

Abstract
There is some evidence to suggest that acetazolamide may improve obstructive sleep apnoea (OSA). However, how acetazolamide affects the key traits causing OSA remains uncertain. We aimed to investigate the effect of acetazolamide on the traits contributing to OSA and its severity. Acetazolamide (500 mg twice daily) was administered for 1 week to 13 OSA subjects. Pharyngeal anatomy/collapsibility, loop gain (LG), upper-airway muscle responsiveness (gain) and the arousal threshold were determined using multiple 3 min ‘CPAP pressure drops’: pharyngeal anatomy/collapsibility was quantified as the ventilation at CPAP = 0. LG was defined as the ratio of the ventilatory overshoot to the preceding reduction in ventilation. Upper-airway gain was taken as the ratio of the increase in ventilation to the increase in ventilatory drive across the drop. Arousal threshold was quantified as the level of ventilatory drive associated with arousal. The apnoea-hypopnoea index (AHI) was assessed on separate nights using standard polysomnography. Acetazolamide reduced the median [interquartile range] LG (3.4 [2.4–5.4]versus 2.0 [1.4–3.5]; P < 0.05) and NREM AHI (50 [36–57]versus 24 [13–42] events h−1; P < 0.05), but did not significantly alter pharyngeal anatomy/collapsibility, upper-airway gain, or arousal threshold. There was a modest correlation between the percentage reduction in LG and the percentage reduction in AHI (r = 0.660, P = 0.05). Our findings suggest that acetazolamide can improve OSA, probably due to reductions in the sensitivity of the ventilatory control system. Identification of patients who may benefit from reductions in LG alone or in combination with other therapies to alter the remaining traits may facilitate pharmacological resolution of OSA in the future.
Key points
Obstructive sleep apnoea (OSA) probably results from the interaction of key pathophysiological traits including compromised pharyngeal anatomy, inadequate upper-airway muscle function, high ventilatory response to a change in ventilation (high loop gain), and a low arousal threshold.
Because the standard therapy with positive airway pressure is often poorly tolerated, alternative options have long been sought which have included various pharmacological interventions.
Acetazolamide may be a useful therapeutic tool, yet there have been few studies examining how it affects the traits causing OSA.
Our study demonstrates that acetazolamide reduces loop gain by approximately 40% in individuals with OSA, but has little impact on the remaining OSA traits.
The marked reduction in loop gain with acetazolamide suggests that acetazolamide may be of therapeutic benefit when used alone or in combination with other therapies to treat individuals whose loop gain is known to contribute to OSA.
Introduction
Obstructive sleep apnoea (OSA) is a common disorder characterised by repetitive pharyngeal collapse during sleep, with its pathogenesis attributed to the interaction of at least four key physiological traits: (i) a compromised pharyngeal anatomy, (ii) the inadequate ability of the upper airway muscles to stiffen/dilate the airway in response to an increase in ventilatory drive, (iii) a hypersensitive ventilatory control system (i.e. high loop gain, LG) defined by a large ventilatory response to a change in ventilation, and (iv) a low arousal threshold (Smith et al. 1988; Younes et al. 2001; White, 2005; Eckert et al. 2011; Wellman et al. 2011). Continuous positive airway pressure (CPAP) is the most common treatment but is often poorly tolerated; only ∼50% of patients diagnosed with OSA continue therapy beyond 3 months (Kribbs et al. 1993; Engleman & Wild, 2003). Given this limitation, an alternative approach for non-adherent patients may be via the pharmacological manipulation of one or more pathogenic traits (Heinzer et al. 2008; Eckert et al. 2011; Wellman et al. 2011).
To date, several studies have assessed pharmacological agents for the treatment of OSA. The respiratory stimulant acetazolamide, a carbonic anhydrase inhibitor that produces a metabolic acidosis yielding an increase in baseline ventilation, has received some attention as a potential treatment for OSA. This potential is suggested by its utility to attenuate central sleep apnoea in healthy individuals at high altitude and in heart-failure patients (Sutton et al. 1979; Javaheri, 2006). In OSA patients, acetazolamide improves OSA severity in some, but not all, individuals studied (Sharp et al. 1985; Tojima et al. 1988; Whyte et al. 1988; Sakamoto et al. 1995). Such disparities are likely to relate to differences in the underlying pathophysiological traits between individuals and how acetazolamide affects the various traits responsible for OSA.
Based on the understanding that central sleep apnoea is a high LG ventilatory control disorder (Cherniack, 1981; Khoo et al. 1982; Francis et al. 2000; Solin et al. 2000; Javaheri, 2005; Topor et al. 2007; Kee et al. 2010), acetazolamide is likely to be an alternative means to reduce LG which may reduce OSA severity in selected patients. Investigations examining the mechanisms by which acetazolamide may alter LG have focused primarily on the ventilatory responses to hypercapnic and hypoxic stimuli (controller gain). However, ventilatory stability is determined by the entire feedback loop controlling ventilation, which also incorporates the degree to which 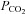 and
and 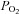 are altered by ventilation (plant gain). Thus, the potential for acetazolamide to alter LG in OSA patients remains unknown. Furthermore, it is also unknown whether acetazolamide alters the remaining key traits (pharyngeal anatomy, upper airway responsiveness, arousal threshold) contributing to OSA pathogenesis.
are altered by ventilation (plant gain). Thus, the potential for acetazolamide to alter LG in OSA patients remains unknown. Furthermore, it is also unknown whether acetazolamide alters the remaining key traits (pharyngeal anatomy, upper airway responsiveness, arousal threshold) contributing to OSA pathogenesis.
Accordingly, the current study aimed to investigate the effect of acetazolamide on four key OSA traits, and to determine how alterations in these traits affect OSA severity. We used our recently published technique to measure the four traits before and after acetazolamide administration, based on repeated ‘drops’ in CPAP levels during sleep (Wellman et al. 2011). Each trait is measured in a way that allows model-based predictions of the presence/absence of OSA. Based on previous data showing an improvement in central sleep apnoea with acetazolamide, we hypothesised that acetazolamide would reduce OSA severity predominantly by lowering LG, and would have minimal effects on the remaining traits. Preliminary results of this analysis have been published in abstract form (Edwards et al. 2010).
Methods
Participants
Thirteen CPAP-treated OSA subjects aged 50 ± 2 years (mean ± SEM) with body mass index (BMI) of 34.2 ± 1.9 kg m−2 were recruited from the sleep laboratory at Brigham and Women's Hospital. All subjects had a history of OSA with an apnoea/hypopnoea index (AHI) >10 events h−1 during supine non-rapid eye movement (NREM) sleep and a documented CPAP use of >5 hours per night for at least two months prior to the study. Written, informed consent was given before participation in the study, which was approved by the Human Research Committee at Brigham and Women's Hospital and conformed to the standards set by the Declaration of Helsinki. Subjects were excluded if they were taking any medication known to influence breathing, sleep/arousal or muscle physiology. Additionally, subjects were excluded if they had a history of renal failure, neuromuscular disease or other major neurological disorders, uncontrolled diabetes, heart failure, central sleep apnoea/Cheyne–Stokes respiration, uncontrolled hypertension, thyroid disease, or any other unstable medical condition. Female subjects were screened to ensure that they were not pregnant. Four of the subjects were on medication to control hypertension (hydrochlorothiazide, Diltiazem and Lisinopril), and two subjects were receiving drugs for the treatment of gastro-oesophageal reflux.
An important consideration in our experimental design was whether to study subjects who are newly diagnosed with OSA that are yet to start CPAP therapy or OSA subjects who are currently treated with CPAP. A major issue with studying CPAP-naive OSA subjects is that we cannot be sure that any changes we observe in the OSA traits are attributed solely to administration of the drug. More specifically, if OSA (and associated hypoxia and sleep deprivation) is known to influence the traits, and acetazolamide improves sleep apnoea severity, then in untreated subjects it would be unclear how much of acetazolamide's influence on the traits was due to improved OSA rather than a direct effect of the pharmacological treatment. Thus we chose a study design (i.e. using CPAP-treated OSA subjects) which allowed us to control for the known confounding effects of OSA (and associated intermittent hypoxia and sleep fragmentation) on the physiological traits.
Experimental design and protocol
Subjects underwent a clinical polysomnography (PSG) to measure OSA severity and a research PSG to measure four OSA traits (consecutive nights) under both baseline and acetazolamide conditions. The order of the baseline and acetazolamide studies was randomly assigned with a ≥1 week washout between conditions. For the acetazolamide studies, subjects took 500 mg of slow-release acetazolamide twice daily for 7 days while still on CPAP, and the clinical and research PSGs were performed on days 6–7.
Clinical PSG
Subjects were asked to sleep supine for the majority of the night with the standard clinical montage that included electroencephalogram (EEG), electrooculogram, sub-mental and leg electromyogram, electrocardiogram, nasal pressure and thermistor, respiratory effort (piezo-electric bands placed around the chest and abdomen), body position, and arterial oxygen saturation monitored at the finger. Data were collected and stored using the Alice digital PSG system (Philips Respironics, Murrysville, PA, USA). Sleep state, arousals and respiratory events were scored by a blinded sleep technician according to standard AASM Criteria (AASM et al. 2001). Specifically, a hypopnoea was defined by the presence of either a >50% reduction in airflow or a >30% reduction in airflow that was accompanied by a >3% desaturation in oxygen or the presence of an arousal. Hypopnoeas were classed as obstructive if flow limitation was present (defined as a characteristic peak–plateau or obvious flattening in nasal pressure) or central if no obvious flow limitation occurred. AHIs were then calculated for both supine NREM and REM sleep although correlations between the traits and AHI were carried out for NREM sleep only as the traits were measured during NREM sleep (see below).
Research PSG
Both subjective (patient-reported usage) and objective (downloaded from subject's machine) measures of CPAP compliance over the last month were taken to document adherence. On the acetazolamide night, side-effects were noted as well as the subjective opinion of whether patients ‘slept better, worse, or the same when taking the drug’. Sleep electrodes were attached similarly to the clinical PSG and were fitted with a nasal mask (Gel Mask; Respironics, Murrysville, PA, USA) attached to a pneumotachometer (model 3700A; Hans-Rudolph, Kansas City, MO, USA) for measuring airflow. Mask pressure was measured from a port in the mask (Validyne, Northridge, CA, USA). CO2 was continuously recorded from a catheter placed inside the nostril with a capnograph (Vacumed, Ventura, CA, USA). The mask was connected to a positive/negative pressure source (Philips-Respironics, Murrysville, PA, USA) to enable rapid switching between CPAP levels. At baseline, CPAP was provided at a level that abolished flow limitation and snoring, with flow limitation defined as a characteristic peak–plateau or obvious flattening in inspiratory airflow. All signals were sampled at 125 Hz and displayed using Nihon Kohden (Tokyo, Japan) and Spike2 software (Cambridge Electronic Design Ltd, Cambridge, UK).
Measuring the traits responsible for OSA
The method for measuring and modelling the traits responsible for OSA have been described previously (Wellman et al. 2011). Briefly, during stable NREM sleep 3 min CPAP ‘drops’ to subtherapeutic levels were repeatedly performed. When CPAP is initially dropped (Fig. 1A), the airway becomes partially obstructed and ventilation is reduced (Fig. 1B). The reduction in ventilation leads to an increase in ventilatory drive (red dashed line, Fig. 1B). In response to the increase in drive (labelled as x, Fig. 1B), the subjects may activate their upper airway muscles and partly reopen the airway, allowing for ventilation to recover somewhat (labelled as z, Fig. 1B). The ratio of z to x is the upper airway gain (UAG), and is taken as a measure of the ability of the airway to stiffen/dilate in response to an increase in ventilatory drive; a positive value indicates that an increase in ventilatory drive ‘buys back’ (recovers) ventilation, whereas a negative value indicates that increased drive is associated with reduced airflow (i.e. the airway becomes smaller while drive rises from one breath to the next). The UAG was calculated from CPAP drops that did not end in arousal. These responses were then averaged to determine a mean upper airway gain for the subject. The increase in ventilatory drive accumulated during the drop (x) is determined by rapidly reopening the airway with therapeutic CPAP and measuring the overshoot in ventilation. The ratio of this ventilatory response (x) to the net reduction in ventilation (y; Fig. 1B) provides a measure of steady-state LG (where LG = x/y). The time course of the rise in ventilatory drive during each drop is determined by transforming ventilation into ventilatory drive based on three parameters: LG, delay and a time constant. These parameters are quantified using the time course of ventilation following the return to the therapeutic pressure (Wellman et al. 2011) and also allow ‘dynamic’ LG to be calculated (see Supplemental Material). In order for a drop to be considered for LG measurement, ventilation during the last 60 s of the drop had to be significantly lower than eupnoeic ventilation on optimum CPAP and no arousals could occur during this interval. To establish the mechanism responsible for changes in LG with acetazolamide, steady-state plant gain and controller gain were obtained; plant gain was given by the reciprocal of the slope of the metabolic hyperbola during NREM sleep, and controller gain was defined as LG/plant gain.
Figure 1. Technique for determining and modelling the physiological traits using CPAP drops.
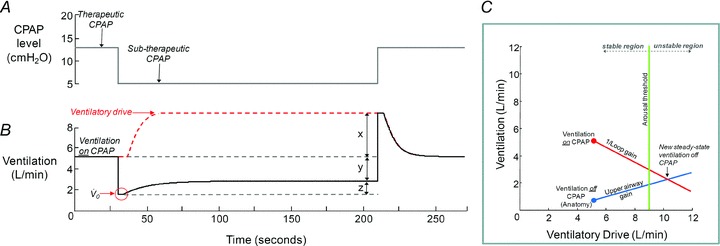
To determine the four physiological traits, CPAP is dropped to a subtherapeutic level (A) which causes partial airway obstruction and a reduction in ventilation (B). Using the time course in ventilation, we calculate the pharyngeal anatomy/collapsibility (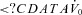 ), upper airway gain (z/x), loop gain (x/y) and arousal threshold (if an arousal occurs); see text for details. C depicts how these four traits can be incorporated into a model to predict the susceptibility towards OSA. In this example, the new steady state (the intersection of the diagonal lines) occurs in the unstable region (i.e. right of the arousal threshold) and thus OSA is predicted to be present.
), upper airway gain (z/x), loop gain (x/y) and arousal threshold (if an arousal occurs); see text for details. C depicts how these four traits can be incorporated into a model to predict the susceptibility towards OSA. In this example, the new steady state (the intersection of the diagonal lines) occurs in the unstable region (i.e. right of the arousal threshold) and thus OSA is predicted to be present.
The ‘passive’ anatomy (i.e. pharyngeal collapsibility) was determined by plotting mask pressure versus ventilation for the 2nd–3rd breaths of all the pressure drops throughout the entire night. The data were fitted with a regression line and the ventilation at zero mask pressure (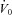 ) was used to measure pharyngeal anatomy/collapsibility. Finally, if an arousal occurred during a drop, then the arousal threshold could be quantified as the level of ventilatory drive immediately preceding the arousal. In order to be counted as a respiratory-related arousal (i.e. driven by increases in respiratory drive and not a spontaneous arousal), we required the presence of flow limitation on the preceding breaths and that the arousal had to occur no later than 90 s after the beginning of the CPAP drop.
) was used to measure pharyngeal anatomy/collapsibility. Finally, if an arousal occurred during a drop, then the arousal threshold could be quantified as the level of ventilatory drive immediately preceding the arousal. In order to be counted as a respiratory-related arousal (i.e. driven by increases in respiratory drive and not a spontaneous arousal), we required the presence of flow limitation on the preceding breaths and that the arousal had to occur no later than 90 s after the beginning of the CPAP drop.
Measurements of pharyngeal anatomy/collapsibility, UAG, LG, and the arousal threshold were then averaged and displayed using our model (Wellman et al. 2011). Figure 1C depicts how all four traits can be incorporated into a model to predict the susceptibility towards OSA. Ventilatory drive is represented on the x-axis and ventilation on the y-axis. The red circle represents the ventilation ‘on’ therapeutic CPAP (e.g. 5.1 l min−1) and is equal to resting ventilatory drive (i.e. the ventilatory drive requirements at rest are met by ventilation). The slope of the red line is the reciprocal of the calculated LG, which shows how ventilatory drive increases as ventilation falls. The blue dot represents the ventilation ‘off’ CPAP and the slope of the blue line indicates how ventilation is altered by increases in ventilatory drive (i.e. upper-airway gain). The point of intersection between the LG and upper-airway gain lines is the new, predicted steady-state ventilation ‘off’ CPAP. The green line represents the ventilatory drive threshold that when reached causes an arousal from sleep; to the left of this threshold breathing would be in the stable region but unstable if it was to the right. What the model says is that if CPAP were to be removed in this individual, ventilation (at the eupnoeic level of ventilatory drive) would fall to 0.7 l min−1. As ventilatory drive increases, the individual will increase the ventilation along the UAG slope until it reaches the arousal threshold (green line) or intersects with the 1/LG line. If the subject were to reach the arousal threshold before the 1/LG line (as seen in Fig. 1C), that is the intersection occurs in the unstable region, then the subject would arouse and the process will start again (i.e. this individual would have OSA). If, however, the arousal threshold was to the right of the intersection, then stable breathing may be achieved without arousal (i.e. no OSA is present).
Statistical analysis
Student's paired t test or Wilcoxon's signed-rank tests were used to assess the effects of acetazolamide as appropriate and were performed using SigmaPlot (Systat Software Inc., San Jose, CA, USA). Changes in the OSA traits with acetazolamide and the NREM AHI were assessed using linear regression. P < 0.05 was considered significant. Values are presented as means ± SEM or medians [interquartile range].
Results
Following acetazolamide, 12 subjects reported the presence of paresthesias and taste disturbances and three people reported nocturia as side-effects of the treatment. Four subjects described no difference in their sleep quality while receiving acetazolamide whereas the remaining subjects felt they slept better. One subject developed hypokalaemia while taking acetazolamide (possibly due to an interaction with hydrochlorothiazide) and this person's participation was discontinued and the data excluded from the analysis presented. Table 1 shows the effect of acetazolamide administration on the ventilatory parameters and electrolytes. As expected, acetazolamide significantly increased minute ventilation (through an increase in tidal volume), and reduced end-tidal CO2 and serum HCO3−. Objective CPAP adherence while receiving acetazolamide during the week of the study did not differ from their previous compliance in the month prior to enrolment (5.5 ± 0.5 vs. 5.9 ± 0.4 h per night). There was no significant difference in BMI (34.2 ± 1.9 vs. 33.9 ± 2.0 kg m−2; P = 0.32) or neck circumference measured either standing (40.9 ± 0.9 vs. 40.5 ± 1.4 cm; P = 0.58) or supine (43.0 ± 1.5 vs. 42.9 ± 1.1 cm; P = 0.87) between the two conditions.
Table 1.
Effect of acetazolamide on respiratory variables (during stable NREM sleep on CPAP) and electrolytes
| Variable (n = 12) | Baseline | Acetazolamide |
|---|---|---|
| Respiratory variables | ||
| Minute ventilation (l min−1) | 7.2 ± 0.3 | 8.4 ± 0.4* |
| Tidal volume (l) | 0.55 ± 0.03 | 0.71 ± 0.06* |
| Respiratory rate (breaths min−1) | 14.4 ± 0.7 | 13.9 ± 0.7* |
| End-tidal CO2 (mmHg) | 40.2 ± 0.8 | 32.5 ± 1.0* |
| Plasma electrolytes | ||
| [HCO3−] (mmol l−1) | 23.8 ± 0.7 | 19.3 ± 0.6* |
| [Na+] (mmol l−1) | 139.5 | 138.5 |
| [137.5–140.0] | [137.3–141.0] | |
| [K+] (mmol l−1) | 4.3 ± 0.1 | 3.6 ± 0.1* |
| [Cl−] (mmol l−1) | 102.5 ± 0.7 | 108.6 ± 0.9* |
| BUN (mg dl−1) | 16.0 [15.0–19.5] | 20.0 [17.3–23.8]* |
| Creatinine (mg dl−1) | 0.98 ± 0.05 | 1.15 ± 0.05* |
| BUN/creatinine ratio | 17.6 ± 1.3 | 18.7 ± 1.4 |
Values are means ± SEM or medians [interquartile range].
Significant difference compared with the baseline condition. Electrolyte measurements during baseline and acetazolamide studies were obtained from a venous blood draw in the evening prior to the research night.
Effect of acetazolamide on the OSA traits
The four traits were estimated from 27.9 ± 1.8 and 33.2 ± 1.7 CPAP drops per subject per night on the baseline and acetazolamide nights, respectively. An example trace illustrating the calculation of three traits from one individual is shown in Fig. 2. The effect of acetazolamide on the traits is shown in Table 2, with the overall model diagrams depicted in Fig. 3A and B. As a group, acetazolamide significantly reduced the median LG (P < 0.05) by 41% (Fig. 4A). Further analysis shows that this reduction is primarily driven by a reduction in plant gain (4.5 ± 0.4 vs. 2.6 ± 0.3 mmHg l−1min−1; P = 0.001) as controller gain remained unchanged (1.0 ± 0.2 vs. 1.0 ± 0.2 l min−1mmHg−1; P = 0.9). Importantly, acetazolamide did not alter the remaining OSA traits despite a trend towards an increased arousal threshold (P = 0.06); consistent with this trend, there was no reduction in the difference between resting ventilation and the arousal threshold (i.e. arousal reserve) between baseline (3.4 ± 1.1 l min−1) and acetazolamide nights (3.2 ± 1.3 l min−1). Individual data regarding acetazolamide's effect on all traits as well as the model diagrams for two typical individuals are provided in the Supplemental Material. When the data for the four traits were combined, our OSA model revealed that the acetazolamide-induced reduction in LG shortens the distance between the projected steady-state ventilation off CPAP (intersection of the LG and UAG lines, see Fig. 4B) and the arousal threshold by 2.3 l min−1 (∼64%).
Figure 2. An example of a CPAP drop.
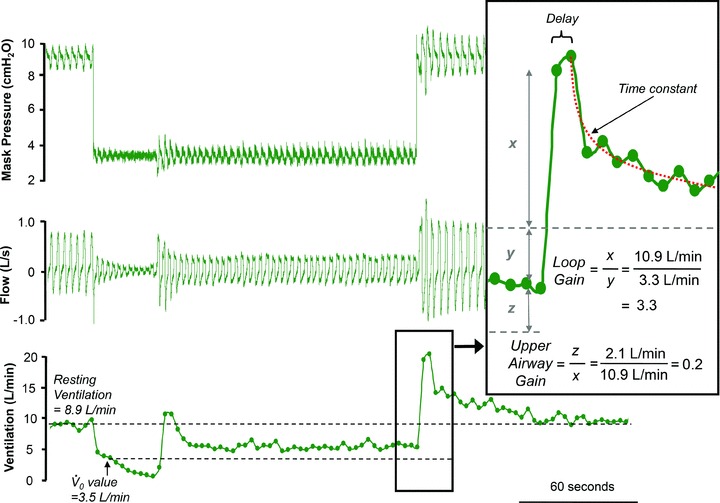
A representative example of a CPAP drop taken from one subject. The top panel shows the CPAP level being dropped from 9 to 3.5 cmH2O. The middle panel shows the reduction in ventilatory flow associated with the CPAP drop. The bottom panel shows the minute ventilation. When CPAP is dropped, minute ventilation falls from 8.9 to 3.5 l min−1. To estimate the pharyngeal anatomy or 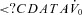 , minute ventilation immediately after the drop is plotted on a graph against mask pressure. To estimate LG, the overshoot in ventilation (x) is divided by the amount ventilation was reduced (y). In this example, LG (x/y) is 3.3 and the upper airway gain (z/x) is +0.2 (inset panel). A delay and time constant is also calculated from the ventilatory overshoot which is used for the calculation of ventilatory drive and dynamic LG (see Supplemental Material). In this example, there was no EEG arousal, such that this drop could not be used to determine arousal threshold.
, minute ventilation immediately after the drop is plotted on a graph against mask pressure. To estimate LG, the overshoot in ventilation (x) is divided by the amount ventilation was reduced (y). In this example, LG (x/y) is 3.3 and the upper airway gain (z/x) is +0.2 (inset panel). A delay and time constant is also calculated from the ventilatory overshoot which is used for the calculation of ventilatory drive and dynamic LG (see Supplemental Material). In this example, there was no EEG arousal, such that this drop could not be used to determine arousal threshold.
Table 2.
Effect of acetazolamide on the pathological traits causing OSA
| Trait | Baseline | Acetazolamide | P-value |
|---|---|---|---|
| Loop gain | 3.4 [2.4–5.4] | 2.0 [1.4–3.5] | <0.05 |
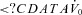 (l min−1) (l min−1) |
3.5 [3.1–4.4] | 4.8 [2.5–5.3] | 0.15 |
| Upper-airway gain | 0.25 [–0.26–0.55] | 0.27 [–0.27–0.58] | 0.65 |
| Arousal threshold (l min−1) | 10.5 [10.1–11.8] | 12.5 [9.8–15.5] | 0.06 |
Data represent medians [interquartile ranges].
Figure 3. Overall effect of acetazolamide on the physiological traits.
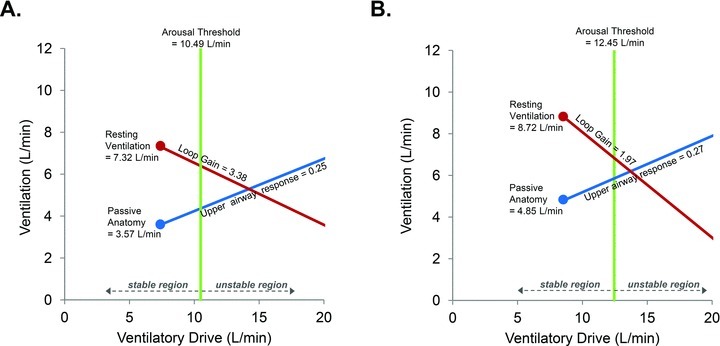
Median data of resting ventilation and the four physiological traits at baseline (A) and during acetazolamide treatment (B) from all 12 subjects. Administration of acetazolamide significantly increased resting ventilation (P < 0.001) during stable NREM sleep and reduced LG (P < 0.05). However, administration of acetazolamide did not significantly alter the remaining traits. Note that the reduction in LG decreases the distance between the steady-state intersection and the arousal threshold from 3.6 l min−1 to 1.2 l min−1 (i.e. the steady-state intersection is closer to the stable region). However, the intersection has not crossed over into the stable region, which may explain the ability of acetazolamide to reduce, but not eliminate, OSA.
Figure 4. Individual effects of acetazolamide on loop gain and the severity of sleep-disordered breathing.
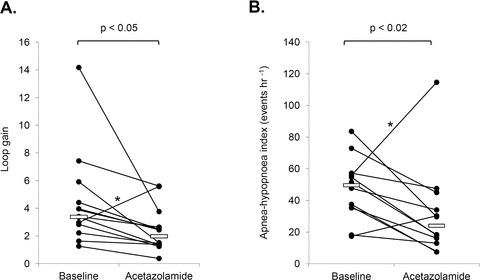
A, individual data comparing steady-state LG during baseline and acetazolamide conditions. Note that the steady-state LG was reduced in all but one subject. B, acetazolamide significantly reduced the median apnoea-hypopnoea index (AHI) from 49.6 [35.5–56.6] to 24.1 [12.9–42.3] events per hour, during supine NREM sleep. Median values for each condition are indicated by the solid white bars. Note that the one patient whose LG doubled with acetazolamide also exhibited a similar rise in AHI (asterisk).
Effect of acetazolamide on sleep and obstructive sleep apnoea severity
Table 3 summarises the effects of acetazolamide on the subjects’ sleep quality and architecture. Acetazolamide did not alter total sleep time, sleep efficiency, duration of REM and NREM sleep, or sleep stage percentages (except for a small increase in the proportion of time spent in NREM Stage 2). There were minimal central apnoeas/hypopnoeas or slow-wave sleep (NREM 3 and 4) during both studies (<1% of total sleep time). While mean overnight oxygen saturation was increased on acetazolamide, the nadir saturation was unchanged. Importantly, acetazolamide lowered the median supine NREM AHI by 51% (Fig. 4B), primarily due to a reduction in the number of obstructive hypopnoeas (Table 4). Note that all but two individuals showed an improvement in their AHI. Interestingly, the percentage reduction in LG was modestly correlated with the percentage reduction in AHI (r = 0.660, P = 0.05). The reduction in AHI was not limited to NREM sleep as a similar reduction was also seen in REM sleep (∼35% reduction); thus the overall AHI was reduced by ∼47% (data not shown). Despite trends for acetazolamide to increase the length of the apnoeas/hypopnoeas, this was only significant for NREM-related hypopnoeas (Table 4).
Table 3.
Effect of acetazolamide on sleep-disordered breathing
| Variable (n = 12) | Baseline | Acetazolamide |
|---|---|---|
| Total sleep time (min) | 339.8 ± 11.1 | 337.7 ± 11.0 |
| REM duration (min) | 35.0 ± 4.3 | 31.6 ± 4.1 |
| NREM duration (min) | 304.8 ± 12.3 | 306.1 ± 12.8 |
| % Sleep efficiency (TST/TIB) | 80.5 ± 2.2 | 82.6 ± 2.4 |
| % REM (TST) | 10.4 ± 1.3 | 9.4 ± 1.2 |
| % Stage 1 (TST) | 37.6 ± 2.9 | 32.3 ± 3.2 |
| % Stage 2 (TST) | 51.5 ± 2.0 | 57.9 ± 2.6* |
| % Stage 3 (TST) | 0 [0–0.2] | 0 [0–0] |
| % Stage 4 (TST) | 0 [0–0] | 0 [0–0] |
Mean overnight 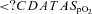 (%) (%) |
94.5 ± 0.4 | 96.0 ± 0.4* |
Lowest 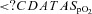 (%) (%) |
81.0 [77.5–82.8] | 83.5 [79.3–87.0] |
| Total arousal index (events h−1) | 41.1 ± 3.2 | 34.1 ± 2.7* |
| Respiratory arousal (without desaturation) | 2.7 [1.5–6.1] | 3.7 [2.0–7.2] |
| Respiratory arousal (with desaturation) | 23.2 ± 3.4 | 13.8 ± 3.3* |
| Leg movement arousals | 0.1 [0–0.4] | 0 [0–0.7] |
| Spontaneous arousal index (events h−1) | 13.6 ± 1.7 | 14.9 ± 2.1 |
| Leg movements (events h−1) | 0.4 [0–6.8] | 0.6 [0–7.8] |
Group data from the clinical PSG during the baseline and acetazolamide conditions. Data were collected in the supine posture in the absence of CPAP. Values are means ± SEM or medians [interquartile range].
Significant difference compared with the baseline condition.
Table 4.
Effect of Acetazolamide on NREM and REM sleep-disordered breathing events and durations
| Index (events h−1) | Event duration (s) | |||
|---|---|---|---|---|
| Sleep stage | Baseline | Acetazolamide | Baseline | Acetazolamide |
| NREM sleep | ||||
| Overall AHI | 49.6 [35.5–56.6] | 24.0 [12.9–42.3]* | 32.3 ± 2.7 | 37.7 ± 2.8* |
| Central apnoeas | 0 [0–0.2] | 0 [0–0] | 0 [0–15.1] | 0 [0–0] |
| Obstructive apnoeas | 6.0 ± 2.1 | 5.7 ± 2.8 | 23.8 [4.1–39.1] | 26.9 [17.3–43.4] |
| Mixed apnoeas | 0 [0–1.1] | 0 [0–0.2] | 0 [0–28.3] | 0 [0–13.0] |
| Overall hypopnoeas | 33.9 [23.5–50.0] | 17.9 [12.7–29.9]* | 32.1 ± 2.6 | 38.2 ± 2.7* |
| – obstructive | 33.9 [19.9–48.9] | 17.6 [12.7–28.9]* | ||
| – central | 0.3 [0–1.9] | 0.2 [0–0.4] | ||
| REM sleep | ||||
| Overall AHI | 43.2 [31.5–55.7] | 28.0 [18.5–35.0]* | 33.4 ± 3.1 | 38.1 ± 3.7 |
| Central apnoeas | 0 [0–0] | 0 [0–0] | 0 [0–0] | 0 [0–0] |
| Obstructive apnoeas | 9.7 ± 3.6 | 5.2 ± 2.0 | 15.4 [0–29.1] | 15.2 [0–35.3] |
| Mixed apnoeas | 0 [0–1.7] | 0 [0–0] | 0 [0–13.0] | 0 [0–0] |
| Overall hypopnoeas | 30.4 [13.4–47.9] | 23.8 [13.9–28.6] | 35.0 ± 3.3 | 38.9 ± 3.5 |
| – obstructive | 30.4 [10.5–46.6] | 23.8 [13.9–27.8] | ||
| – central | 0 [0–1.9] | 0 [0–0] | ||
Data represent medians [interquartile ranges] or means ± SEM.
Significant difference between baseline and acetazolamide
Discussion
The major novel finding of our study was that acetazolamide reduced loop gain (LG) by 41%, with minimal effects on the remaining traits. Importantly, as acetazolamide did not worsen the other traits, it implies that the failure of acetazolamide to improve OSA in some patients is unlikely to be due to a worsening of other key OSA traits. Our study also demonstrated that administration of acetazolamide for 7 days was also associated with a 51% improvement in the AHI. The acetazolamide-induced reduction in LG reflects a smaller increase in ventilatory drive in response to an upper-airway induced reduction in ventilation, thereby making it easier for an individual to reach stable breathing without arousing. Importantly, our finding that the reduction in LG was associated with a reduction in AHI suggests that acetazolamide improves OSA by reducing LG. This finding also highlights the importance of LG in the pathogenesis of OSA (Younes et al. 2001; Salloum et al. 2009).
Methodological considerations
A potential limitation of our study is that we did not use a placebo control to blind subjects to acetazolamide treatment, but this was partially rectified by randomising subjects to whether they received acetazolamide on their first or second study. In addition, objective measurements were made during sleep, minimising the potential effects of participant or investigator expectations. The lack of placebo and blinding are unlikely to have affected our results given that we found almost identical reductions in AHI as seen in a previous randomised double-blind, placebo controlled study in OSA patients with similar demographics and dosage used as in current study (Whyte et al. 1988).
It could be argued that the reduction in NREM AHI was simply a product of the observed increase in the length of the respiratory events following acetazolamide administration, and thus the perceived improvement is misleading. However, if a patient had on average one event every 64 s (based on an apnoea/hypopnoea lasting approximately 32 s followed by a hyperpnoea of a similar length), then the AHI would be 58 events h−1 if the patient cycled continuously. As we saw acetazolamide lengthen the apnoea/hypopnoea by 5 s, we would now expect events to occur every 74 s which would only decrease the AHI to 48 events h−1 (17% reduction). As we observed a 3-fold larger reduction in the AHI, we believe the improvement in OSA severity was not simply related to an increase in apnoea/hypopnoea length. In addition, we would emphasise that the minor prolongation of events led to no worsening of severity of the oxygen desaturations.
A final concern was that the observed improvement in AHI (driven by a reduction in hypopnoeas) may have reflected a shortcoming of the scoring system rather than a clinically relevant reduction in the number of events. Specifically, hypopnoeas may still have occurred on acetazolamide but were no longer scored as a respiratory event because they were not associated with an arousal or desaturation (as individuals are now sitting on the flat region of the oxygen desaturation curve where small decrements in 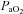 are unlikely to cause
are unlikely to cause 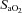 to decrease). In order to assess the effect of our scoring system on our findings, we performed an alternative analysis which characterises events using only a 30% reduction in nasal airflow without the need for arousal or desaturation (see Supplemental Material). Importantly, the alternative analysis demonstrated that acetazolamide still significantly improved the AHI by a magnitude similar to that observed with the original scoring system. Thus we can be confident that acetazolamide is truly reducing the number of events and improving OSA.
to decrease). In order to assess the effect of our scoring system on our findings, we performed an alternative analysis which characterises events using only a 30% reduction in nasal airflow without the need for arousal or desaturation (see Supplemental Material). Importantly, the alternative analysis demonstrated that acetazolamide still significantly improved the AHI by a magnitude similar to that observed with the original scoring system. Thus we can be confident that acetazolamide is truly reducing the number of events and improving OSA.
Effect of acetazolamide on loop gain
The current study is the first to provide data to our knowledge regarding the effect of acetazolamide on the LG of the ventilatory system in humans during sleep. Previous investigations have mostly been in awake healthy individuals and have focused on how acetazolamide affects only a single component of the ventilatory control feedback loop, CO2 sensitivity (a component of controller gain), with the results being disparate. Studies in sleeping dogs (Nakayama et al. 2002), healthy individuals living at sea level (White et al. 1982; Bashir et al. 1990; Teppema & Dahan, 1999) or at altitude (Rivera-Ch et al. 2008) have reported no change in CO2 sensitivity. Although we did not measure the awake ventilatory sensitivity to CO2 using traditional methods, our calculations of controller gain during sleep support these previous findings that suggest that CO2 sensitivity remains unchanged with acetazolamide. However, other animal studies report that acetazolamide reduces the sensitivity of peripheral and central chemoreceptors (Wagenaar et al. 1998; Teppema et al. 2001; Yamauchi et al. 2007). Conversely, the only study conducted in OSA subjects suggests acetazolamide increases CO2 sensitivity, which would tend to raise LG (Tojima et al. 1988). The differential effects on controller gain in previous studies may be due to several factors: differences in the dosage, the species/strain studied, the method for measuring controller gain and the use of anaesthetics that may have altered respiratory control. A recent study in rabbits measured acetazolamide's effect on both components of the overall LG, controller gain and plant gain (Kiwull-Schone et al. 2006). Interestingly this study reported a 35% reduction in LG, which was driven by a 27% reduction in controller gain and a 12% reduction in plant gain. Despite this finding, the majority of studies in humans suggest that controller gain remains unchanged, suggesting the reduction in LG must be driven by reductions in plant gain, an effect expected based on the reduction in 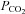 and increase in ventilation (Khoo et al. 1982; Francis et al. 2000). Our finding that the reduction in LG was primarily driven by the reduction in plant gain confirms this notion.
and increase in ventilation (Khoo et al. 1982; Francis et al. 2000). Our finding that the reduction in LG was primarily driven by the reduction in plant gain confirms this notion.
Supplemental oxygen has been shown to selectively reduce LG (measured using proportional assist ventilation) and AHI by approximately 50% in OSA patients with a high LG but has no effect in those with a low LG (Wellman et al. 2008), suggesting that reducing LG may be of benefit only in those with a high LG. In contrast, the current study demonstrates that acetazolamide significantly reduces LG and improves OSA severity regardless of whether LG is high or low (see Supplemental Material). The disparity between our previous and current study may be attributed to the scope of oxygen to lower LG. Previous studies in untreated OSA patients (Loewen et al. 2009; Salloum et al. 2009) suggest that a high LG is predominantly driven by an elevated controller gain, and we speculate that supplemental oxygen acts to lower controller gain and therefore LG in high LG patients. In those with low LG, controller gain is likely to be low and is therefore unlikely to be reduced further with oxygen therapy. Acetazolamide on the other hand, by working predominantly via lowering plant gain, reduces the overall LG and AHI regardless of the initial LG. Thus, our study demonstrates that any intervention that effectively reduces LG should have a beneficial effect on OSA severity.
Loop gain as an acquired pathophysiological trait
Two recent findings raised the possibility that increased LG may be a consequence of OSA rather than a pathophysiological trait. Loewen et al. (2009) demonstrated that CPAP therapy for 1 month in previously undiagnosed OSA patients reduced dynamic CO2 sensitivity back to the level of control subjects. Likewise, Salloum et al. (2009) showed that elevated CO2 sensitivity and steady-state LG are normalised by CPAP therapy, supporting the view that elevated LG is an acquired rather than inherent trait, and raising doubt as to the causal influence of LG in OSA. The current study measured LG in OSA patients already compliant with CPAP therapy, such that any acquired abnormality should have subsided and the baseline LG values reflect the ‘inherent’ pathophysiology. Indeed, our baseline LG values (median = 3.4, mean = 4.0) are remarkably similar to those of Salloum et al. (2009) following CPAP (mean = 3.9). Our demonstration that acetazolamide reduces LG and AHI provides further evidence for a causal role of LG in the pathophysiology of OSA. In aggregate, these data suggest that elevated LG is both an acquired and pathophysiological trait, constituting a positive feedback mechanism whereby OSA elevates LG, which in turn worsens OSA severity until the cycle is broken with clinical intervention.
Effect of acetazolamide on 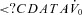 , GUA and arousal threshold
, GUA and arousal threshold
In the current study, we found no statistical improvement in our measurement of upper airway collapsibility (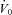 ). Acetazolamide may have been expected to improve
). Acetazolamide may have been expected to improve 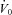 in two ways. First, acetazolamide increases baseline ventilatory drive, which is expected to stimulate the upper airway muscles (stiffen the airway) and activate the diaphragm at baseline. Although the passive anatomy per se may not be improved, the ventilation achieved off CPAP (at eupnoeic drive) might be improved in some individuals. Second, as acetazolamide is a mild diuretic, we considered that this effect may also improve upper airway collapsibility by reducing upper airway oedema and the rostral fluid shift observed overnight (Redolfi et al. 2009), although the impact that fluid shift and upper airway oedema contributes to OSA pathogenesis in obese individuals has been recently challenged (Jafari & Mohsenin, 2011). Given that we found no change in supine neck circumference with acetazolamide and no statistical improvement in
in two ways. First, acetazolamide increases baseline ventilatory drive, which is expected to stimulate the upper airway muscles (stiffen the airway) and activate the diaphragm at baseline. Although the passive anatomy per se may not be improved, the ventilation achieved off CPAP (at eupnoeic drive) might be improved in some individuals. Second, as acetazolamide is a mild diuretic, we considered that this effect may also improve upper airway collapsibility by reducing upper airway oedema and the rostral fluid shift observed overnight (Redolfi et al. 2009), although the impact that fluid shift and upper airway oedema contributes to OSA pathogenesis in obese individuals has been recently challenged (Jafari & Mohsenin, 2011). Given that we found no change in supine neck circumference with acetazolamide and no statistical improvement in 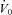 , the upper airway effects of acetazolamide appear to be minor.
, the upper airway effects of acetazolamide appear to be minor.
In addition, acetazolamide did not alter the responsiveness or effectiveness of the upper airway muscles (UAG). There was a trend for the arousal threshold to be increased, but this failed to reach significance. Notably the magnitude of the median increase in the arousal threshold almost paralleled the observed increase in minute ventilation (1.9 l min−1vs. 1.4 l min−1, respectively), such that there was clearly no change in the amount of additional ventilatory drive (above eupnoeic ventilatory drive) required for arousal (P = 0.79). This finding has important treatment implications, as one might have expected that any agent that increases ventilatory drive may also reduce the ‘arousal reserve’ and thereby increase the propensity to arousal. Importantly, our observation that 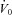 , GUA and arousal threshold remain unchanged with acetazolamide, even in those subjects whose AHI fell the least (see Supplemental Material); on this basis, we conclude that the failure to improve OSA in non-responders with acetazolamide is not the consequence of worsening key OSA traits.
, GUA and arousal threshold remain unchanged with acetazolamide, even in those subjects whose AHI fell the least (see Supplemental Material); on this basis, we conclude that the failure to improve OSA in non-responders with acetazolamide is not the consequence of worsening key OSA traits.
Clinical implications for the treatment of OSA
A common limitation of previous studies examining the effect of acetazolamide as a treatment for OSA is that they were performed in largely unselected populations. We believe a strength of the modelling approach used in this study is that it can provide better matching of patients with treatment. Importantly, the model could be used to make predictions about who might potentially benefit from an agent that lowers LG, such as acetazolamide. As an example, consider the model diagram shown in Fig. 3A and imagine that it is the actual model of a patient. If this patient took acetazolamide which lowered the LG by 41% (see Fig. 3B), then the steady state point would not be brought into the stable region. Thus, another agent might be needed to completely treat the OSA. For example, a sedative to increase the arousal threshold might also be needed. Studies from our laboratory have shown that the arousal threshold can be increased pharmacologically with either eszopiclone (Eckert et al. 2011) or trazodone (Heinzer et al. 2008) by approximately 28% and 48% respectively. If we conservatively assume we can increase the arousal threshold by 30% in addition to reducing the LG, the new steady-state point would now sit to the left of the arousal threshold (Fig. 5A). Alternatively, an improvement in the anatomy could be made; Schwartz and colleagues demonstrated that a 17% reduction in BMI decreased the Pcrit by 5.5 cmH2O (Schwartz et al. 1991). Such a reduction corresponds to an increase in 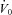 of approximately 2.5 l min−1 (if normal values for breath timing are used to convert peak flow to ventilation), which would also be predicted to stabilise breathing in this example (Fig. 5B). This concept is strengthened by an observation that when acetazolamide is given as an adjunctive therapy to individuals that have also had a uvulopalatopharyngoplasty (to improve anatomical compromise), the improvement in AHI with combined therapy is far greater than that achieved with acetazolamide or uvulopalatopharyngoplasty alone (Inoue et al. 1999). We consider that our model-based approach will allow clinicians to counsel patients more informatively on the potential success of any drug or therapy to resolve OSA.
of approximately 2.5 l min−1 (if normal values for breath timing are used to convert peak flow to ventilation), which would also be predicted to stabilise breathing in this example (Fig. 5B). This concept is strengthened by an observation that when acetazolamide is given as an adjunctive therapy to individuals that have also had a uvulopalatopharyngoplasty (to improve anatomical compromise), the improvement in AHI with combined therapy is far greater than that achieved with acetazolamide or uvulopalatopharyngoplasty alone (Inoue et al. 1999). We consider that our model-based approach will allow clinicians to counsel patients more informatively on the potential success of any drug or therapy to resolve OSA.
Figure 5. Potential effects of manipulating other traits in addition to loop gain on OSA severity.
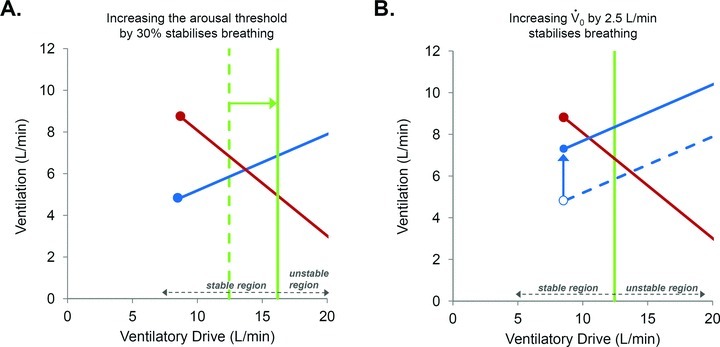
If, in addition to reducing LG by 41% with acetazolamide (taken from Fig. 3A), the arousal threshold was also increased by a small amount (e.g. 30%) with a sedative (A), or if the 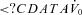 can be improved by approximately 2.5 l min−1 with weight loss (B), then the model predicts that this would move the steady-state intersection to the left of the arousal threshold and into the stable region (i.e. stable breathing is now possible). Therefore, our model allows us to speculate that such combination therapy could achieve a complete resolution of OSA.
can be improved by approximately 2.5 l min−1 with weight loss (B), then the model predicts that this would move the steady-state intersection to the left of the arousal threshold and into the stable region (i.e. stable breathing is now possible). Therefore, our model allows us to speculate that such combination therapy could achieve a complete resolution of OSA.
Concluding remarks
We have observed that acetazolamide approximately halved LG without significantly worsening the other pathogenic traits causing OSA. Such a magnitude of change means that acetazolamide is a useful tool for lowering LG and thus potentially treating OSA, either alone or in combination with another agent in selected patients. Moreover, our results are consistent with the causal role of LG in OSA pathogenesis in that a lowering of LG with acetazolamide produced a reduction in OSA severity. Furthermore, our results have highlighted the utility of an integrated model for determining which trait or combination of traits should be altered, and by how much, to eliminate OSA. The concept of combination therapy as a novel method for the future treatment of OSA thus indeed looks promising. However, further work is clearly needed using well-designed trials to document the effectiveness of this approach.
Acknowledgments
The authors would like to thank Miss Lauren Hess and Mrs Karen Stevenson for their laboratory assistance. This work was supported by the National Institutes of Health: 5R01HL048531-16, R01 HL085188-02, R01 HL090897-01A2, K24 HL 093218-01A1 and P01 HL 095491 and the American Heart Association: 0840159N, 0575028N. B.A.E. is supported by the Thoracic Society of Australia and New Zealand/Allen and Hanbury's Respiratory Research Fellowship. S.A.S is supported by an American Heart Association fellowship (11POST7360012). D.J.E. is supported by American Heart Association and a NHMRC of Australia Overseas Biomedical Fellowship (510392).
Glossary
Abbreviations
- AHI
apnoea–hypopnoea index
- CPAP
continuous positive airway pressure
- LG
Loop gain
- NREM
non-rapid eye movement
- OSA
obstructive sleep apnoea
- PSG
polysomnography
- REM
rapid eye movement
- TIB
time in bed
- TST
total sleep time
- UAG
upper airway gain
Author contributions
B.A.E, A.W, A.M, D.P.W and D.J.E contributed to the conception and design of the experiments. B.A.E, S.A.S, D.J.E, D.P.W, J.P.B, R.L.O, A.M and A.W. were involved in the collection, analysis and interpretation of data. B.A.E, S.A.S, D.J.E, D.P.W, J.P.B, R.L.O, A.M and A.W. were involved with drafting the article or critically reviewing it for important intellectual content. All authors have approved the final version of this article. The study was conducted in the Sleep Disorders Research Program Laboratory, Division of Sleep Medicine, Brigham and Women's Hospital, Boston.
Disclosures
D.J.E. is a consultant for Apnex Medical. A.M. is a consultant for Philips Respironics, Sepracor, Ethicon, Medtronic, SHC, SGS, Novartis, Apnex, Pfizer, Cephalon, Galleon and Merck. A.W. is a consultant for Philips Respironics. D.P.W. is the chief medical officer for Philips Respironics. All other authors have no conflicts to disclose, and do not have a financial relationship with a commercial entity that has an interest in the subject of this article.
References
- AASM; ESRS; JSSR; LASS. International Classification of Sleep Disorders, Revised: Diagnostic and Coding Manual. American Academy of Sleep Medicine; 2001. [Google Scholar]
- Bashir Y, Kann M, Stradling JR. The effect of acetazolamide on hypercapnic and eucapnic/poikilocapnic hypoxic ventilatory responses in normal subjects. Pulm Pharmacol. 1990;3:151–154. doi: 10.1016/0952-0600(90)90046-l. [DOI] [PubMed] [Google Scholar]
- Cherniack NS. Respiratory dysrhythmias during sleep. New Engl J Med. 1981;305:325–330. doi: 10.1056/NEJM198108063050606. [DOI] [PubMed] [Google Scholar]
- Eckert DJ, Owens RL, Kehlmann GB, Wellman A, Rahangdale S, Yim-Yeh S, White DP, Malhotra A. Eszopiclone increases the respiratory arousal threshold and lowers the apnoea/hypopnoea index in obstructive sleep apnoea patients with a low arousal threshold. Clin Sci (Lond) 2011;120:505–514. doi: 10.1042/CS20100588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards BA, Eckert DJ, Jordan AS, Malhotra A, Hess L, Stevenson K, White DP, Wellman A. Effect of acetazolamide in obstructive sleep apnea. Am J Respir Crit Care Med. 2010;181:A4199. [Google Scholar]
- Engleman HM, Wild MR. Improving CPAP use by patients with the sleep apnoea/hypopnoea syndrome (SAHS) Sleep Med Rev. 2003;7:81–99. doi: 10.1053/smrv.2001.0197. [DOI] [PubMed] [Google Scholar]
- Francis DP, Willson K, Davies LC, Coats AJ, Piepoli M. Quantitative general theory for periodic breathing in chronic heart failure and its clinical implications. Circulation. 2000;102:2214–2221. doi: 10.1161/01.cir.102.18.2214. [DOI] [PubMed] [Google Scholar]
- Heinzer RC, White DP, Jordan AS, Lo YL, Dover L, Stevenson K, Malhotra A. Trazodone increases arousal threshold in obstructive sleep apnoea. Eur Respir J. 2008;31:1308–1312. doi: 10.1183/09031936.00067607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Takata K, Sakamoto I, Hazama H, Kawahara R. Clinical efficacy and indication of acetazolamide treatment on sleep apnea syndrome. Psychiatry Clin Neurosci. 1999;53:321–322. doi: 10.1046/j.1440-1819.1999.00551.x. [DOI] [PubMed] [Google Scholar]
- Jafari B, Mohsenin V. Overnight rostral fluid shift in obstructive sleep apnea: does it affect the severity of sleep-disordered breathing? Chest. 2011;140:991–997. doi: 10.1378/chest.11-0044. [DOI] [PubMed] [Google Scholar]
- Javaheri S. Central sleep apnea in congestive heart failure: prevalence, mechanisms, impact, and therapeutic options. Semin Respir Crit Care Med. 2005;26:44–55. doi: 10.1055/s-2005-864206. [DOI] [PubMed] [Google Scholar]
- Javaheri S. Acetazolamide improves central sleep apnea in heart failure: a double-blind, prospective study. Am J Respir Crit Care Med. 2006;173:234–237. doi: 10.1164/rccm.200507-1035OC. [DOI] [PubMed] [Google Scholar]
- Kee K, Sands SA, Edwards BA, Berger PJ, Naughton MT. Positive airway pressure in congestive heart failure. In: Berry RB, editor. Sleep Medicine Clinics: Positive airway Pressure Therapy. Elsevier; 2010. pp. 393–405. [Google Scholar]
- Khoo MCK, Kronauer RE, Strohl KP, Slutsky AS. Factors inducing periodic breathing in humans: a general model. J Appl Physiol. 1982;53:644–659. doi: 10.1152/jappl.1982.53.3.644. [DOI] [PubMed] [Google Scholar]
- Kiwull-Schone H, Teppema L, Wiemann M, Kiwull P. Loop gain of respiratory control upon reduced activity of carbonic anhydrase or Na+/H+ exchange. Adv Exp Med Biol. 2006;580:239–244. doi: 10.1007/0-387-31311-7_37. discussion 351–239. [DOI] [PubMed] [Google Scholar]
- Kribbs NB, Pack AI, Kline LR, Smith PL, Schwartz AR, Schubert NM, Redline S, Henry JN, Getsy JE, Dinges DF. Objective measurement of patterns of nasal CPAP use by patients with obstructive sleep apnea. Am Rev Respir Dis. 1993;147:887–895. doi: 10.1164/ajrccm/147.4.887. [DOI] [PubMed] [Google Scholar]
- Loewen A, Ostrowski M, Laprairie J, Atkar R, Gnitecki J, Hanly P, Younes M. Determinants of ventilatory instability in obstructive sleep apnea: inherent or acquired? Sleep. 2009;32:1355–1365. doi: 10.1093/sleep/32.10.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama H, Smith CA, Rodman JR, Skatrud JB, Dempsey JA. Effect of ventilatory drive in carbon dioxide sensitivity below eupnea during sleep. Am J Respir Crit Care Med. 2002;165:1251–1259. doi: 10.1164/rccm.2110041. [DOI] [PubMed] [Google Scholar]
- Redolfi S, Yumino D, Ruttanaumpawan P, Yau B, Su MC, Lam J, Bradley TD. Relationship between overnight rostral fluid shift and obstructive sleep apnea in nonobese men. Am J Respir Crit Care Med. 2009;179:241–246. doi: 10.1164/rccm.200807-1076OC. [DOI] [PubMed] [Google Scholar]
- Rivera-Ch M, Huicho L, Bouchet P, Richalet JP, Leon-Velarde F. Effect of acetazolamide on ventilatory response in subjects with chronic mountain sickness. Respir Physiol Neurobiol. 2008;162:184–189. doi: 10.1016/j.resp.2008.06.010. [DOI] [PubMed] [Google Scholar]
- Sakamoto T, Nakazawa Y, Hashizume Y, Tsutsumi Y, Mizuma H, Hirano T, Mukai M, Kotorii T. Effects of acetazolamide on the sleep apnea syndrome and its therapeutic mechanism. Psychiatry Clin Neurosci. 1995;49:59–64. doi: 10.1111/j.1440-1819.1995.tb01858.x. [DOI] [PubMed] [Google Scholar]
- Salloum A, Rowley JA, Mateika JH, Chowdhuri S, Omran Q, Badr MS. Increased propensity for central apnea in patients with obstructive sleep apnea: effect of nCPAP. Am J Respir Crit Care Med. 2009;181:189–193. doi: 10.1164/rccm.200810-1658OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz AR, Gold AR, Schubert N, Stryzak A, Wise RA, Permutt S, Smith PL. Effect of weight loss on upper airway collapsibility in obstructive sleep apnea. Am Rev Respir Dis. 1991;144:494–498. doi: 10.1164/ajrccm/144.3_Pt_1.494. [DOI] [PubMed] [Google Scholar]
- Sharp JT, Druz WS, D'Souza V, Diamond E. Effect of metabolic acidosis upon sleep apnea. Chest. 1985;87:619–624. doi: 10.1378/chest.87.5.619. [DOI] [PubMed] [Google Scholar]
- Smith PL, Wise RA, Gold AR, Schwartz AR, Permutt S. Upper airway pressure-flow relationships in obstructive sleep apnea. J Appl Physiol. 1988;64:789–795. doi: 10.1152/jappl.1988.64.2.789. [DOI] [PubMed] [Google Scholar]
- Solin P, Roebuck T, Johns DP, Walters EH, Naughton MT. Peripheral and central ventilatory responses in central sleep apnea with and without congestive heart failure. Am J Respir Crit Care Med. 2000;162:2194–2200. doi: 10.1164/ajrccm.162.6.2002024. [DOI] [PubMed] [Google Scholar]
- Sutton JR, Houston CS, Mansell AL, McFadden MD, Hackett PM, Rigg JR, Powles AC. Effect of acetazolamide on hypoxemia during sleep at high altitude. New Engl J Med. 1979;301:1329–1331. doi: 10.1056/NEJM197912133012406. [DOI] [PubMed] [Google Scholar]
- Teppema LJ, Dahan A. Acetazolamide and breathing. Does a clinical dose alter peripheral and central CO2 sensitivity? Am J Respir Crit Care Med. 1999;160:1592–1597. doi: 10.1164/ajrccm.160.5.9903088. [DOI] [PubMed] [Google Scholar]
- Teppema LJ, Dahan A, Olievier CN. Low-dose acetazolamide reduces CO2–O2 stimulus interaction within the peripheral chemoreceptors in the anaesthetised cat. J Physiol. 2001;537:221–229. doi: 10.1111/j.1469-7793.2001.0221k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tojima H, Kunitomo F, Kimura H, Tatsumi K, Kuriyama T, Honda Y. Effects of acetazolamide in patients with the sleep apnoea syndrome. Thorax. 1988;43:113–119. doi: 10.1136/thx.43.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topor ZL, Vasilakos K, Younes M, Remmers JE. Model based analysis of sleep disordered breathing in congestive heart failure. Respir Physiol Neurobiol. 2007;155:82–92. doi: 10.1016/j.resp.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Wagenaar M, Teppema L, Berkenbosch A, Olievier C, Folgering H. Effect of low-dose acetazolamide on the ventilatory CO2 response during hypoxia in the anaesthetized cat. Eur Respir J. 1998;12:1271–1277. doi: 10.1183/09031936.98.12061271. [DOI] [PubMed] [Google Scholar]
- Wellman A, Eckert DJ, Jordan AS, Edwards BA, Passaglia CL, Jackson AC, Gautam S, Owens RL, Malhotra A, White DP. A method for measuring and modeling the physiological traits causing obstructive sleep apnea. J Appl Physiol. 2011;110:1627–1637. doi: 10.1152/japplphysiol.00972.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman A, Malhotra A, Jordan AS, Stevenson KE, Gautam S, White DP. Effect of oxygen in obstructive sleep apnea: Role of loop gain. Respir Physiol Neurobiol. 2008;162:144–151. doi: 10.1016/j.resp.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DP. Pathogenesis of obstructive and central sleep apnea. Am J Respir Crit Care Med. 2005;172:1363–1370. doi: 10.1164/rccm.200412-1631SO. [DOI] [PubMed] [Google Scholar]
- White DP, Zwillich CW, Pickett CK, Douglas NJ, Findley LJ, Weil JV. Central sleep apnea. Improvement with acetazolamide therapy. Arch Int Med. 1982;142:1816–1819. [PubMed] [Google Scholar]
- Whyte KF, Gould GA, Airlie MA, Shapiro CM, Douglas NJ. Role of protriptyline and acetazolamide in the sleep apnea/hypopnea syndrome. Sleep. 1988;11:463–472. doi: 10.1093/sleep/11.5.463. [DOI] [PubMed] [Google Scholar]
- Yamauchi M, Dostal J, Strohl KP. Acetazolamide protects against posthypoxic unstable breathing in the C57BL/6J mouse. J Appl Physiol. 2007;103:1263–1268. doi: 10.1152/japplphysiol.01287.2006. [DOI] [PubMed] [Google Scholar]
- Younes M, Ostrowski M, Thompson W, Leslie C, Shewchuk W. Chemical control stability in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 2001;163:1181–1190. doi: 10.1164/ajrccm.163.5.2007013. [DOI] [PubMed] [Google Scholar]