

Abstract
The hairy/enhancer-of-split related with YRPW motif 1 (HEY1) is a member of the basic-helix-loop-helix-Orange (bHLH-O) family of transcriptional repressors that mediate Notch signaling. Several cancer-related pathways also regulate HEY1 expression, and HEY1 itself acts as an indirect positive regulator of the p53 tumor suppressor protein and a negative regulator of androgen receptor activity. In this study we show how a naturally occurring non-synonymous polymorphism at codon 94 of HEY1, which results in a substitution of leucine by methionine (Leu94Met), converts HEY1 from an androgen receptor corepressor to an androgen receptor co-activator without affecting its intrinsic transcriptional repressive domains. The polymorphism Leu94Met also abolishes HEY1-mediated activation of p53 and suppresses the ability of HEY1 to induce p53-dependent cell-cycle arrest and aberrant cell differentiation in human osteosarcoma U2OS cells. Moreover, expression of HEY1, but not of the variant Leu94Met, confers sensitivity to p53-activating chemotherapeutic drugs on U2OS cells. In addition, we have identified motifs in HEY1 that are critical for the regulation of its subcellular localization and analysed how mutations in those motifs affect both HEY1 and HEY1-Leu94Met functions. These findings suggest that the polymorphism Leu94Met in HEY1 radically alters its biological activities and may affect oncogenic processes.
Keywords: HEY1, androgen receptor, p53, Notch, single-nucleotide polymorphism
Introduction
The hairy/enhancer-of-split related with YRPW motif (HEY) proteins are a family of transcription factors that belong to the vertebrate basic-helix-loop-helix-Orange (bHLH-O) superfamily of transcriptional repressors (Davis and Turner, 2001; Iso et al., 2003). The HEY family consists of HEY1, HEY2 and HEYL, which are encoded by three distinct genes. The expression of HEY1 was initially shown to be directly regulated by Notch signaling (Maier and Gessler, 2000; Nakagawa et al., 2000; Iso et al., 2001a). Upon Notch activation, HEY1 expression increases and it accumulates in the nucleus, leading to transcriptional inhibition of downstream targets. Thus, HEY1 is a critical effector within the Notch signaling pathway during cardiac and vascular development, neurogenesis (Sakamoto et al., 2003), gliogenesis (Satow et al., 2001) and bone development (Zamurovic et al., 2004). Recent studies revealed that HEY1 is also a direct target gene of transforming growth factor-β/Smad signaling (Itoh et al., 2004; Zavadil et al., 2004; Sivertsen et al., 2007). In addition to its roles during embryonic development, HEY1 has also been linked to several cancer-related pathways. HEY1 expression is essential for transforming growth factor-β-dependent epithelial-to-mesenchymal transition, a developmental program of cell plasticity frequently observed in advanced carcinogenesis (Yang and Weinberg, 2008). The retinoblastoma protein/E2 transcription factor cell-cycle pathway can upregulate HEY1 expression in human glioma cells through E2 transcription factor-binding sites present in its promoter (Hulleman et al., 2009). Furthermore, it was found that HEY1 overexpression in neural stem cells promotes proliferation, whereas the impairment of HEY1 expression in glioblastoma cells has the opposite effect. HEY1 is upregulated in human gliomas and its expression in glioblastomas correlates with tumor grade and with survival, suggesting that HEY1 contributes to the proliferation of glioblastoma cells (Hulleman et al., 2009).
Changes in HEY1 expression levels or activity may have a widespread role during oncogenesis, as a genome-wide functional analysis revealed that HEY1 is a positive regulator of the p53 tumor suppressor protein. Ectopic expression of HEY1 activated p53 and induced apoptosis in vivo in different biological models through mechanisms that involved inhibition of murine double minute-2 and post-translational stabilization of p53 (Huang et al., 2004). In addition, we previously showed that HEY1 functions as a specific corepressor of the androgen receptor (AR), providing a mechanism for crosstalk between Notch and androgen-signaling pathways. Furthermore, an expression analysis revealed that the majority of prostate cancer samples showed total exclusion of HEY1 from the nucleus, raising the possibility that abnormal HEY1 subcellular distribution may have a role in the aberrant hormonal responses observed in prostate cancer (Belandia et al., 2005).
Interestingly, it was recently described that a naturally occurring non-synonymous single-nucleotide polymorphism (SNP) in HEY1 that changes the leucine at residue 94 to methionine in the bHLH domain abolishes its ability to repress the human dopamine transporter promoter, although the molecular mechanisms responsible for the lack of activity and the physiological consequences are still unknown (Fuke et al., 2005). Allele frequency data for HEY1-L94M SNP (SNP ID rs11553421), obtained by the International HapMap Project, indicates that it is present in approximately 1% of all studied populations (Asian, European and African).
We analysed how the L94M polymorphism affects the role of HEY1 as a regulator of the AR and p53 pathways. Our study has uncovered profound differences in HEY1 biological action caused by the single amino acid substitution L94M with a potential effect on cancer risk and cancer treatment response.
Results
A human non-synonymous SNP converts HEY1 from an AR repressor to an AR activator
We have previously shown that HEY1 is an AR corepressor (Belandia et al., 2005 and Figure 1b). To test the possible effects on HEY1 biological activity caused by the polymorphism L94M, we generated this variant by site-directed mutagenesis (Figure 1a) and tested the effect of L94M expression on the ability of AR to stimulate transcription from androgen-responsive luciferase reporter genes in the transfected cells. Surprisingly, the substitution L94M increased the activity of the AR in both COS-1 and U2OS cells, transforming HEY1 from an AR repressor to a potent AR activator (Figure 1b and Supplementary Figure 1a).
Figure 1.

Characterization of the hairy/enhancer-of-split related with YRPW motif 1 (HEY1) L94M variant. (a) Schematic representation of full-length HEY1, mutants and enhanced green fluorescent protein (EGFP) fusions used in this study. (b) L94M enhances androgen receptor (AR)-mediated hormone-dependent transactivation. COS-1 cells were co-transfected with 100 ng of TAT-GRE-E1B-LUC, 25 ng of expression vector for AR and increasing amounts of expression vector for HEY1 or the variant L94M (100 or 200 ng). After transfection, the cells were washed and, when required, incubated for 24 h in the presence of vehicle (white bars) or 10 nM dihydrotestosterone (DHT) (black bars). Subsequently, cell lysates were assayed using a dual-luciferase reporter system. The normalized values are expressed relative to the activity of AR alone in the presence of DHT. The results shown represent the averages of the results of at least three independent experiments assayed in duplicate + s.d. Inset: Western blot analysis confirmed that both HEY1 variants are expressed at similar levels. (c) L94M retains functional transcriptional repression domains. COS-1 cells were co-transfected with 200 ng of pGL2-Lex-Gal-Luc reporter and expression vectors for LexA-VP16 (100 ng) and GAL4-DNA-binding domain (DBD) fusion proteins (100 ng) as indicated. Data were obtained and presented as described for (b). The results shown represent the averages of the results of three independent experiments assayed in duplicate + s.d. *P<0.001 as compared with values for controls (Student’s t-test).
HEY1 has intrinsic transcriptional repressive activity (Iso et al., 2001b; Belandia et al., 2005). We fused the L94M variant to Gal4-DNA-binding domain and analysed its ability to repress in trans the activity of the VP16 transcription factor fused to a LexA-DNA-binding domain. We observed a similar strong repression of Lex-Gal-LUC reporter gene expression for both HEY1 variants (Figure 1c), indicating that L94M retains functional transcriptional repression domains. Notably, the L94M substitution does not affect the interaction between HEY1 and the AR or the nuclear receptor co-activator, steroid receptor co-activator 1 (Supplementary Figure 1b). These observations indicate that the polymorphism L94M profoundly changes HEY1 regulation of AR but it does not affect either its physical interaction or its intrinsic repressive activity.
The SNP L94M alters HEY1 protein–protein interaction specificity
The residue L94 is located at the beginning of Helix 2 within the HLH domain, and is conserved between all members of the bHLH-O superfamily, including hairy and enhancer of split 1 (HES1), and the less closely related bHLH transcription factors such as the Mycassociated factor X (Figure 2a). The second helix seems to be the main determinant of the homo- or hetero-dimerization surface between bHLH proteins (Ferre-D’Amare et al., 1993). Moreover, the leucine is exposed in the surface of the helix orientated toward the equivalent leucine residue present in the opposite monomer, suggesting that changes in this residue may alter the specificity of the interaction between HLH proteins. To address whether alterations in this residue affect the interaction between HEY1 and other bHLH proteins, we performed glutathione S-transferase pulldown experiments. HEY1 forms homodimers but it is also able to form heterodimers with HES1 (Iso et al., 2001b). Glutathione S-transferase-HEY1 interacted in a similar manner both with itself and with the variant L94M but it showed only a weak interaction with HES1 (Figure 2b). Strikingly, the single L94M substitution changed the specificity of the interactions, facilitating the formation of the heterodimer HES1–HEY1, having no noticeable effect on interaction with wild-type HEY1, but showing only a relatively weak interaction with another L94M monomer (Figure 2b). In addition, the L94M substitution also greatly enhanced the ability of HEY1 to interact with HEY2 (Figure 2c). These findings support the idea that the radical changes in the function of L94M as a modulator of AR transcriptional activity may be caused by preferential interactions with different bHLH partners.
Figure 2.

The single-nucleotide polymorphism (SNP) L94M favors the formation of heterodimers with hairy and enhancer of split 1 (HES1) and hairy/enhancer-of-split related with YRPW motif 1 (HEY2). (a) The helix-loop-helix domain of HEY1, HEY2, HEYL, HES1 and Myc-associated factor X (MAX) were aligned by ClustalW2 and formatted with BOXSHADE. Identical amino acids are in black, and conserved residues are in gray. The asterisk indicates the conserved leucine residue at position 94 in HEY1. (b) Whole-cell extracts from COS-1 cells previously transfected with expression vectors for flag-tagged HEY1, the variant L94M or HES1 were incubated with glutathione S-transferase (GST) fusion proteins of HEY1 or the variant L94M coupled with Sepharose beads. The associated proteins were detected by western blotting using anti-Flag antibody. (c) Whole-cell extracts from COS-1 cells previously transfected with expression vector for Flag-tagged HEY2 were incubated with GST fusion proteins of HEY1 or the variant L94M coupled with Sepharose beads. The associated proteins were detected by western blotting using anti-Flag antibody.
The variant L94M does not require nuclear localization to exert its positive effects on AR activity
In view of our previous observations indicating that abnormal HEY1 subcellular distribution may have a role in the aberrant hormonal responses observed in prostate cancer (Belandia et al., 2005), we tested whether normal nuclear localization is required for L94M coactivating function. First, we performed mutational analysis to confirm that a putative classical monopartite nuclear localization signal (NLS), which is present in the HEY1 basic domain (49ARKRRR54; Figure 1a), was involved in the regulation of HEY1 subcellular trafficking. We generated HEY1 and L94M mutants containing three point mutations within the putative NLS in the context of the full-length protein (HEY1-K51A/R53G/R51A and L94M-K51A/R53G/R51A) and examined subcellular localization using indirect immunofluorescence. As previously reported, wild-type HEY1 accumulates in the nucleus (Figure 3a, Iso et al., 2001a), and the L94M variant presented identical nuclear expression (Figure 3c), suggesting that the polymorphism does not affect HEY1 subcellular localization. The substitutions within the putative NLS cause a greater tendency to distribute into the cytoplasm for both HEY1 variants (Figures 3b and d), suggesting that the putative NLS contributes to the nuclear localization of HEY1 and L94M. To confirm this, we fused a 13-amino acid peptide derived from HEY1 sequence containing the NLS (amino acids 46–58) to enhanced green fluorescent protein (EGFP; Figure 1a) and examined the subcellular localization of the fusion protein. The enhanced green fluorescent protein alone was largely cytoplasmic (Figure 3e) but the fusion of the short peptide containing the HEY1 NLS dramatically changed the staining pattern, leading to a clear nuclear localization of enhanced green fluorescent protein (Figure 3f). Thus, our results indicate that the sequence 49ARKRRR54 present in HEY1 is a bona fide NLS.
Figure 3.

Characterization of the hairy/enhancer-of-split related with YRPW motif 1 (HEY1) nuclear localization signal. COS-1 cells were transfected with expression vectors for Flag-tagged HEY1 (a), the variant L94M (c), the triple-point mutants HEY1-K51A/ R53G/R51A (b) and L94M-K51A/R53G/R51A (d), enhanced green fluorescent protein (EGFP) (e) or EGFP-nuclear localization signal (NLS) (f) and assayed by indirect immunofluorescence with anti-Flag antibody, or direct EGFP fluorescence, as described in Materials and methods. The first column shows the indirect immunofluorescence with anti-Flag antibody or the direct EGFP fluorescence (green), the second column shows 4,6-diamidino-2-phenylindole (DAPI) staining of DNA (blue) and the third column shows the merge image indicating the degree of colocalization of green and blue fluorescence. Bars, 20 μm.
To determine how abnormal subcellular location affects the ability of both HEY1 variants to modulate AR transcriptional activity, we performed functional analyses with androgen-responsive luciferase reporters. Unexpectedly, the NLS-mutant (cytoplasmic) HEY1 also behaved as an AR transcriptional co-activator (Figure 4). In accordance with this result, mutation of the NLS did not affect significantly the co-activation potential of the L94M variant on AR transcriptional activity (Figure 4), indicating that, conversely to its repressive activity, HEY1 positive transcriptional effects do not require nuclear localization of the protein.
Figure 4.
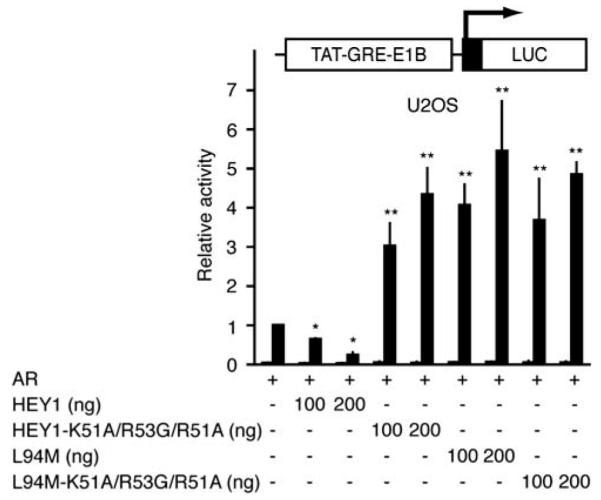
The effects of mutations in the nuclear localization signal of hairy/enhancer-of-split related with YRPW motif 1 (HEY1) or the variant L94M on the modulation of androgen receptor (AR)-mediated transactivation. U2OS cells were co-transfected with 100 ng of TAT-GRE-E1B-LUC, 25 ng of expression vector for AR and increasing amounts of expression vector for HEY1, L94M, the triple-point mutant HEY1-K51A/R53G/R51A or the triple-point mutant L94M-K51A/R53G/R51A (100 or 200 ng). The results shown represent the averages of the results of three independent experiments assayed in duplicate + s.d. *P<0.001 as compared with values for controls (Student’s t-test). **P<0.01 as compared with values for controls (Student’s t-test). After transfection, the cells were washed and incubated for 24 h in the presence of vehicle (white bars) or 10 nM DHT (black bars). Subsequently, cell lysates were assayed using a dual-luciferase reporter system. Data are presented as described for Figure 1b.
The SNP L94M inhibits the ability of HEY1 to enhance p53 transcriptional activity
Co-transfection of HEY1 with artificial p53-dependent reporters increases p53 activity (Huang et al., 2004). We performed transfection experiments in p53-competent U2OS cells co-transfecting HEY1 with several natural p53-responsive luciferase reporters. Two of the reporters tested (p21 and BAX) were moderately activated by HEY1 co-transfection (1.5 to twofold) but one (p53-inducible gene 3 (PIG3)) showed high induction (>12-fold) in the presence of HEY1 (Figure 5a). Hence, we chose the PIG3-luciferase reporter for further characterization of the functional connection between HEY1 and p53. To analyse the requirement of direct p53 binding to the PIG3 reporter for HEY1-positive effects, we used a mutated reporter lacking the p53-binding site in PIG3 (PIG3-delF; Contente et al., 2002). Removal of the p53-binding site almost completely abolished the reporter induction upon HEY1 cotransfection (Figure 5b). Moreover, co-transfection of an expression plasmid for the oncoprotein E6, which targets p53 for proteasome-mediated degradation, eliminated the positive effects of HEY1 on p53 activity (Figure 5c). We then performed similar transfection experiments using the isogenic HCT116 p53+/+ and p53−/− cells, which are genetically identical except for the p53 status. HEY1 overexpression greatly enhanced the activity of the PIG3 reporter in the p53+/+ cells, but caused only a modest induction in p53-negative cells (Figure 5d). Expression of p53 in HCT116 p53−/− cells dramatically induced PIG3 reporter activity, and cotransfection of HEY1 further stimulated the p53-responsive reporter (Figure 5d). These findings indicate that HEY1 effects are mediated directly by p53 binding to its cognate response element in the PIG3 reporter. To assess how the change of L94M affects HEY1-positive modulation of the p53-responsive reporter, HEY1 or the L94M variant were transfected into U2OS cells and tested for their ability to stimulate luciferase activity. The strong activation induced by HEY1 was severely impaired by the single amino acid substitution, L94M (Figure 5e). Thus, again, this minor change in HEY1 protein sequence has profound effects on its biological functions. Further, the positive effect of HEY1 on p53 transcriptional activity requires nuclear localization of the protein, as transfection of the HEY1 cytoplasmic mutant (HEY1-K51A/R53G/R51A) failed to significantly enhance the PIG3-luciferase reporter (Figure 5e), implying that HEY1-positive effects on p53 transcriptional activity require nuclear localization of the protein.
Figure 5.

Hairy/enhancer-of-split related with YRPW motif 1 (HEY1) positively regulates p53-dependent transactivation in a promoter-specific manner. (a) HEY1 greatly enhances p53-inducible gene 3 (PIG3)-luciferase reporter activity. U2OS cells were transfected with 100 ng of p21-Luc, BAX-Luc or PIG3-Luc in the presence or absence of 200 ng of expression vector for HEY1. (b) Deletion of the p53-binding site in PIG3 impairs the HEY1-positive effect. U2OS cells were transfected with 100 ng of PGL3, PIG3-Luc or PIG3 delF-Luc and 200 ng of expression vector for HEY1. (c) E6-mediated p53 degradation abolishes HEY1-positive effect on PIG3-luciferase reporter. U2OS cells were transfected with 100 ng PIG3-Luc and expression vectors for HEY1 (200 ng) or E6 (200 ng). (d) HEY1-positive effect on PIG3 promoter depends on the p53 status. HCT116 p53+/+ and p53−/− cells were transfected with 100 ng PIG3-Luc and expression vectors for HEY1 (200 ng) and p53 (5 ng) as indicated. (e) The polymorphic change L94M and mutations affecting the nuclear localization signal (NLS) impair HEY1-positive effect on PIG3. U2OS cells were transfected with 100 ng PIG3-Luc and 200 ng of transfection vectors for HEY1, the variant L94M or the triple-point mutant K51A/R53G/R51A. After transfection, the cells were washed and incubated for 24 h. Subsequently, cell lysates were assayed using a dual-luciferase reporter system. Normalized values are expressed relative to the activity of the reporter in the absence of HEY1 (a), relative to the activity of the empty pGL3-Luc reporter in the presence of HEY1 (b) or relative to the activity of PIG3 reporter in the absence of other vectors (c–e). The results shown represent the averages of results of at least two independent experiments assayed in duplicate + s.d.
The SNP L94M inhibits HEY1 ability to induce p53-dependent cell-cycle arrest
HEY1 expression induces p53-mediated apoptosis in zebrafish and chick embryos (Huang et al., 2004), and p53-dependent growth arrest in Ewing’s sarcoma family tumor cells (Ban et al., 2008). As HEY1 enhances p53 transcriptional activity in U2OS cells, we examined whether HEY1 overexpression can recapitulate p53-induced phenotypes. For this purpose, we generated tetracycline-inducible pools of U2OS cells stably expressing either HEY1 (U2OS-HEY1) or L94M (U2OS-L94M) (Figure 6a). Induction of HEY1 expression inhibited cell proliferation (Figure 6b, left panel). In contrast, induction of L94M expression did not affect cell proliferation (Figure 6b, right panel). HEY1-induced growth arrest was reversible even after 9 days of tetracycline treatment (data not shown). Immunoblot analysis showed a moderate but highly reproducible transient increase in p53 protein levels after 8 h of induction of wild-type HEY1, but not L94M, which was no longer detected at 24 h after induction (Figures 6c and d). The stability of p53 protein is regulated by murine double minute-2 and, in agreement with previously published results (Huang et al., 2004), immunoblot analysis showed that wild-type HEY1 expression reduces murine double minute-2 protein levels (Figure 6c). Interestingly, levels of p21 protein, a key transcriptional target of p53 involved in the control of cell cycle, were markedly decreased by HEY1 expression (Supplementary Figure 2d). However, expression of L94M did not decrease murine double minute-2 (Figure 6c) or p21 protein levels (Supplementary Figure 2d). Fluorescence-activated cell sorting analysis showed that HEY1 expression, but not L94M expression, results in alterations in cell-cycle distribution in U2OS cells that do not fit with typical p53-induced G1 or G2 phase arrest. Wild-type HEY1 expression reduces the percentage of cells in G1 phase, increases the populations in S and G2 phase and a small fraction of cells appears in the sub-G1 fraction (Figure 6e). No polyploid populations were detected. HEY1 effects on cell proliferation are accompanied by striking changes in cell morphology, indicating neuron-like differentiation (Supplementary Figure 2a), as well as downregulation of expression of components of the Notch pathway (Notch1, Jagged1 and HES1) and RUNX2, a master regulator of osteoblast differentiation (Karsenty, 2008) (Supplementary Figure 2b). Further analysis of mRNA expression for several cell-cycle regulatory genes showed that most were downregulated by HEY1 but not by L94M (Supplementary Figure 2c). Taken together, these observations suggest that growth arrest coincides with inhibition of Notch signaling and abnormal cellular differentiation. To analyse whether HEY1 effects on cell proliferation depend on p53 activation, U2OS-HEY1 cells were transfected with p53-specific small interfering RNA (siRNA) (sip53) or nonspecific control siRNA, and cell proliferation after induction of HEY1 was determined at different time points. As expected, tetracycline induction had an inhibitory effect on U2OS-HEY1 cell proliferation transfected with control siRNA (Figure 6f). However, U2OS-HEY1 cells transfected with p53-specific siRNA, which efficiently reduced endogenous p53 levels (Figure 6g), continued to proliferate during the entire 6-day monitored period (Figure 6f). This result indicates that p53 is essential to achieve HEY1-mediated growth arrest.
Figure 6.

The single-nucleotide polymorphism (SNP) L94M inhibits the ability of hairy/enhancer-of-split related with YRPW motif 1 (HEY1) to induce p53-dependent cell-cycle arrest. (a) Immunoblot analysis of HEY1 and L94M expression in stable U2OS cell lines expressing inducible HEY1 (U2OS-HEY1) or the variant L94M (U2OS-L94M) treated with 1 μg/ml tetracycline (Tet) for 1, 2 or 4 h. The protein levels of β-actin are shown as a loading control. (b) Parental U2OS cells, U2OS-HEY1 cells (left panel) or U2OS-L94M cells (right panel) were treated either with vehicle or 1 μg/ml tetracycline (Tet). Cell proliferation was monitored at different time points by using 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) assay. The results shown represent the averages of the results of two independent experiments assayed in quadruplicate ± s.d. (c) Immunoblot analysis of p53, murine double minute-2 (MDM2) and β-actin in U2OS-HEY1 and U2OS-L94M cells treated with 1 μg/ml tetracycline (Tet) for 4 or 8h. (d) Immunoblot analysis of p53 and β-actin in U2OS-HEY1 and U2OS-L94M cells treated with 1 μg/ml tetracycline (Tet) for 24 h. (e) Flow cytometry analysis of U2OS-HEY1 cells or U2OS-L94M cells treated either with vehicle or 1 μg/ml tetracycline (Tet) for 72 h. A representative experiment is shown. The right panels show the percentage of sub-G1, G0/G1, S and G2/M cells. The results shown represent the averages of the results of two independent experiments assayed in duplicate + s.d. (f) U2OS-HEY1 cells transfected with p53-specific small interfering RNA (siRNA) (sip53) or nonspecific control siRNA (siControl) were treated either with vehicle or 1 μg/ml tetracycicline (Tet). Cell proliferation was monitored at different time points by using MTS assay. The results shown represent the averages of results of two independent experiments assayed in quadruplicate±s.d. (g) Immunoblot analysis of p53 and β-actin in U2OS-HEY1 cells transfected with specific p53 or control siRNAs.
Expression of HEY1 but not L94M sensitizes U2OS cells to p53-activating chemotherapeutic drugs
p53 status has a key role in the sensitivity to chemotherapy (reviewed in Lu and El-Deiry, 2009). To determine whether HEY1 modulation of p53 activity might influence the response of U2OS cells to chemotherapeutic agents, we performed cytotoxicity assays using cisplatin and doxorubicin. Induction of HEY1 expression in U2OS-HEY1 cells resulted in an increase in sensitivity to both drugs (Figure 7). However, induction of L94M expression in U2OS-L94M cells did not change the sensitivity of U2OS cells to cisplatin or doxorubicin (Figure 7). Thus, a single amino acid substitution in HEY1 has profound effects on the cellular response to anticancer drugs.
Figure 7.
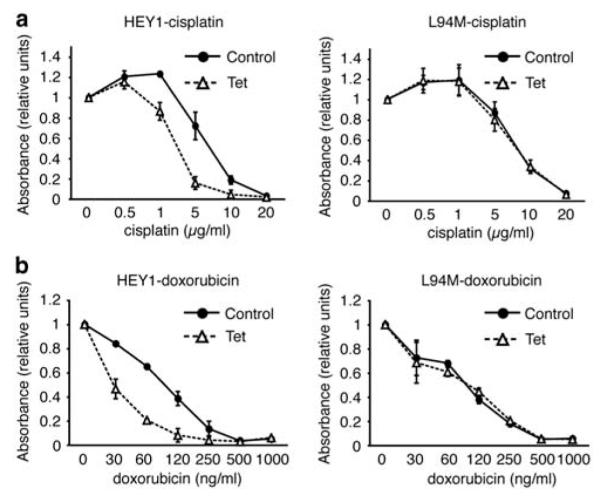
The expression of hairy/enhancer-of-split related with YRPW motif 1 (HEY1) but not L94M leads to increased sensitivity to cisplatin and doxorubicin. U2OS-HEY1 cells (left panels) or U2OS-L94M cells (right panels) were treated with 1 μg/ml tetracycline (Tet) to induce expression of HEY1 or L94M or with vehicle (Control). After 8 h, the cells were cultured with varying concentrations of cisplatin (a) or doxorubicin (b) for another 72 h in the presence and absence of tetracycline. Subsequently, cell viability was assayed using an 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS)-based assay. Data were plotted relative to the drug-free controls. The results shown represent the averages of results of two independent experiments assayed in quadruplicate±s.d.
Discussion
Cancers arise as a result of the acquisition of a series of DNA mutations that confer a growth advantage upon the cells in which they have occurred (Knudson, 2001). In addition, most genes possess some level of polymorphism (Sachidanandam et al., 2001) that may also influence cancer susceptibility (Hunter and Crawford, 2006) or response to cancer therapy (Undevia et al., 2005). The p53 protein is a sequence-specific transcription factor that has a crucial role in tumor suppression by inducing apoptosis or cell-cycle arrest in response to cellular damage. Several polymorphisms have already been identified that influence the function of the p53 pathway (reviewed in Pietsch et al., 2006; Whibley et al., 2009). Polymorphisms can potentially alter the coding sequence or the promoter activity of genes encoding p53, p53-induced target genes or p53-regulatory proteins (Whibley et al., 2009). In this study we show that a single amino acid change caused by a non-synonymous SNP in HEY1 drastically reduces its effects as an indirect p53 activator. In addition, this single amino acid substitution has even more dramatic effects on the role of HEY1 as an AR transcriptional corepressor: we found that the variant L94M not only lost all repressive functions on AR, but also behaved as a potent transcriptional co-activator of AR.
The lack of detailed knowledge regarding the molecular mechanisms by which HEY1 exerts its corepressor function on each of its described target transcription factors makes it difficult to explain how a single amino acid substitution causes such profound functional effects. Both HEY1 variants have similar intrinsic transcriptional repressive activity when fused to a heterologous DNA-binding domain, in agreement with the presence of several independent repressive domains in HEY1 (Iso et al., 2001b; Belandia et al., 2005). Further characterization of HEY1 and L94M biological properties has provided us with several clues regarding the possible cause of the functional differences. First, as predicted for its location in the putative dimerization surface, the SNP L94M changes the specificity of the interaction between HEY1 and other known bHLH partners, such as HES1 and HEY2. This observation, together with the fact that the motif responsible for nuclear localization, and consequently DNA binding, are dispensable for L94M-positive effects on AR transcriptional activity, suggest that L94M functions by sequestering other repressor proteins, presumably bHLH proteins, required in the nucleus for the repression of AR by HEY1. The positive effects of the cytoplasmic HEY1 mutant on AR activity may also reflect a nuclear depletion of AR corepressors. We have performed assays by either overexpressing HES1 or inhibiting its expression with RNA interference, but we failed to detect any modulation of L94M-positive effects on AR activity (data not shown). However, there are many other bHLH proteins in the cell that may interact with HEY1. Further studies are required to discover novel functional interactions between HEY1 and other cellular proteins that could be altered by the polymorphism L94M. In contrast, the indirect positive effects of HEY1 on p53 function require nuclear localization of the protein, suggesting that this effect depends on the repression of as-yet-uncharacterized target genes. Microarray analysis of global changes in gene expression caused by induction of HEY1 expression in U2OS-HEY1 cells will help to unravel the p53-regulatory pathways responsible for HEY1-dependent p53 activation.
The p53-dependent transcriptional program is extremely flexible and the cellular response depends on the nature of the p53-activating stimuli, the cellular context and the duration of the activation signal (reviewed in Espinosa, 2008). In keeping with this, HEY1-dependent activation of p53 has been described to induce apoptosis in vivo in zebrafish and avian developmental systems (Huang et al., 2004), although it can also promote cell-cycle arrest in Ewing’s sarcoma family tumor cell lines (Ban et al., 2008). Induction of HEY1 expression in U2OS cells has pleiotropic effects. We observed a complete inhibition of cell proliferation accompanied by a reduction in the expression of components of the Notch pathway and the osteoblast differentiation factor Runx2. Activation of p53 in osteoblast progenitor cells inhibits both proliferation and Runx2 expression (Lengner et al., 2006). Hence, HEY1 effects may reflect, at least in part, alteration in the osteoblast-like differentiation of U2OS cells caused by p53 activation. Although the detailed molecular mechanisms responsible for the growth arrest and the differentiation events are still uncharacterized, RNA interference experiments indicated that p53 is an essential mediator of HEY1 effects. However, induction of HEY1 in U2OS cells does not entirely recapitulate well-characterized p53-mediated tumor suppressor activities, such as G1- or G2-phase arrest or induction of apoptosis. It has been recently reported that inhibition of the Notch pathway prevents osteosarcoma growth by modulation of cell-cycle regulatory genes (Tanaka et al., 2009) and that downregulation of Notch1 or Jagged-1 induces cell growth inhibition and S-phase arrest in prostate cancer cells (Zhang et al., 2006). Thus, the observed phenotype might result from the combined modulation of two highly complex and context-dependent regulatory axes, the Notch and p53 pathways.
Another significant consequence of HEY1 expression, which was not observed upon L94M expression, was the sensitization of U2OS osteosarcoma cells to p53-activating chemotherapeutic drugs. This effect may reflect the decreased expression of p21 in the cells. p21 is a major player in cell-cycle control but it is also an inhibitor of apoptosis, and p21 repression sensitizes tumor cells to apoptosis by anticancer drugs (reviewed in Gartel and Radhakrishnan, 2005). Moreover, ablation of Notch3 in human hepatocellular carcinoma cell lines enhances the cell death-promoting effect of doxorubicin through p53-dependent mechanisms that inhibit both HES1 and p21 expression (Giovannini et al., 2009).
In view of our findings showing that this single amino acid change in HEY1 dramatically affects several biological activities, it may be potentially relevant to extend its polymorphic study to a wider population. These studies might be particularly valuable for populations with diseases related to alterations in Notch- and p53-dependent signaling pathways, or directly associated with alterations in HEY1-dependent gene regulation. Among the Notch-associated diseases are several types of cancer, including highly prevalent diseases such as prostate cancer (Leong and Gao, 2008; Villaronga et al., 2008). Approximately half of human cancers express wild-type p53, and it has been suggested that the p53 pathway in those tumor cells might be compromised by alterations upstream or downstream of p53. A genetic study of the possible presence of the HEY1 L94M SNP in p53-positive tumors will help to test the attractive hypothesis that this polymorphism may contribute to oncogenic transformation in a fraction of those tumors. It is noteworthy that the suppression of Notch signaling is a critical mechanism by which the Ewing’s sarcoma-FLI1 oncogene inhibits p53 activity in p53-positive Ewing’s sarcoma family tumors. The presence of HEY1 L94M SNP provides an additional putative mechanism for alteration of the functional crosstalk between Notch and p53 pathways. Finally, exclusion of HEY1 from the nucleus may have a multiple oncogenic role in prostate tumor cells, eliminating at the same time a molecular mechanism for repression of AR and an activation mechanism for p53, and inducing aberrant AR activation by sequestering other AR co-repressors.
Materials and methods
Plasmids
Plasmids are described in the Supplementary material.
Cell culture and transient transfections
COS-1 and U2OS cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. At 24 h before transfection, the cells were plated in 24-well plates in phenol red-free medium with 10% dextran charcoalstripped serum. Cells were transfected using Lipofectamine 2000 (Invitrogen, Prat de Llobregat, Spain). Transfected plasmids are detailed in the figure legends. pRL-TK (Promega, Alcobendas, Spain) was used as an internal control for transfection efficiency. Cell extracts were assayed for luciferase activity as described previously (Belandia et al., 2005).
Antibodies and immunoblotting
Immunoblotting was performed as previously described (Belandia et al., 2005). The antibodies used are described in the Supplementary material.
Glutathione S-transferase-pull-down assays
Protein–protein interaction assays were performed as previously described (Belandia et al., 2005).
Fluorescence analysis
COS-1 cells grown on coverslips were transfected with 100 ng of the plasmids indicated in Figure 3. After 36 h, cells were fixed with 4% paraformaldehyde in phosphate buffered saline (PBS) for 15 min and permeabilized in 0.1% Triton X-100 in PBS for 5 min. To perform indirect immunofluorescence, cells were incubated in 3% bovine serum albumin in PBS for 30 min. Primary immunostaining with mouse anti-Flag antibody (1:500) was carried out at room temperature for 1 h. Secondary immunostaining with Alexa Fluor 488 goat antimouse antibody (Invitrogen-A11029) was performed at room temperature for 1 h. DNA was counterstained with 4,6-diamidino-2-phenylindole.
Generation of U2OS cells expressing tetracycline-inducible HEY1 or L94M
Lentiviral vectors encoding HEY1 or the variant L94M were used to generate lentivirus expressing HEY1 or L94M by the ViraPower T-REx system following manufacturer’s instructions (Invitrogen). U2OS cells were co-transduced with Tet-repressor-lentivirus and either HEY1-or L94M-lentivirus. Selection of stably co-transduced cells was achieved with zeocin (1000 μg/ml) and blasticidin (4 μg/ml). To induce HEY1 or L94M expression, 1 μg/ml of tetracycline was added to the media.
Proliferation assays
U2OS cells or U2OS cells expressing tetracycline-inducible HEY1 or L94M were plated into 96-well plates (2500 cells/ well). After 24 h, 1 μg/ml of tetracycline was added and cell proliferation was measured at different time points. Medium and tetracycline were replaced every 3 days. Cell growth was determined in quadruplicates using the CellTiter One-Solution-Assay (Promega) and reading absorbance at 490 nm. siRNA was used to knockdown p53 expression. A synthetic 21-nucleotide duplex siRNA against p53 (p53-F; 5′-GACUCCAGUGGUAAUCUACUU-3′, p53-R; 5′-GUAGAUUACCA CUGGAGUCUU-3′) or a control ON-TARGETplus Non-Targeting siRNA (Dharmacon, Lafayette, CO, USA) were transfected with Lipofectamine 2000 (Invitrogen) in 60-mm dishes. After 24 h in culture with siRNA, the cells were split and proliferation was determined as described above.
Cell-cycle analysis
The cells were collected, washed with PBS and fixed with ice-cold 70% ethanol for at least 1 h. Cells were washed twice with PBS, treated for 30 min at 37 °C with 20 μg/ml RNase A (Sigma-Aldrich, St Louis, MO, USA), stained with 20 μg/ml propidium iodide and analysed on an FACScan (BD Bioscences, San Agustin del Guadalix, Spain). The percentage of cells in cell-cycle phases was calculated using ModFit LT (BD Bioscences).
Cytotoxicity assays
U2OS cells expressing tetracycline-inducible HEY1 or L94M were plated into 96-well plates (5000 cells/well). HEY1 or L94M expression was induced by adding 1 μg/ml of tetracycline and 8 h later were incubated with varying concentrations of cisplatin or doxorubicin for 72 h. Cell viability was determined in quadruplicate using the CellTiter One-Solution-Assay (Promega) and reading absorbance at 490 nm.
Supplementary Material
Acknowledgements
We thank Ana Aranda for her continuous support and Linn Markert for technical assistance. This study was supported by the MICINN (SAF2007-62642, SAF2006-07785), the Fundación de Investigación Médica Mutua Madrileña, the FIS (RD06/0020/0036), the CRESCENDO (FP-018652) and Cancer Research UK (C11509/A8570).
Footnotes
Conflict of interest The authors declare no conflict of interest.
References
- Ban J, Bennani-Baiti IM, Kauer M, Schaefer KL, Poremba C, Jug G, et al. EWS-FLI1 suppresses NOTCH-activated p53 in Ewing’s sarcoma. Cancer Res. 2008;68:7100–7109. doi: 10.1158/0008-5472.CAN-07-6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belandia B, Powell SM, Garcia-Pedrero JM, Walker MM, Bevan CL, Parker MG. Hey1, a mediator of notch signaling, is an androgen receptor corepressor. Mol Cell Biol. 2005;25:1425–1436. doi: 10.1128/MCB.25.4.1425-1436.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contente A, Dittmer A, Koch MC, Roth J, Dobbelstein M. A polymorphic microsatellite that mediates induction of PIG3 by p53. Nat Genet. 2002;30:315–320. doi: 10.1038/ng836. [DOI] [PubMed] [Google Scholar]
- Davis RL, Turner DL. Vertebrate hairy and enhancer of split related proteins: transcriptional repressors regulating cellular differentiation and embryonic patterning. Oncogene. 2001;20:8342–8357. doi: 10.1038/sj.onc.1205094. [DOI] [PubMed] [Google Scholar]
- Espinosa JM. Mechanisms of regulatory diversity within the p53 transcriptional network. Oncogene. 2008;27:4013–4023. doi: 10.1038/onc.2008.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre-D’Amare AR, Prendergast GC, Ziff EB, Burley SK. Recognition by Max of its cognate DNA through a dimeric b/HLH/Z domain. Nature. 1993;363:38–45. doi: 10.1038/363038a0. [DOI] [PubMed] [Google Scholar]
- Fuke S, Sasagawa N, Ishiura S. Identification and characterization of the Hesr1/Hey1 as a candidate trans-acting factor on gene expression through the 3′ non-coding polymorphic region of the human dopamine transporter (DAT1) gene. J Biochem. 2005;137:205–216. doi: 10.1093/jb/mvi020. [DOI] [PubMed] [Google Scholar]
- Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 2005;65:3980–3985. doi: 10.1158/0008-5472.CAN-04-3995. [DOI] [PubMed] [Google Scholar]
- Giovannini C, Gramantieri L, Chieco P, Minguzzi M, Lago F, Pianetti S, et al. Selective ablation of Notch3 in HCC enhances doxorubicin’s death promoting effect by a p53 dependent mechanism. J Hepatol. 2009;50:969–979. doi: 10.1016/j.jhep.2008.12.032. [DOI] [PubMed] [Google Scholar]
- Huang Q, Raya A, DeJesus P, Chao SH, Quon KC, Caldwell JS, et al. Identification of p53 regulators by genome-wide functional analysis. Proc Natl Acad Sci USA. 2004;101:3456–3461. doi: 10.1073/pnas.0308562100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulleman E, Quarto M, Vernell R, Masserdotti G, Colli E, Kros JM, et al. A role for the transcription factor HEY1 in glioblastoma. J Cell Mol Med. 2009;13:136–146. doi: 10.1111/j.1582-4934.2008.00307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter KW, Crawford NP. Germ line polymorphism in metastatic progression. Cancer Res. 2006;66:1251–1254. doi: 10.1158/0008-5472.CAN-05-3705. [DOI] [PubMed] [Google Scholar]
- Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol. 2003;194:237–255. doi: 10.1002/jcp.10208. [DOI] [PubMed] [Google Scholar]
- Iso T, Sartorelli V, Chung G, Shichinohe T, Kedes L, Hamamori Y. HERP, a new primary target of Notch regulated by ligand binding. Mol Cell Biol. 2001a;21:6071–6079. doi: 10.1128/MCB.21.17.6071-6079.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iso T, Sartorelli V, Poizat C, Iezzi S, Wu HY, Chung G, et al. HERP, a novel heterodimer partner of HES/E(spl) in Notch signaling. Mol Cell Biol. 2001b;21:6080–6089. doi: 10.1128/MCB.21.17.6080-6089.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh F, Itoh S, Goumans MJ, Valdimarsdottir G, Iso T, Dotto GP, et al. Synergy and antagonism between Notch and BMP receptor signaling pathways in endothelial cells. EMBO J. 2004;23:541–551. doi: 10.1038/sj.emboj.7600065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsenty G. Transcriptional control of skeletogenesis. Annu Rev Genomics Hum Genet. 2008;9:183–196. doi: 10.1146/annurev.genom.9.081307.164437. [DOI] [PubMed] [Google Scholar]
- Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1:157–162. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- Lengner CJ, Steinman HA, Gagnon J, Smith TW, Henderson JE, Kream BE, et al. Osteoblast differentiation and skeletal Supplementary Information accompanies the paper on the development are regulated by Mdm2-p53 signaling. J Cell Biol. 2006;172:909–921. doi: 10.1083/jcb.200508130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong KG, Gao WQ. The Notch pathway in prostate development and cancer. Differentiation. 2008;76:699–716. doi: 10.1111/j.1432-0436.2008.00288.x. [DOI] [PubMed] [Google Scholar]
- Lu C, El-Deiry WS. Targeting p53 for enhanced radio- and chemo-sensitivity. Apoptosis. 2009;14:597–606. doi: 10.1007/s10495-009-0330-1. [DOI] [PubMed] [Google Scholar]
- Maier MM, Gessler M. Comparative analysis of the human and mouse Hey1 promoter: Hey genes are new Notch target genes. Biochem Biophys Res Commun. 2000;275:652–660. doi: 10.1006/bbrc.2000.3354. [DOI] [PubMed] [Google Scholar]
- Nakagawa O, McFadden DG, Nakagawa M, Yanagisawa H, Hu T, Srivastava D, et al. Members of the HRT family of basic helix-loop-helix proteins act as transcriptional repressors downstream of Notch signaling. Proc Natl Acad Sci USA. 2000;97:13655–13660. doi: 10.1073/pnas.250485597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietsch EC, Humbey O, Murphy ME. Polymorphisms in the p53 pathway. Oncogene. 2006;25:1602–1611. doi: 10.1038/sj.onc.1209367. [DOI] [PubMed] [Google Scholar]
- Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- Sakamoto M, Hirata H, Ohtsuka T, Bessho Y, Kageyama R. The basic helix-loop-helix genes Hesr1/Hey1 and Hesr2/Hey2 regulate maintenance of neural precursor cells in the brain. J Biol Chem. 2003;278:44808–44815. doi: 10.1074/jbc.M300448200. [DOI] [PubMed] [Google Scholar]
- Satow T, Bae SK, Inoue T, Inoue C, Miyoshi G, Tomita K, et al. The basic helix-loop-helix gene hesr2 promotes gliogenesis in mouse retina. J Neurosci. 2001;21:1265–1273. doi: 10.1523/JNEUROSCI.21-04-01265.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivertsen EA, Huse K, Hystad ME, Kersten C, Smeland EB, Myklebust JH. Inhibitory effects and target genes of bone morphogenetic protein 6 in Jurkat TAg cells. Eur J Immunol. 2007;37:2937–2948. doi: 10.1002/eji.200636759. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Setoguchi T, Hirotsu M, Gao H, Sasaki H, Matsunoshita Y, et al. Inhibition of Notch pathway prevents osteosarcoma growth by cell cycle regulation. Br J Cancer. 2009;100:1957–1965. doi: 10.1038/sj.bjc.6605060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Undevia SD, Gomez-Abuin G, Ratain MJ. Pharmacokinetic variability of anticancer agents. Nat Rev Cancer. 2005;5:447–458. doi: 10.1038/nrc1629. [DOI] [PubMed] [Google Scholar]
- Villaronga MA, Bevan CL, Belandia B. Notch signaling: a potential therapeutic target in prostate cancer. Curr Cancer Drug Targets. 2008;8:566–580. doi: 10.2174/156800908786241096. [DOI] [PubMed] [Google Scholar]
- Whibley C, Pharoah PD, Hollstein M. p53 polymorphisms: cancer implications. Nat Rev Cancer. 2009;9:95–107. doi: 10.1038/nrc2584. [DOI] [PubMed] [Google Scholar]
- Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Zamurovic N, Cappellen D, Rohner D, Susa M. Coordinated activation of notch, Wnt, and transforming growth factor-beta signaling pathways in bone morphogenic protein 2-induced osteogenesis. Notch target gene Hey1 inhibits mineralization and Runx2 transcriptional activity. J Biol Chem. 2004;279:37704–37715. doi: 10.1074/jbc.M403813200. [DOI] [PubMed] [Google Scholar]
- Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004;23:1155–1165. doi: 10.1038/sj.emboj.7600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang Z, Ahmed F, Banerjee S, Li Y, Sarkar FH. Down-regulation of Jagged-1 induces cell growth inhibition and S phase arrest in prostate cancer cells. Int J Cancer. 2006;119:2071–2077. doi: 10.1002/ijc.22077. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.