulating in human plasma, enabling the peptide to exert the marked protective effect observed in this study, as an autocrine or paracrine agent under pathophysiological conditions in vivo, especially in response to chronic ischaemia and/or oxidative stress. It is less certain that augmented transcriptional expression of the IMD gene would occur sufficiently rapidly to enable large amounts of IMD to be generated within minutes to hours after ischaemic insult. Although we observed 2- to 3-fold increase in IMD protein levels in HMVECs within 4 h of ischaemia relative to cells maintained for 4 h under normoxia, mRNA level was not increased significantly within this time. Furthermore, IMD protein levels were not greater, and indeed were slightly reduced, relative to cells maintained under ischaemic conditions for 4 h, in HMVECs subjected to 3 h ischaemia and 1 h simulated reperfusion. These data are suggestive of augmented processing of a preformed, stored precursor, in response to acute ischaemic insult and oxidative stress; as stores of the precursor become exhausted, continued supply of IMD then becomes dependent subsequently on further expression of mRNA encoding the precursor.
CRLR and RAMP1–3 expression was detected at both the mRNA and protein level in each of the human cell types studied, consistent with previous reports of mRNA expression in human vasculature (Kamitani et al. 1999; Sams et al. 2000; Oliver et al. 2002; Jansen-Olesen et al. 2003; Nikitenko et al. 2006; Albertin et al. 2010) including endothelium and smooth muscle; CRLR mRNA expression in human microvascular endothelial cells is upregulated by hypoxia (Nikitenko et al. 2006). Specifically in the human heart, mRNA encoding these receptor components has been detected in coronary arteries (Frayon et al. 2000; Hasbak et al. 2003), myocardial trabeculae (Saetrum et al. 2000) and cardiomyocytes. Rat cardiomyocytes and fibroblasts also express CRLR and RAMPs 2 and 3 (Nishikimi & Matsuoka, 2005). At the protein level, CRLR has been detected by Western blotting in human left ventricular cardiomyocytes and macrovascular and microvascular endothelial cells (Hagner et al. 2003). Although expression of CRLR and RAMPs is consistent with the presence of functional CGRP1, AM1 and AM2 receptors on human cells (Hasbak et al. 2003), it should be noted that additional functions for the RAMPs, independent of CRLR, are starting to emerge in mammals: RAMPs can interact with the calcitonin receptor protein CT to form amylin AMY1-3 receptors (Udawela et al. 2004) and influence VIP/PACAP receptor–effector coupling (Sexton et al. 2006), and RAMP3 can interact with secretin receptors (Harikumar et al. 2009). The abundance of individual RAMPs has often varied between studies and it cannot necessarily be presumed that the most abundant RAMP implies a predominant involvement of the associated CGRP/AM receptor subtype. Limited functional data are available in human tissue and pharmacological studies have been hampered by lack of availability of highly selective agonists and antagonists for each receptor subtype. IMD itself interacts non-selectively at CGRP1, AM1 and AM2 receptors (Bell and McDermott, 2008) although additional independent receptors for IMD have been postulated in rodent studies (Taylor et al. 2006; Owji et al. 2008; Zeng et al. 2009). Although CGRP1 receptors have been implicated in functional responses to CGRP in human coronary arteries, an additional novel functional receptor for these peptides has been proposed in distal coronary artery (Gupta et al. 2006). Lack of antagonism by CGRP8-37 of the action of IMD to reduce caspase activity in HAECs, together with a lack of inhibition of caspase activity by AM in the same assay, has also prompted speculation of the existence of novel, IMD-specific receptors (Pearson et al. 2009). In the present study, the protective effect of IMD against ischaemia–reperfusion injury was abolished by siRNA to CRLR, confirming the requirement for the CRLR protein. Viewed together with the abolition of the protective effect similarly by siRNA to RAMP2, but not by siRNAs to RAMP1 or RAMP3, these data are strongly supportive of the involvement of AM1 receptors predominantly in the protection afforded by IMD. AM1 receptors are known to couple to a range of downstream signalling pathways in rodent tissues including cAMP, K+ channels and phosphatidylinositol 3-kinase (Bell & McDermott, 2008). It is important to identify in future studies the mechanisms downstream of the AM1 receptor responsible for the protective effects.
Unfortunately it is not easy to obtain adult human tissue for isolation of cardiomyocytes of sufficient quantity and quality to undertake similar experiments. In the present study, v-HCFs were more susceptible to the deleterious effects of ischaemia–reperfusion injury than vascular cells, and derived more protection from IMD. Significant differences occur in antioxidant capacity in different vascular cell types (Serrano et al. 2006) as well as in the same cell type but located at different points throughout the circulation (Mahadevan et al. 2006). The greater sensitivity of ventricular cardiac fibroblasts to ischaemic injury could be explained by unique metabolic properties of endothelial cells such as greater nitric oxide production, glycolytic activity and glutathione peroxidase and reductase activities (Dobrina & Rossi, 1983). Fibroblasts are likely to behave similarly to other cardiac non-vascular cells, including cardiomyocytes. Infusion of IMD during reperfusion could therefore represent an attractive treatment option in the early management of acute coronary syndrome to salvage myocardial tissue. Recent reports of the successful delivery of the IMD gene, packaged in an adenoviral vector, to ischaemic rat hindlimb and kidney (Hagiwara et al. 2008; Smith et al. 2009) open up the exciting possibility of a similar approach of adenoviral-mediated gene delivery of IMD or the receptor components RAMP2/CRLR to offset the deleterious consequences of chronic or intermittent ischaemia in humans.
Acknowledgments
This study was funded by a project grant awarded to D.B., M.C. and M.H. from Heart Research UK.
Glossary
Abbreviations
- AM
adrenomedullin
- CGRP
calcitonin gene-related peptide
- CRLR
calcitonin receptor-like receptor
- DNPH
2,4-dinitrophenylhydrazine
- HAEC
human aortic endothelial cell
- HASMC
human aortic smooth muscle cell
- HMVEC
human cardiac microvascular endothelial cell (HMVEC)
- IMD
intermedin
- RAMP
receptor activity-modifying protein
- ROS
reactive oxygen species
- v-HCF
human ventricular cardiac fibroblast
Author contributions
D.B. conceived the study and obtained funding, designed experimental protocols and wrote the paper. M.C. was responsible for the day to day running of the project, establishment of the methods for measurement of the specific experimental endpoints, preparation of the figures and generation of some of the data. M.F., L.S., L.D. and A.O'R. were undergraduate students undertaking short-term research projects within the laboratory at the time of the study and who each contributed to the generation of some of the data. V.J. provided technical assistance, especially in regard to tissue culture protocols, and assisted with data analysis. M.H. provided input to enhance the clinical relevance of the study undertaken in human cells, based on expertise in managing patients presenting with acute coronary syndromes.
References
- Albertin G, Sorato E, Oselladore B, Mascarin A, Tortorella C, Guidolin D. Involvement of vascular endothelial growth factor signaling in CLR/RAMP1 and CLR/RAMP2-mediated pro-angiogenic effect of intermedin on human vascular endothelial cells. Int J Mol Med. 2010;26:289–294. doi: 10.3892/ijmm_00000464. [DOI] [PubMed] [Google Scholar]
- Bell D, McDermott BJ. Intermedin (adrenomedullin-2): a novel counter-regulatory peptide in the cardiovascular and renal systems. Br J Pharmacol. 2008;153(Suppl 1):S247–S262. doi: 10.1038/sj.bjp.0707494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell D, Zhao Y, McCoy FP, Devine A, McDermott BJ. Expression of the counter-regulatory peptide intermedin is augmented in the presence of oxidative stress in hypertrophied cardiomyocytes. Cell Physiol Biochem. 2008;21:409–420. doi: 10.1159/000129633. [DOI] [PubMed] [Google Scholar]
- Bell D, Zhao Y, McCoy FP, Devine AB, McDermott BJ. Differential effects of an anti-oxidant intervention on cardiomyocyte expression of adrenomedullin and intermedin and their receptor components in chronic nitric oxide deficiency. Cell Physiol Biochem. 2007;20:269–282. doi: 10.1159/000107513. [DOI] [PubMed] [Google Scholar]
- Cai H. Hydrogen peroxide regulation of endothelial function: origins, mechanisms, and consequences. Cardiovasc Res. 2005;68:26–36. doi: 10.1016/j.cardiores.2005.06.021. [DOI] [PubMed] [Google Scholar]
- Cai Y, Xu MJ, Teng X, Zhou YB, Chen L, Zhu Y, Wang X, Tang CS, Qi YF. Intermedin inhibits vascular calcification by increasing the level of matrix γ-carboxyglutamic acid protein. Cardiovasc Res. 2010;85:864–873. doi: 10.1093/cvr/cvp366. [DOI] [PubMed] [Google Scholar]
- Campbell M, Anderson P, Trimble ER. Glucose lowers the threshold for human aortic vascular smooth muscle cell migration: inhibition by protein phosphatase-2A. Diabetologia. 2008;51:1068–1080. doi: 10.1007/s00125-008-0962-7. [DOI] [PubMed] [Google Scholar]
- Chen L, Kis B, Hashimoto H, Busija DW, Takei Y, Yamashita H, Ueta Y. Adrenomedullin 2 protects rat cerebral endothelial cells from oxidative damage in vitro. Brain Res. 2006;1086:42–49. doi: 10.1016/j.brainres.2006.02.128. [DOI] [PubMed] [Google Scholar]
- Dobrina A, Rossi F. Metabolic properties of freshly isolated bovine endothelial cells. Biochim Biophys Acta. 1983;762:295–301. doi: 10.1016/0167-4889(83)90084-8. [DOI] [PubMed] [Google Scholar]
- Dong F, Taylor MM, Samson WK, Ren J. Intermedin (adrenomedullin-2) enhances cardiac contractile function via a protein kinase C- and protein kinase A-dependent pathway in murine ventricular myocytes. J Appl Physiol. 2006;101:778–784. doi: 10.1152/japplphysiol.01631.2005. [DOI] [PubMed] [Google Scholar]
- Frayon S, Cueille C, Gnidehou S, Vernejoul deMC, Garel JM. Dexamethasone increases RAMP1 and CRLR mRNA expressions in human vascular smooth muscle cells. Biochem Biophys Res Commun. 2000;270:1063–1067. doi: 10.1006/bbrc.2000.2552. [DOI] [PubMed] [Google Scholar]
- Fujisawa Y, Nagai Y, Miyatake A, Miura K, Nishiyama A, Kimura S, Abe Y. Effects of adrenomedullin 2 on regional hemodynamics in conscious rats. Eur J Pharmacol. 2007;558:128–132. doi: 10.1016/j.ejphar.2006.11.043. [DOI] [PubMed] [Google Scholar]
- Gupta S, Mehrotra S, Villalon CM, Garrelds IM, Vries deR, Kats vanJP, Sharma HS, Saxena PR, Maassenvandenbrink A. Characterisation of CGRP receptors in human and porcine isolated coronary arteries: evidence for CGRP receptor heterogeneity. Eur J Pharmacol. 2006;530:107–116. doi: 10.1016/j.ejphar.2005.11.020. [DOI] [PubMed] [Google Scholar]
- Hagiwara M, Bledsoe G, Yang ZR, Smith RS, Jr, Chao L, Chao J. Intermedin ameliorates vascular and renal injury by inhibition of oxidative stress. Am J Physiol Renal Physiol. 2008;295:F1735–F1743. doi: 10.1152/ajprenal.90427.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagner S, Haberberger R, Hay DL, Facer P, Reiners K, Voigt K, McGregor GP. Immunohistochemical detection of the calcitonin receptor-like receptor protein in the microvasculature of rat endothelium. Eur J Pharmacol. 2003;481:147–151. doi: 10.1016/j.ejphar.2003.09.030. [DOI] [PubMed] [Google Scholar]
- Hagner S, Stahl U, Knoblauch B, McGregor GP, Lang RE. Calcitonin receptor-like receptor: identification and distribution in human peripheral tissues. Cell Tissue Res. 2002;310:41–50. doi: 10.1007/s00441-002-0616-x. [DOI] [PubMed] [Google Scholar]
- Harikumar KG, Simms J, Christopoulos G, Sexton PM, Miller LJ. Molecular basis of association of receptor activity-modifying protein 3 with the family B G protein-coupled secretin receptor. Biochemistry. 2009;48:11773–11785. doi: 10.1021/bi901326k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasbak P, Saetrum OO, Eskesen K, Schifter S, Arendrup H, Longmore J, Edvinsson L. Investigation of CGRP receptors and peptide pharmacology in human coronary arteries. Characterization with a nonpeptide antagonist. J Pharmacol Exp Ther. 2003;304:326–333. doi: 10.1124/jpet.102.037754. [DOI] [PubMed] [Google Scholar]
- Hirose T, Totsune K, Mori N, Morimoto R, Hashimoto M, Nakashige Y, Metoki H, Asayama K, Kikuya M, Ohkubo T, Hashimoto J, Sasano H, Kohzuki M, Takahashi K, Imai Y. Increased expression of adrenomedullin 2/intermedin in rat hearts with congestive heart failure. Eur J Heart Fail. 2008;10:840–849. doi: 10.1016/j.ejheart.2008.06.020. [DOI] [PubMed] [Google Scholar]
- Jansen-Olesen I, Jorgensen L, Engel U, Edvinsson L. In-depth characterization of CGRP receptors in human intracranial arteries. Eur J Pharmacol. 2003;481:207–216. doi: 10.1016/j.ejphar.2003.09.021. [DOI] [PubMed] [Google Scholar]
- Jia YX, Yang JH, Pan CS, Geng B, Zhang J, Xiao Y, Zhao J, Gerns H, Yang J, Chang JK, Wen JK, Tang CS, Qi YF. Intermedin1–53 protects the heart against isoproterenol-induced ischemic injury in rats. Eur J Pharmacol. 2006;549:117–123. doi: 10.1016/j.ejphar.2006.07.054. [DOI] [PubMed] [Google Scholar]
- Jolly L, March JE, Kemp PA, Bennett T, Gardiner SM. Mechanisms involved in the regional haemodynamic effects of intermedin (adrenomedullin 2) compared with adrenomedullin in conscious rats. Br J Pharmacol. 2009;157:1502–1513. doi: 10.1111/j.1476-5381.2009.00306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani S, Asakawa M, Shimekake Y, Kuwasako K, Nakahara K, Sakata T. The RAMP2/CRLR complex is a functional adrenomedullin receptor in human endothelial and vascular smooth muscle cells. FEBS Lett. 1999;448:111–114. doi: 10.1016/s0014-5793(99)00358-0. [DOI] [PubMed] [Google Scholar]
- Kindt F, Wiegand S, Loser C, Nilles M, Niemeier V, Hsu SY, Steinhoff M, Kummer W, Gieler U, Haberberger RV. Intermedin: a skin peptide that is downregulated in atopic dermatitis. J Invest Dermatol. 2007;127:605–613. doi: 10.1038/sj.jid.5700576. [DOI] [PubMed] [Google Scholar]
- Mahadevan VS, Campbell M, McKeown PP, Bayraktutan U. Internal mammary artery smooth muscle cells resist migration and possess high antioxidant capacity. Cardiovasc Res. 2006;72:60–68. doi: 10.1016/j.cardiores.2006.06.022. [DOI] [PubMed] [Google Scholar]
- McDermott BJ, McWilliams S, Smyth K, Kelso EJ, Spiers JP, Zhao Y, Bell D, Mirakhur RK. Protection of cardiomyocyte function by propofol during simulated ischemia is associated with a direct action to reduce pro-oxidant activity. J Mol Cell Cardiol. 2007;42:600–608. doi: 10.1016/j.yjmcc.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Morimoto R, Satoh F, Murakami O, Totsune K, Suzuki T, Sasano H, Ito S, Takahashi K. Expression of adrenomedullin2/intermedin in human brain, heart, and kidney. Peptides. 2007;28:1095–1103. doi: 10.1016/j.peptides.2007.01.018. [DOI] [PubMed] [Google Scholar]
- Nagoshi Y, Kuwasako K, Cao YN, Imamura T, Kitamura K, Eto T. Tumor necrosis factor-α downregulates adrenomedullin receptors in human coronary artery smooth muscle cells. Peptides. 2004;25:1115–1121. doi: 10.1016/j.peptides.2004.04.010. [DOI] [PubMed] [Google Scholar]
- Nikitenko LL, Blucher N, Fox SB, Bicknell R, Smith DM, Rees MC. Adrenomedullin and CGRP interact with endogenous calcitonin-receptor-like receptor in endothelial cells and induce its desensitisation by different mechanisms. J Cell Sci. 2006;119:910–922. doi: 10.1242/jcs.02783. [DOI] [PubMed] [Google Scholar]
- Nishikimi T, Matsuoka H. Cardiac adrenomedullin: its role in cardiac hypertrophy and heart failure. Curr Med Chem Cardiovasc Hematol Agents. 2005;3:231–242. doi: 10.2174/1568016054368241. [DOI] [PubMed] [Google Scholar]
- Oliver KR, Wainwright A, Edvinsson L, Pickard JD, Hill RG. Immunohistochemical localization of calcitonin receptor-like receptor and receptor activity-modifying proteins in the human cerebral vasculature. J Cereb Blood Flow Metab. 2002;22:620–629. doi: 10.1097/00004647-200205000-00014. [DOI] [PubMed] [Google Scholar]
- Owji AA, Chabot JG, Dumont Y, Quirion R. Adrenomedullin-2/intermedin induces cAMP accumulation in dissociated rat spinal cord cells: evidence for the existence of a distinct class of sites of action. J Mol Neurosci. 2008;35:355–361. doi: 10.1007/s12031-008-9062-x. [DOI] [PubMed] [Google Scholar]
- Pan CS, Yang JH, Cai DY, Zhao J, Gerns H, Yang J, Chang JK, Tang CS, Qi YF. Cardiovascular effects of newly discovered peptide intermedin/adrenomedullin 2. Peptides. 2005;26:1640–1646. doi: 10.1016/j.peptides.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Pearson LJ, Yandle TG, Nicholls MG, Evans JJ. Intermedin (adrenomedullin-2): a potential protective role in human aortic endothelial cells. Cell Physiol Biochem. 2009;23:97–108. doi: 10.1159/000204098. [DOI] [PubMed] [Google Scholar]
- Roh J, Chang CL, Bhalla A, Klein C, Hsu SY. Intermedin is a calcitonin/calcitonin gene-related peptide family peptide acting through the calcitonin receptor-like receptor/receptor activity-modifying protein receptor complexes. J Biol Chem. 2004;279:7264–7274. doi: 10.1074/jbc.M305332200. [DOI] [PubMed] [Google Scholar]
- Saetrum OO, Hasbak P, Vries deR, Saxena PR, Edvinsson L. Positive inotropy mediated via CGRP receptors in isolated human myocardial trabeculae. Eur J Pharmacol. 2000;397:373–382. doi: 10.1016/s0014-2999(00)00233-8. [DOI] [PubMed] [Google Scholar]
- Sams A, Knyihar-Csillik E, Engberg J, Szok D, Tajti J, Bodi I, Edvinsson L, Vecsei L, Jansen-Olesen I. CGRP and adrenomedullin receptor populations in human cerebral arteries: in vitro pharmacological and molecular investigations in different artery sizes. Eur J Pharmacol. 2000;408:183–193. doi: 10.1016/s0014-2999(00)00781-0. [DOI] [PubMed] [Google Scholar]
- Serrano MC, Pagani R, Manzano M, Comas JV, Portoles MT. Mitochondrial membrane potential and reactive oxygen species content of endothelial and smooth muscle cells cultured on poly(epsilon-caprolactone) films. Biomaterials. 2006;27:4706–4714. doi: 10.1016/j.biomaterials.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Sexton PM, Morfis M, Tilakaratne N, Hay DL, Udawela M, Christopoulos G, Christopoulos A. Complexing receptor pharmacology: modulation of family B G protein-coupled receptor function by RAMPs. Ann N Y Acad Sci. 2006;1070:90–104. doi: 10.1196/annals.1317.076. [DOI] [PubMed] [Google Scholar]
- Smith RS, Jr, Gao L, Bledsoe G, Chao L, Chao J. Intermedin is a new angiogenic growth factor. Am J Physiol Heart Circ Physiol. 2009;297:H1040–H1047. doi: 10.1152/ajpheart.00404.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JQ, Teng X, Cai Y, Tang CS, Qi YF. Activation of Akt/GSK-3β signaling pathway is involved in intermedin(1–53) protection against myocardial apoptosis induced by ischemia/reperfusion. Apoptosis. 2009;14:1299–1307. doi: 10.1007/s10495-009-0398-7. [DOI] [PubMed] [Google Scholar]
- Takei Y, Inoue K, Ogoshi M, Kawahara T, Bannai H, Miyano S. Identification of novel adrenomedullin in mammals: a potent cardiovascular and renal regulator. FEBS Lett. 2004;556:53–58. doi: 10.1016/s0014-5793(03)01368-1. [DOI] [PubMed] [Google Scholar]
- Taylor MM, Bagley SL, Samson WK. Intermedin/adrenomedullin-2 inhibits growth hormone release from cultured, primary anterior pituitary cells. Endocrinology. 2006;147:859–864. doi: 10.1210/en.2005-0949. [DOI] [PubMed] [Google Scholar]
- Townsend PA, Cutress RI, Carroll CJ, Lawrence KM, Scarabelli TM, Packham G, Stephanou A, Latchman DS. BAG-1 proteins protect cardiac myocytes from simulated ischemia/reperfusion-induced apoptosis via an alternate mechanism of cell survival independent of the proteasome. J Biol Chem. 2004;279:20723–20728. doi: 10.1074/jbc.M400399200. [DOI] [PubMed] [Google Scholar]
- Udawela M, Hay DL, Sexton PM. The receptor activity modifying protein family of G protein coupled receptor accessory proteins. Semin Cell Dev Biol. 2004;15:299–308. doi: 10.1016/j.semcdb.2003.12.019. [DOI] [PubMed] [Google Scholar]
- Hoek VandenT, Becker LB, Shao ZH, Li CQ, Schumacker PT. Preconditioning in cardiomyocytes protects by attenuating oxidant stress at reperfusion. Circ Res. 2000;86:541–548. doi: 10.1161/01.res.86.5.541. [DOI] [PubMed] [Google Scholar]
- Yang JH, Cai Y, Duan XH, Ma CG, Wang X, Tang CS, Qi YF. Intermedin 1–53 inhibits rat cardiac fibroblast activation induced by angiotensin II. Regul Pept. 2009;158:19–25. doi: 10.1016/j.regpep.2009.05.012. [DOI] [PubMed] [Google Scholar]
- Yang JH, Jia YX, Pan CS, Zhao J, Ouyang M, Yang J, Chang JK, Tang CS, Qi YF. Effects of intermedin(1–53) on cardiac function and ischemia/reperfusion injury in isolated rat hearts. Biochem Biophys Res Commun. 2005;327:713–719. doi: 10.1016/j.bbrc.2004.12.071. [DOI] [PubMed] [Google Scholar]
- Yang JH, Ma CG, Cai Y, Pan CS, Zhao J, Tang CS, Qi YF. Effect of intermedin1–53 on angiotensin II-induced hypertrophy in neonatal rat ventricular myocytes. J Cardiovasc Pharmacol. 2010;56:45–52. doi: 10.1097/FJC.0b013e3181ddc785. [DOI] [PubMed] [Google Scholar]
- Zeng Q, Yuan Y, Wang X, Wu HM, Fan L, Qi YF, Tang CS, Cai Y, Pan CS. Upregulated expression of intermedin and its receptor in the myocardium and aorta in spontaneously hypertensive rats. Peptides. 2009;30:391–399. doi: 10.1016/j.peptides.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Zhang HY, Jiang W, Liu JY, Li Y, Chen CL, Xin HB, Huang DJ. Intermedin is upregulated and has protective roles in a mouse ischemia/reperfusion model. Hypertens Res. 2009;32:861–868. doi: 10.1038/hr.2009.120. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Bell D, Smith LR, Zhao L, Devine AB, McHenry EM, Nicholls DP, McDermott BJ. Differential expression of components of the cardiomyocyte adrenomedullin/intermedin receptor system following blood pressure reduction in nitric oxide-deficient hypertension. J Pharmacol Exp Ther. 2006;316:1269–1281. doi: 10.1124/jpet.105.092783. [DOI] [PubMed] [Google Scholar]