

Abstract
The present study examined the role of endogenous noradrenaline on glial and neuronal plasticity in the spinal cord in rats after peripheral nerve injury. An intrathecal injection of dopamine-β-hydroxylase antibody conjugated to saporin (DβH-saporin) completely depleted noradrenergic axons in the spinal cord and also reduced noradrenergic neurons in the locus coeruleus (A6) and A5 noradrenergic nucleus in the brainstem and noradrenergic axons in the paraventricular nucleus of the hypothalamus. DβH-saporin treatment itself did not alter mechanical withdrawal threshold, but enhanced mechanical hypersensitivity and intrathecal clonidine analgesia after L5–L6 spinal nerve ligation (SNL). In the spinal dorsal horn of SNL rats, DβH-saporin treatment increased choline acetyltransferase (ChAT) immunoreactivity as well as immunoreactivity in microglia of ionized calcium binding adaptor molecule 1[IBA1] and in astrocytes of glial fibrillary acidic protein [GFAP], and brain derived nerve growth factor (BDNF) content. DβH-saporin treatment did not, however alter the fractional release of acetylcholine from terminals by dexmedetomidine after nerve injury. These results suggest that endogenous tone of noradrenergic fibers is not necessary for the plasticity of α2-adrenoceptor analgesia and glial activation after nerve injury, but might play an inhibitory role on glial activation.
Perspective
This study demonstrates that endogenous noradrenaline modulates plasticity of glia and cholinergic neurons in the spinal cord after peripheral nerve injury and hence influences the pathophysiology of spinal cord changes associated with neuropathic pain.
Keywords: neuropathic pain, noradrenaline, acetylcholine, brain-derived neurotrophic factor, microglia, astrocytes
Introduction
Bulbospinal noradrenergic pathways have been shown to inhibit pain transmission 37. In both normal and neuropathic pain states, noradrenaline, released by descending noradrenergic axons activates α2-adrenoceptors to produce acute antinociception via reduction of neurotransmitter release from primary afferent terminals 27 and hyperpolarization of second order spinal dorsal horn neurons 35. Some of these effects are direct, but others reflect activation of cholinergic signaling 30, 31. We previously demonstrated that α2-adrenoceptor agonists, clonidine and dexmedetomidine, inhibit KCl-evoked acetylcholine release in spinal cord slices and synaptosomes in normal rats 15, 28, consistent with this classical inhibitory action of α2-adrenoceptors. In contrast, after peripheral nerve injury, activation of α2-adrenoceptors by dexmedetomidine results in Gs-protein mediated facilitation of acetylcholine release from the spinal dorsal horn synaptosomes 15, consistent with increased cholinergic dependency of α2-adrenoceptor-mediated analgesia after nerve injury 30, 31.
In normal animals, depletion of noradrenergic fibers in the spinal cord by the neurotoxins such as N-2-chloroethyl-N-ethyl-2-bromobenzylamine hydrochloride (DSP4) and 6-hydroxydopamine (6-OHDA) enhances clonidine analgesia, associated with denervation super-sensitivity of postsynaptic spinal α2-adrenoceptors 32, 33, 39. However, the role of these fibers, which release noradrenaline, ATP, and neuropeptide-Y on neuronal and glial plasticity associated with neuropathic pain states has not been fully tested. One goal of the current study was to test whether depletion of spinal noradrenergic axons by an intrathecal injection of dopamine-β-hydroxylase antibody conjugated to saporin (DβH-saporin) affects clonidine analgesia, ChAT immunoreactivity in the dorsal horn, and the facilitatory effect of dexmedetomidine on acetylcholine release from synaptosomes in rats after L5–L6 spinal nerve ligation (SNL). We hypothesized that denervation supersensitivity might result in an increased fractional release of acetylcholine from spinal cord synaptosomes after nerve injury in DβH-saporin treated animals.
Peripheral nerve injury increases brain-derived neurotrophic factor (BDNF) content in the spinal dorsal horn 16, 26 and the most likely sources of spinal BDNF after nerve injury are the central terminals of primary afferents and resident microglia 4, 11, 16, 34. We recently reported that blockade of BDNF-tropomyosine receptor kinase B (trkB) signaling by spinal infusion of BNDF antibody or repeated intrathecal injection of trk inhibitor K252a reduces choline acetyltransferase (ChAT) immunoreactivity in the dorsal horn and also abolishes the shift from inhibition to facilitation by dexmedetomidine of acetylcholine release 15, 17. These results suggest that BDNF-trkB signaling is essential for maintenance and functional change of cholinergic neurons in the spinal cord after nerve injury, and that this plasticity in cholinergic neurons is important for the α2-adrenoceptor-mediated analgesia in neuropathic pain.
Activation of spinal glia also participates in neuropathic hypersensitivity 5. Whether the products released by descending noradrenergic fibers alter this response is not known, but stimulation of α2-adrenoceptors reduces activation of microglia and astrocytes in the spinal cord after peripheral nerve injury or chronic inflammation 10, 41. Peripheral nerve injury enhances spinal noradrenergic inhibition by increasing content and basal release of noradrenaline in the spinal dorsal horn 14, 16. We therefore hypothesized that spinal noradrenergic fibers, perhaps by the release of noradrenaline, modulate glial activity and BDNF production in the spinal cord after nerve injury. A final goal of the current study was to test whether depletion of spinal noradrenergic fibers reduces expression of glial activation markers and BDNF content in the spinal cord in SNL rats and hence, as described above, the shift in direct effects of α2-adrenoceptor activation on cholinergic terminals from inhibition to excitation.
Materials and Methods
Animals
Male Sprague-Dawley rats (weighing 180–250 g) from Harlan Industries (Indianapolis, IN, USA), housed under a 12-h light-dark cycle with food and water ad libitum, were used. All experiments were approved by Animal Care and Use Committee at Wake Forest University (Winston Salem, NC, USA).
Surgical preparations and DβH-saporin treatment
Animals were anesthetized with 2% isoflurane in oxygen and intrathecal catheterization was performed as previously described 43. A small puncture was made in the atlanto-occipital membrane of the cisterna magnum and a polyethylene catheter (external diameter; 0.23 mm, internal volume; 6 μL, ReCathCo LLC, Allison Park, PA, USA), 7.5 cm, was inserted so that the caudal tip reached the lumbar enlargement of the spinal cord. At least 5 days after intrathecal catheterization, animals received an intrathecal injection of mouse anti-DβH-saporin (Advanced Targeting Systems, San Diego, CA, USA, 5 μg in 10 μl of saline) or saporin (Advanced Targeting Systems, 5 μg in 10 μl of saline) followed by 10 μl of saline. One week after injection, SNL was performed as previously described 23. Briefly, under anesthesia with 2% isoflurane, the right L6 transverse process was removed and the right L5 and L6 spinal nerves were tightly ligated using 5–0 silk suture. When animals were sacrificed to collect the spinal cord, the placement of catheter was verified. No catheter misplacement was observed.
Behavioral test
The person performing the behavioral test was blinded to drug and treatment. Paw withdrawal threshold in response to calibrated von Frey filaments (Stoelting, Wood Dale, IL, USA) was determined using an up–down statistical method 3. Filaments were applied to the bending point for 5 s, and a brisk paw withdrawal was considered a positive response. At 12 days after SNL, saporin and DβH-saporin treated animals received an intrathecal injection of clonidine (Sigma Chemical CO., St. Louis, MO, USA, 15 μg in 10 μl of saline) with or without atropine (Sigma Chemical CO., 30 μg in 10 μl of saline) followed by 10 μl of saline 1 h prior to the measurement. Doses of clonidine and atropine were determined from our previous studies 30, 31. At 14 days after SNL, spinal cord tissues from those animals were used for following experiments.
Immunohistochemistry
Immunostaining for ChAT and DβH was performed in the brain and/or spinal cord, as we previously described 15, 17. Briefly, brain and L4–L6 spinal cord sections were incubated at 4 °C with a goat anti-ChAT antibody (1:200, AB144P, Millipore, Billerica, MA, USA) or a mouse monoclonal anti-DβH antibody (1:500, MAB308, Chemicon International Inc., Temecula, CA, USA) followed by biotinylated donkey anti-goat or anti-mouse IgG (1:200, Vector Laboratories, Burlingame, CA, USA) in 1.5 % normal donkey serum (NDS, Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA), processed using Elite Vectastain ABC kit (Vector Laboratories), and then developed by the standard glucose oxidase-nickel method. For labeling of microglia and astrocytes, sections were incubated at 4°C with anti-ionized calcium binding adaptor molecule 1 (rabbit anti-IBA1, 1:2000, Wako Chemicals, Richmond, VA, USA)) or anti-glial fibrillary acidic protein (rabbit anti-GFAP, 1:2000, Dako, Carpinteria, CA, USA) for 24 hr followed by CY3 conjugated donkey anti-rabbit IgG (1:600, Jackson Immunoresearch) for 2 hours in 1% NDS.
Images of the spinal cord were captured on a Nikon E600 epifluorescence microscope fitted with a CCD digital camera using a 10× objective at a resolution of 1,600 × 1,200 pixels. Quantification for each immunostaining was performed in 4–5 randomly selected L4–L6 spinal cord sections from each rat. Image analysis software (Image J; NIH Image, National Institutes of Health, Bethesda, MD) was used to quantify changes in immunofluorescence (GFAP and IBA1) or immunodensity (ChAT). In Image J, the upper and lower threshold optical densities were adjusted to encompass and match the immunoreactivity, providing an image with immunoreactive material appearing as red pixels. For quantification of ChAT immunoreactivity, the number of pixels of ChAT-immunoreactivity within the defined threshold and within the total area of the dorsal horn containing lamina I to IV were quantified per section. All sections for a given antibody reaction were reacted on the same day in the same reaction, using a custom designed plate. The image thresholds were determined and applied to all sections. Data are expressed as percentage of ChAT-immunoreactive pixels compared to the total number of pixels in the sampled area. For quantification of IBA1 and GFAP immunofluorescence, a fixed area (250 × 250 μm2) was positioned in the middle one third of the mediolateral extent of the spinal cord dorsal horn and the number of pixels of IBA1- or GFAP-immunoreactive cells within the threshold value was quantified. Data are expressed as number of pixels in the area. To limit variability in immunohistochemical measurements, all groups of rats were sacrificed and processed on the same day and the same threshold value was applied to all images for a given antibody. Images in figure 6 were pseudocolored with same adjustments to brightness or contrast to distinguish labeling of different proteins. The observer quantifying the sections was blind to the treatment. Negative controls were obtained for each antibody by omitting the primary antibody.
Figure 6.

DβH-saporin treatment increased glial markers in the spinal dorsal horn at 3 days following SNL. Photomicrographs depict GFAP and IBA1 immunoreactivity in the ipsilateral spinal dorsal horn from normal (A, B), SNL-saporin (C, D), and SNL-DβH-saporin treated (E, F) rats. Scale bar =100 μm. Quantification of GFAP (G) and IBA1 (H) immunoreactivity in the ipsilateral and contralateral spinal dorsal horn from normal (N), SNL-saporin(S-sap), and SNL-DβH-saporin (S-DβH) treated rats. Values are mean ± S.E of n=4–6 in each group.*p<0.01 vs. normal. #p<0.05 vs. saporin.
Acetylcholine release from synaptosomes
Preparation of crude synaptosomes from the L4–L6 spinal dorsal horn and measurement of [3H]-Acetylcholine release were performed as previously described 15. Each synaptosome preparation contained 3 unilateral dorsal quadrants of spinal cord from 3 SNL rats. Briefly, after loading with 1 μM choline chloride (combination of both triturated and unlabeled choline) for 20 min at 37°C, synaptosomes were perfused in temperature controlled perfusion chambers (SF-12, Brandel, Gaithersburg, MD) with Krebs buffer (in mM: 124 mM NaCl, 3 mM KCl, 2 mM MgSO4, 2 mM CaCl2, 1.25 mM KH2PO4, 25 mM NaHCO3, and 10 mM glucose, pH 7.35, 0.67 ml/min) for 25 min to remove free radioactivity, and then fractions were collected every 5 min for 20 min. After a 10 min baseline collection, synaptosomes were perfused with 10 nM dexmedetomidine alone for 2 min, then stimulated with 12 mM KCl-Krebs buffer (in mM: 115 NaCl, 12 KCl, 2 MgSO4, 2 CaCl2, 1.25 KH2PO4, 25 NaHCO3, and 10 glucose, pH 7.4) containing dexmedetomidine for 3 min. Concentrations of KCl and dexmedetomidine were determined from our previous studies 15, 17. An inhibitor of the acetylcholine transporter, hemicholinium-3 (10 μM), was present during perfusion to prevent reuptake of acetylcholine. [3H]-Acetylcholine release from synaptosomes in each fraction was measured by a liquid scintillation counter (LS6500, Beckman Coulter Inc., Fullerton, CA, USA). All chemicals were purchased from Sigma Chemical Co. except [3H]-choline (Perkin Elmer, Boston, MA, USA).
BDNF assay
The dorsal quadrants of the spinal cord ipsi- and contralateral to SNL were collected from saporin and DβH-saporin treated animals. Tissues were weighed and homogenized in ice cold homogenization buffer (mammalian cell lysis kit, Sigma Chemical CO) containing 1 mM phenylmethanesulfonyl fluoride (Sigma Chemical CO) and 1 % glycerol. The homogenates were centrifuged at 14,000×G for 30 min and the resulting supernatants were used to measure BDNF concentration using ChemiKine BDNF ELISA kit (Millipore) according to the manufacturer’s instructions.
Statistical analyses
Data were normally distributed and are presented as mean ± SE. Differences among groups were determined using one or two-way ANOVA as appropriate. P< 0.05 was considered significant.
Results
Effects of spinal noradrenaline depletion on hypersensitivity and clonidine analgesia after nerve injury
Consistent with previously reports 22, 25, intrathecal injection of DβH-saporin but not saporin alone completely depleted DβH-immunoreactive axons in the spinal dorsal horn (Fig 1 A, B) and also reduced DβH-immunoreactive neurons in the locus coeruleus (Fig 1 C, D) and A5 noradrenergic nuclei (Fig 1 E, F) as well as DβH-immunoreactive axons in the paraventricular nucleus (Fig 1 G, H). Similar results were observed in 3 saporin and 3 DβH-saporin treated rats (Images not shown). Animals tolerated both saporin and DβH-saporin treatments with no significant change in withdrawal threshold values in the right paw at 7 days after injection (Table 1). At 12 days after SNL, withdrawal threshold values were significantly lower than baseline in both saporin and DβH-saporin treated animals (p<0.01). DβH-saporin treated animals showed slightly but significantly lower withdrawal threshold values compared to saporin treated animals (p<0.05), as previously reported 22. In both saporin and DβH-saporin treated animals, intrathecal injection of clonidine (15 μg) significantly increased withdrawal threshold in the paw ipsilateral to SNL compared to pre-clonidine values (p<0.01) and co-administration of intrathecal atropine (30 μg) completely blocked the analgesia from clonidine (Fig. 2). DβH-saporin treated animals showed enhancement of peak analgesic efficacy of clonidine compared to saporin treated animals (p<0.05).
Figure 1.

Photomicrographs of DβH immunoreactive axons and neurons in the ipsilateral spinal dorsal horn (A, B), locus coeruleus (C, D), A5 noradrenergic nucleus (E, F), and paraventricular nucleus (G, H) of saporin and DβH-saporin treated SNL rats at 14 days after surgery. Scale bar in A, B, E, and F = 100 μm; in C, D, G, and H = 250 μm.
Table 1.
Effect of DβH-saporin on hypersensitivity after SNL
| Baseline | 7 days after injection | 12 days after SNL | |
|---|---|---|---|
| Saporin (n=16) | 18.0 ± 0.9 | 17.4 ± 1.0 | 2.0 ± 0.3* |
| DβH-saporin (n=16) | 18.0 ± 0.9 | 16.3 ± 1.0 | 1.1 ± 0.2*# |
Mean values of right paw withdrawal threshold (g) ±SE.
p<0.01 vs baseline.
p<0.05 vs saporin.
Figure 2.

DβH-saporin treatment enhanced the antihypersensitivity effect of clonidine in SNL rats. Effect of intrathecal clonidine (15 μg) with or without atropine (30 μg) was tested in saporin and DβH-saporin treated SNL rats at 12 days after surgery (values are mean ± S.E of n=8 in each group). Post-clonidine withdrawal threshold values were measured 1 h following clonidine administration. *p<0.01 vs. pre-clonidine. #p<0.05 vs. clonidine alone. $p<0.05 vs. saporin.
ChAT immunoreactivity and acetylcholine release from synaptosomes in the spinal dorsal horn
Figure 3 A and B depict ChAT immunoreactivity in the lumbar spinal dorsal horn in SNL rats treated with saporin or DβH-saporin. The vast majority of ChAT immunoreactivity in the spinal dorsal horn in all treatment groups was found in axons but a few ChAT-positive cells were also observed. DβH-saporin treated animals showed a trend towards higher ChAT immunoreactivity in the spinal dorsal horn ipsilateral (p=0.051) and contralateral (p<0.05) to SNL compared to saporin treated animals (Fig. 3C).
Figure 3.

DβH-saporin treatment increased ChAT immunoreactivity in the spinal dorsal horn 14 days following SNL. (A, B) Photomicrographs depict ChAT immunoreactivity in the ipsilateral spinal dorsal horn from saporin (A) and DβH-saporin treated (B) rats. Scale bar =100 μm. (C) Quantification of ChAT immunoreactivity in the ipsilateral and contralateral spinal dorsal horn from saporin and DβH-saporin treated rats (values are mean ± S.E of n=4 in each group).
[3H]-acetylcholine release from synaptosomes evoked by 12 mM KCl with or without dexmedetomidine did not differ between saporin and DβH-saporin treated animals. In both saporin and DβH-saporin treated animals, [3H]-acetylcholine release increased about 25–35 % from the baseline by 12 mM KCl and dexmedetomidine significantly enhanced KCl-evoked [3H]-acetylcholine release compared to KCl alone in synaptosomes both ipsilateral and contralateral to SNL surgery (Fig. 4; p<0.05). Consistent with our recent observations 15, 17, there was a significant difference in the facilitatory effect of dexmedetomidine on KCl-evoked [3H]-acetylcholine release between synaptosomes ipsilateral and contralateral to SNL in both saporin and DβH-saporin treated animals. We only examined fractional release in these experiments, and did not examine quantitative uptake or release.
Figure 4.

DβH-saporin treatment did not alter the facilitatory effect of dexmedetomidine on KCl-evoked acetylcholine release in spinal dorsal horn synaptosomes from rats after SNL. SNL synaptosomes were prepared from saporin and DβH-saporin treated animals at 14 days after surgery, and treated with 12mM KCl or a combination of 12 mM KCl with 10 nM dexmedetomidine (Dex). Values are mean ± S.E of n=6 in each group. *p<0.05 vs. KCl alone.
BDNF content and glial markers in the lumbar spinal dorsal horn
In spinal dorsal horn in saporin treated animals, BDNF content bilaterally increased at 7 days after SNL surgery compared to normal animals (Fig. 5; p<0.05), and then decreased to normal levels by day 14, consistent with our previous observation 16. DβH-saporin treated animals showed more BDNF content in both sides of spinal dorsal horn at 3 and 7 days after surgery than normal and saporin treated animals (p<0.05). Based on this result, we chose a time point at 3 days after SNL to examine glial activation in the spinal dorsal horn after surgery. Figure 6 A–F depict GFAP (an astrocyte marker) and IBA1 (a microglial marker) immunoreactivity in the spinal dorsal horn in normal and SNL animals treated with saporin and DβH-saporin. At 3 days after SNL, saporin treated animals showed more GFAP and IBA1 immunoreactivity in the spinal cord ipsilateral to surgery compared to the normal (Fig. 6G and H; p<0.05). In the spinal dorsal horn of DβH-saporin treated animals, GFAP immunoreactivity was higher bilaterally and IBA1 immunoreactivity was higher on the ipsilateral side compared to normal and saporin treated animals (p<0.05).
Figure 5.
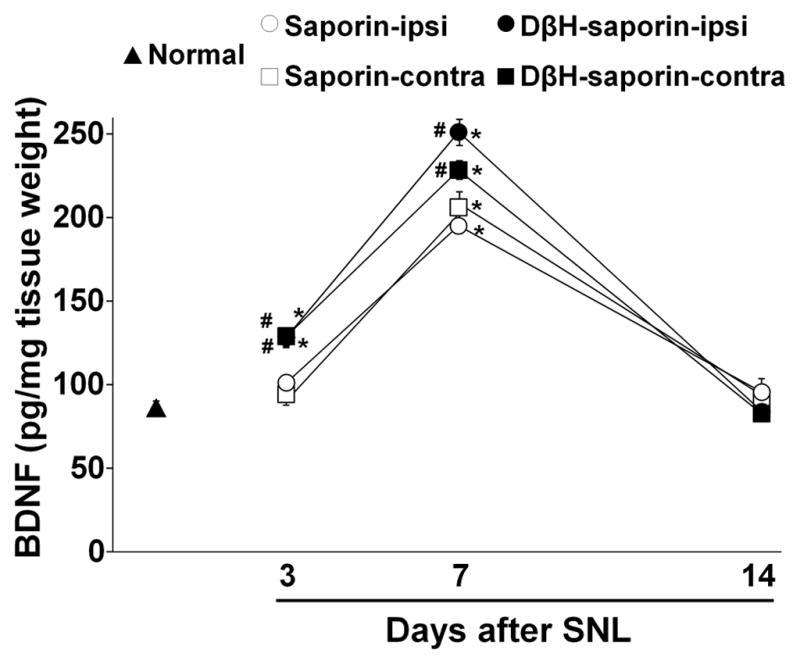
DβH-saporin treatment enhanced upregulation of BDNF in the spinal dorsal horn following SNL. BDNF content in L4–L6 level of ipsilateral (ipsi) and contralateral (contra) spinal dorsal horn was measured from normal, saporin treated SNL (3, 7, 14 days after surgery), and DβH-saporin treated SNL (3, 7, 14 days after surgery) animals. Values are mean ± S.E of n=7–9 in each group.*p<0.01 vs. normal. #p<0.05 vs. saporin.
Discussion
Bulbospinal noradrenergic fibers contain and release NA, ATP, and neuropeptide Y, factors which regulate neuronal and glial activation state and responses. The key finding of the current study is that presence of these fibers is not necessary for NA plasticity in analgesia after nerve injury and a suggestion that they may be important in braking glial activation. Spinal NA regulation of nociception has been studied for 3 decades, but many investigators consider it of minor importance in the pathophysiology of neuropathic pain. This conclusion is derived from the moderate effect of destruction of these noradrenergic pathways on hypersensitivity after nerve injury, 22 (as reproduced in the current study) and from the lack of effect of intrathecally injected α2-adrenoceptor antagonists on this hypersensitivity. 14, 24 These observations are, however, weakened by the inability to see exacerbated hypersensitivity in an already hypersensitive state after NA destruction and by the assumption that NA release in the spinal cord produces an effect which can be immediately reversed with an antagonist. If NA release, as we postulate, alters glial and neuronal plasticity which require hours or days to achieve, one would not necessarily anticipate an acute inhibition of this effect within minutes of administration of a NA antagonist.
In the current study, intrathecal injection of DβH-saporin completely depleted noradrenergic axons in the spinal cord. We also observed partial reduction of noradrenergic neurons and axons in the noradrenergic nuclei (LC and A5) and paraventricular nucleus by this DβH-saporin treatment, consistent with previous observations 22, 25. These results indicate that intrathecal injection of DβH-saporin not only depletes spinal noradrenaline but also reduces supra-spinal noradrenergic innervation to some extent. Clearly, extensive supraspinal noradrenergic denervation has profound effects on various physiologic states, and such extensive denervation or silencing, by intracerebroventricular injection of DβH-saporin or intra-LC injection of lidocaine, reduces hypersensitivity after nerve injury 2. In contrast, more selective ablation of noradrenergic innervation to the spinal cord by intrathecal injection of DβH-saporin as in the current and previous studies 22, results in a small increase rather than decrease in hypersensitivity in rats after spinal nerve ligation. This protective effect at a time of extreme hypersensitivity after nerve injury is consistent with laboratory and clinical observations that activation of LC or spinal noradrenergic pathway reduces hypersensitivity in rats and humans with chronic pain 8, 9, 12–14, 29, 30. Nonetheless, a clear limitation of this study is the partial reduction in supraspinal NA innervation induced by intrathecal DβH-saporin injected, which could have affected behavior and descending influences on glial and neuronal plasticity.
In rodents, activation of microglia and astrocytes in the spinal cord contributes to the development and maintenance of hypersensitivity after peripheral nerve injury 38. Consistent with behavioral observations, depletion of spinal NA in the current study did not affect immunohistochemical markers of activation of microglia and astrocytes in the normal spinal cord 22, but increased expression of these markers after nerve injury. We recognize that static examination of IBA-1 and GFAP do not adequately characterize activation state of glia or their anti- or pro-nociceptive or –inflammatory effects. Nonetheless, the approach used in the current study is widely applied to provide a rough gauge of glial activation. Previous studies demonstrated that α2-adrenoceptor agonists inhibit activation of microglia and astrocytes in the spinal cord in rats after peripheral nerve injury or inflammation 10, 41. We have recently demonstrated that peripheral nerve injury enhances spinal noradrenergic inhibition by increasing content and basal release of noradrenaline in the spinal dorsal horn 14, 16. These results suggest that peripheral nerve injury enhances endogenous spinal noradrenergic tone which, over time, directly or indirectly modulates the degree of glial activation and hypersensitivity, although this degree of endogenous noradrenergic tone itself is not sufficient to relieve neuropathic pain. The current study cannot determine whether increased glial activation in noradrenaline-depleted animals reflects reduced inhibition of primary afferent release or a direct effect on glia.
The most likely sources of BDNF in the spinal cord after peripheral nerve injury include the terminals of primary afferents and microglia. 4, 11, 16, 34 In normal and injured spinal cord, astrocytes can also produce BDNF 6, 7, 19. We and others have shown that peripheral nerve injury increases BDNF content in the primary sensory neurons and the spinal dorsal horn 11, 16, 26, 34. Previous studies demonstrated that depletion of noradrenaline itself does not alter BDNF content or expression in the brain 18, 40, consistent with the lack of effect of noradrenaline depletion on glial activity in the brain in the absence of nerve injury 22. Although the current study did not test whether NA depletion alters BDNF content in the spinal dorsal horn in the absence of injury, these previous observations in the brain suggest that NA depletion itself is unlikely to affect spinal BDNF content. In the current study, DβH-saporin treated SNL animals showed higher BDNF content in the spinal dorsal horn compared to saporin treated SNL animals, consistent with enhanced glial activation in DβH-saporin treated spinal dorsal horn. Although the contribution of primary sensory afferents to the increase in BDNF content in the spinal dorsal horn after nerve injury is possible, these results suggest that depletion of endogenous spinal noradrenaline induces more BDNF production in the spinal dorsal horn likely via enhancement of glial activation after nerve injury.
BDNF plays important roles in survival and axonal growth in cholinergic neurons and also shifts action of α2-adrenoceptors with those neurons from Gi/o-protein mediated inhibition to Gs-protein-mediated facilitation of acetylcholine release 15. Previous studies demonstrated that spinal infusion of BDNF or over-expression of BDNF by gene-transfer increases survival and axonal growth of cholinergic neurons in rats after spinal cord injury20, 42, and that intracerebroventricular infusion of BDNF prevents loss of cholinergic neurons in the brainstem after hypoglossal nerve transection 36. We have recently demonstrated that blockade of BDNF-trkB signaling by spinal infusion of BDNF antibody or repeated intrathecal injection of K252a reduced ChAT-immunoreactivity in the spinal dorsal horn and blocked the shift in α2-adrenoceptor-mediated facilitatory effect on acetylcholine release in the spinal cord 15, 17. The current study confirmed the α2-adrenoceptor-mediated facilitation of acetylcholine release after nerve injury and further demonstrated that DβH-saporin treatment increased cholinergic immunoreactivity in the spinal dorsal horn, consistent with enhancement of BDNF up-regulation after nerve injury by DβH-saporin treatment. Interestingly, in the current study, spinal BDNF level in DβH-saporin treated animals was higher than the saporin treated and normal animals at 3–7 days and returned to normal level at 14 days after surgery, suggesting that initial enhancement of BDNF production is important to increase cholinergic immunoreactivity in the spinal dorsal horn. These results should, however, be qualified, since the methodology employed did not allow us to determine whether increased ChAT-immunoreactivity was associated with increased quantitative release of acetylcholine release, as only fractional release was studied. As such, other interpretations of increased ChAT-immunoreactivity, such as increased antigen availability to the antibody without change in fiber density, are possible.
It may appear paradoxical that clonidine’s behavioral effect was enhanced after nerve injury in animals receiving DβH-saporin, consistent with denervation supersensitivity or increased receptor expression, but α2-adrenoceptor facilitated release of acetylcholine in synaptosomes was not increased. Previous studies showed that depletion of spinal noradrenaline increases α2-adrenoceptor number in the spinal cord or affinity to clonidine in normal animals 21, 32, while peripheral nerve injury itself does not alter neither number nor affinity of α2-adrenoceptors 1. Although the current study does not address whether expression or affinity of α2-adrenoceptors alters in cholinergic neurons by DβH-saporin treatment, this plasticity in α2-adrenoceptors by depletion of spinal noradrenaline may contribute to enhanced antihypersensitive effect of clonidine after nerve injury. As regards acetylcholine release, if one assumes that the increased ChAT immunostaining reflects increased capacity for acetylcholine release, then the same fractional release of acetylcholine release observed with dexmedetomidine in synaptosomal preparations after DβH-saporin as control would quantitatively release more acetylcholine. Neither the relationship between ChAT immunostaining and acetylcholine content nor the quantitative release of acetylcholine were examined in the current study.
In summary, depletion of spinal noradrenergic fibers does not prevent α2-adrenoceptor analgesia and associated plasticity of its interaction with cholinergic terminals, suggesting that tonic activity of these fibers does not drive these plastic responses. NA fiber depletion is associated with increased expression of a markers of glial activation, consistent with acute pharmacologic studies demonstrated glial inhibition by NA receptor activation.
Footnotes
Disclosures
This work was supported by grants DA27690 to KH and NS59574 to JE from the National Institutes of Health, Bethesda, Maryland. The Authors have no conflict of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bantel C, Eisenach JC, Duflo F, Tobin JR, Childers SR. Spinal nerve ligation increases alpha2-adrenergic receptor G-protein coupling in the spinal cord. Brain Res. 2005;1038:76–82. doi: 10.1016/j.brainres.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 2.Brightwell JJ, Taylor BK. Noradrenergic neurons in the locus coeruleus contribute to neuropathic pain. Neuroscience. 2009;160:174–85. doi: 10.1016/j.neuroscience.2009.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 4.Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–21. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 5.DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- 6.Dougherty KD, Dreyfus CF, Black IB. Brain-derived neurotrophic factor in astrocytes, oligodendrocytes, and microglia/macrophages after spinal cord injury. Neurobiol Dis. 2000;7:574–85. doi: 10.1006/nbdi.2000.0318. [DOI] [PubMed] [Google Scholar]
- 7.Dreyfus CF, Dai X, Lercher LD, Racey BR, Friedman WJ, Black IB. Expression of neurotrophins in the adult spinal cord in vivo. J Neurosci Res. 1999;56:1–7. doi: 10.1002/(SICI)1097-4547(19990401)56:1<1::AID-JNR1>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 8.Eisenach JC, DuPen S, Dubois M, Miguel R, Allin D. Epidural clonidine analgesia for intractable cancer pain. The Epidural Clonidine Study Group. Pain. 1995;61:391–9. doi: 10.1016/0304-3959(94)00209-W. [DOI] [PubMed] [Google Scholar]
- 9.Eisenach JC. Muscarinic-mediated analgesia. Life Sci. 1999;64:549–54. doi: 10.1016/s0024-3205(98)00600-6. [DOI] [PubMed] [Google Scholar]
- 10.Feng X, Zhang F, Dong R, Li W, Liu J, Zhao X, Xue Q, Yu B, Xu J. Intrathecal administration of clonidine attenuates spinal neuroimmune activation in a rat model of neuropathic pain with existing hyperalgesia. Eur J Pharmacol. 2009;614:38–43. doi: 10.1016/j.ejphar.2009.04.044. [DOI] [PubMed] [Google Scholar]
- 11.Fukuoka T, Kondo E, Dai Y, Hashimoto N, Noguchi K. Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. J Neurosci. 2001;21:4891–900. doi: 10.1523/JNEUROSCI.21-13-04891.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldstein DJ, Lu Y, Detke MJ, Lee TC, Iyengar S. Duloxetine vs. placebo in patients with painful diabetic neuropathy. Pain. 2005;116:109–18. doi: 10.1016/j.pain.2005.03.029. [DOI] [PubMed] [Google Scholar]
- 13.Hayashida K, DeGoes S, Curry R, Eisenach JC. Gabapentin activates spinal noradrenergic activity in rats and humans and reduces hypersensitivity after surgery. Anesthesiology. 2007;106:557–62. doi: 10.1097/00000542-200703000-00021. [DOI] [PubMed] [Google Scholar]
- 14.Hayashida K, Obata H, Nakajima K, Eisenach JC. Gabapentin acts within the locus coeruleus to alleviate neuropathic pain. Anesthesiology. 2008;109:1077–84. doi: 10.1097/ALN.0b013e31818dac9c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayashida K, Eisenach JC. Spinal alpha2-adrenoceptor-mediated analgesia in neuropathic pain reflects brain-derived nerve growth factor and changes in spinal cholinergic neuronal function. Anesthesiology. 2010;113:406–12. doi: 10.1097/ALN.0b013e3181de6d2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayashida KI, Clayton BA, Johnson JE, Eisenach JC. Brain derived nerve growth factor induces spinal noradrenergic fiber sprouting and enhances clonidine analgesia following nerve injury in rats. Pain. 2008;136:348–55. doi: 10.1016/j.pain.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayashida KI, Eisenach JC. A Tropomyosine Receptor Kinase Inhibitor Blocks Spinal Neuroplasticity Essential for the Anti-Hypersensitivity Effects of Gabapentin and Clonidine in Rats With Peripheral Nerve Injury. J Pain. 2010 doi: 10.1016/j.jpain.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hutter P, Johansson M, Saria A, Humpel C. Acute and chronic noradrenergic regulation of neurotrophin messenger RNA expression in rat hippocampus: evidence from lesions and organotypic cultures. Neuroscience. 1996;70:15–29. doi: 10.1016/0306-4522(95)00346-k. [DOI] [PubMed] [Google Scholar]
- 19.Ikeda O, Murakami M, Ino H, Yamazaki M, Nemoto T, Koda M, Nakayama C, Moriya H. Acute up-regulation of brain-derived neurotrophic factor expression resulting from experimentally induced injury in the rat spinal cord. Acta Neuropathol. 2001;102:239–45. doi: 10.1007/s004010000357. [DOI] [PubMed] [Google Scholar]
- 20.Jakeman LB, Wei P, Guan Z, Stokes BT. Brain-derived neurotrophic factor stimulates hindlimb stepping and sprouting of cholinergic fibers after spinal cord injury. Exp Neurol. 1998;154:170–84. doi: 10.1006/exnr.1998.6924. [DOI] [PubMed] [Google Scholar]
- 21.Janss AJ, Jones SL, Gebhart GF. Effect of spinal norepinephrine depletion on descending inhibition of the tail flick reflex from the locus coeruleus and lateral reticular nucleus in the rat. Brain Res. 1987;400:40–52. doi: 10.1016/0006-8993(87)90651-2. [DOI] [PubMed] [Google Scholar]
- 22.Jasmin L, Boudah A, Ohara PT. Long-term effects of decreased noradrenergic central nervous system innervation on pain behavior and opioid antinociception. J Comp Neurol. 2003;460:38–55. doi: 10.1002/cne.10633. [DOI] [PubMed] [Google Scholar]
- 23.Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–63. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 24.Kim SK, Park JH, Bae SJ, Kim JH, Hwang BG, Min BI, Park DS, Na HS. Effects of electroacupuncture on cold allodynia in a rat model of neuropathic pain: mediation by spinal adrenergic and serotonergic receptors. Exp Neurol. 2005;195:430–6. doi: 10.1016/j.expneurol.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 25.Li X, Conklin D, Ma W, Zhu X, Eisenach JC. Spinal noradrenergic activation mediates allodynia reduction from an allosteric adenosine modulator in a rat model of neuropathic pain. Pain. 2002;97:117–25. doi: 10.1016/s0304-3959(02)00011-8. [DOI] [PubMed] [Google Scholar]
- 26.Miletic G, Miletic V. Increases in the concentration of brain derived neurotrophic factor in the lumbar spinal dorsal horn are associated with pain behavior following chronic constriction injury in rats. Neurosci Lett. 2002;319:137–40. doi: 10.1016/s0304-3940(01)02576-9. [DOI] [PubMed] [Google Scholar]
- 27.Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66:355–474. doi: 10.1016/s0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- 28.Obata H, Li X, Eisenach JC. alpha2-Adrenoceptor activation by clonidine enhances stimulation-evoked acetylcholine release from spinal cord tissue after nerve ligation in rats. Anesthesiology. 2005;102:657–62. doi: 10.1097/00000542-200503000-00027. [DOI] [PubMed] [Google Scholar]
- 29.Obata H, Saito S, Koizuka S, Nishikawa K, Goto F. The monoamine-mediated antiallodynic effects of intrathecally administered milnacipran, a serotonin noradrenaline reuptake inhibitor, in a rat model of neuropathic pain. Anesth Analg. 100:1406–10. doi: 10.1213/01.ANE.0000149546.97299.A2. table of contents, 2005. [DOI] [PubMed] [Google Scholar]
- 30.Pan HL, Chen SR, Eisenach JC. Intrathecal clonidine alleviates allodynia in neuropathic rats: interaction with spinal muscarinic and nicotinic receptors. Anesthesiology. 1999;90:509–14. doi: 10.1097/00000542-199902000-00027. [DOI] [PubMed] [Google Scholar]
- 31.Paqueron X, Conklin D, Eisenach JC. Plasticity in action of intrathecal clonidine to mechanical but not thermal nociception after peripheral nerve injury. Anesthesiology. 2003;99:199–204. doi: 10.1097/00000542-200307000-00030. [DOI] [PubMed] [Google Scholar]
- 32.Post C, Arwestrom E, Minor BG, Wikberg JE, Jonsson G, Archer T. Noradrenaline depletion increases noradrenaline-induced antinociception in mice. Neurosci Lett. 1985;59:105–9. doi: 10.1016/0304-3940(85)90222-8. [DOI] [PubMed] [Google Scholar]
- 33.Post C, Persson ML, Archer T, Minor BG, Danysz W, Sundstrom E. Increased antinociception by alpha-adrenoceptor drugs after spinal cord noradrenaline depletion. Eur J Pharmacol. 1987;137:107–16. doi: 10.1016/0014-2999(87)90188-9. [DOI] [PubMed] [Google Scholar]
- 34.Shen H, Chung JM, Chung K. Expression of neurotrophin mRNAs in the dorsal root ganglion after spinal nerve injury. Brain Res Mol Brain Res. 1999;64:186–92. doi: 10.1016/s0169-328x(98)00314-3. [DOI] [PubMed] [Google Scholar]
- 35.Sonohata M, Furue H, Katafuchi T, Yasaka T, Doi A, Kumamoto E, Yoshimura M. Actions of noradrenaline on substantia gelatinosa neurones in the rat spinal cord revealed by in vivo patch recording. J Physiol. 2004;555:515–26. doi: 10.1113/jphysiol.2003.054932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tuszynski MH, Mafong E, Meyer S. Central infusions of brain-derived neurotrophic factor and neurotrophin-4/5, but not nerve growth factor and neurotrophin-3, prevent loss of the cholinergic phenotype in injured adult motor neurons. Neuroscience. 1996;71:761–71. doi: 10.1016/0306-4522(95)00440-8. [DOI] [PubMed] [Google Scholar]
- 37.Tyce GM, Yaksh TL. Monoamine release from cat spinal cord by somatic stimuli: an intrinsic modulatory system. J Physiol. 1981;314:513–29. doi: 10.1113/jphysiol.1981.sp013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vallejo R, Tilley DM, Vogel L, Benyamin R. The role of glia and the immune system in the development and maintenance of neuropathic pain. Pain Pract. 2010;10:167–84. doi: 10.1111/j.1533-2500.2010.00367.x. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Cheng CY, Wang JY, Lin JC. Enhanced antinociception of clonidine in spontaneously hypertensive rats involves a presynaptic noradrenergic mechanism. Pharmacol Biochem Behav. 1998;59:109–14. doi: 10.1016/s0091-3057(97)00383-3. [DOI] [PubMed] [Google Scholar]
- 40.Windle V, Power A, Corbett D. Norepinephrine depletion facilitates recovery of function after focal ischemia in the rat. Eur J Neurosci. 2007;26:1822–31. doi: 10.1111/j.1460-9568.2007.05799.x. [DOI] [PubMed] [Google Scholar]
- 41.Xu B, Zhang WS, Yang JL, Lu N, Deng XM, Xu H, Zhang YQ. Evidence for suppression of spinal glial activation by dexmedetomidine in a rat model of monoarthritis. Clin Exp Pharmacol Physiol. 2010;37:e158–66. doi: 10.1111/j.1440-1681.2010.05426.x. [DOI] [PubMed] [Google Scholar]
- 42.Xu K, Uchida K, Nakajima H, Kobayashi S, Baba H. Targeted retrograde transfection of adenovirus vector carrying brain-derived neurotrophic factor gene prevents loss of mouse (twy/twy) anterior horn neurons in vivo sustaining mechanical compression. Spine (Phila Pa 1976) 2006;31:1867–74. doi: 10.1097/01.brs.0000228772.53598.cc. [DOI] [PubMed] [Google Scholar]
- 43.Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav. 1976;17:1031–6. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]