

Abstract
Mitochondrial dysfunction is one of the major pathological changes seen in Alzheimer's disease (AD). Amyloid beta-peptide (Aβ), a neurotoxic peptide, accumulates in the brain of AD subjects and mediates mitochondrial and neuronal stress. Therefore, protecting mitochondrion from Aβ-induced toxicity holds potential benefits for halting and treating and perhaps preventing AD. Here, we report that administration of ginsenoside Rg1, a known neuroprotective drug, to primary cultured cortical neurons, rescues Aβ-mediated mitochondrial dysfunction as shown by increases in mitochondrial membrane potential, ATP levels, activity of cytochrome c oxidase (a key enzyme associated with mitochondrial respiratory function), and decreases in cytochrome c release. The protective effects of Rg1 on mitochondrial dysfunction correlate to neuronal injury in the presence of Aβ. This finding suggests that ginsenoside Rg1 may attenuate Aβ-induced neuronal death through the suppression of intracellular mitochondrial oxidative stress and may rescue neurons in AD.
Keywords: Alzheimer's disease, oligomeric beta-amyloid peptide1-42, mitochondria, ginsenoside Rg1
INTRODUCTION
Alzheimer's disease (AD) is the most common form of dementia and affects millions of people worldwide. However, mechanisms of AD remain unclear. One dominant theory in the field since 1992, the amyloid hypothesis, suspects that Aβ42 shifts from fibrils to protofibrils to oligomers to perhaps dimers [1–5]. A large body of recent evidence points to the overproduction and accumulation of Aβ (particularly oligomeric Aβ1-42) as a trigger of the pathological cascade in AD.
Mitochondrial dysfunction is an early pathological feature of AD. It is known that activities of several mitochondrial enzymes are altered in AD [6, 7]. Impairments in energy metabolism and increased oxidative stress occur in AD brain. Recent studies show a significant Aβ accumulation in mitochondria of AD brain and transgenic AD mouse models [8–13]. Aβ-rich mitochondria have deficits in mitochondrial function such as impaired respiratory function, decreased ATP levels, increased opening of the membrane permeability transition pore (mPTP) and reactive oxygen free radicals (ROS), and impaired calcium buffering capacity [7–9, 12, 14–16]. Furthermore, Aβ can directly intervene with mitochondrial and neuronal function. For example, treatment of oligomeric Aβ causes decreased mitochondrial membrane potential, decreased activity of cytochrome c oxidase (CcO), and increased cytochrome c release, resulting in neuronal apoptosis [9]. These studies indicate that Aβ-induced neural mitochondrial dysfunction plays an important role in the pathogenesis of AD [17–20]. Therefore, the inhibition of Aβ-mediated neuronal mitochondrial stress may have potential benefits for halting, treating and/or preventing AD.
Ginseng, on the other hand, has long been used to alleviate many ailments, particularly those associated with aging and memory deterioration [21]. A representative constituent of ginsenoside, Rg1 Fig. (1), may be a candidate neuroprotective agent for treating AD and in fact, has been documented to produce antioxidant effects [22–27]. However, whether ginsenoside Rg1 has any protective effect on Aβ-induced mitochondrial injury has not yet been reported. In this study, we assessed the effects of ginsenoside, Rg1, on mitochondrial function associated with neurotoxicity from oligomeric Aβ in primary cultured cortical neurons. Our results clearly show that Rg1 treatment significantly improves mitochondrial function contributing to decreased neuronal death.
Fig. (1). Structure of ginsenside Rg1.
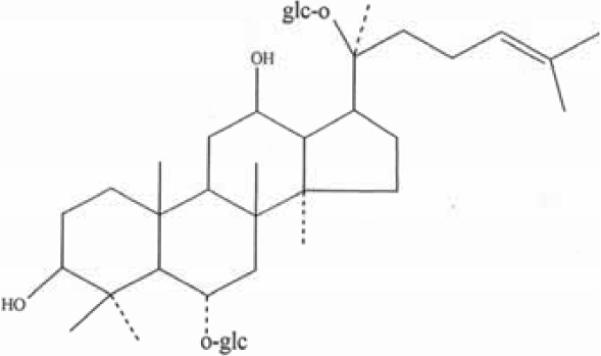
Molecular formula: C42H72O14. Molecular weight of 800.
MATERIALS AND METHODS
Materials
Ginsenoside Rg1 was obtained from the Department of Biochemistry, Norman Bethune Medical College of Jilin University (Jilin, China) in the form of white powder-like crystals, with a melting point of 194–195°C, a molecular weight of 800, general formula C42H72O14 Fig. (1), and a purity of over 98% as documented by reverse phase high-pressure liquid chromatography (HPLC). Aβ1-42 was purchased from QCB (Quality Controlled Biochemicals) (Hopkinton, MA, USA). Oligomeric Aβ1-42 peptide was prepared as described previously, and characterized by atomic force microscopy [9]. Neurobasal Medium, Trypsin-EDTA, L-glutamin, B27 Supplement, Fetal Bovine Serum (FBS) were purchased from Gibco-BRL (Grand Island, NY, USA). POLY-LYSINE, Cytochrome c and dichlorofluorescin diacetate (DCFH-DA) were obtained from Sigma Chemical Co. (St. Louis, MO, USA). The caspase-3 Assay kit was obtained from Clontech Laboratories Inc, (Mountain View, CA, USA) and Function ELISA Cytochrome c and Mitochondrial Fractonation kits from Active Motif (Carlsbad, CA, USA). The Peroxide/Peroxidase Assay Kit was from Molecular Probes, Inc. (Eugene, OR, USA), the JC-1 Mitochondrial Membrance Potential Detection Kit from Cell Technology (Mountain View, CA, USA), the ATP Bioluminescence Assay Kit HS II from Roche company (Indianapolis, IN, USA), and the Apoptosis detection kit from Trevigen company (Gaithersburg, MD, USA).
Cortical Neuron Culture and Treatment
Cortical neurons from C57BL/6 mouse fetuses at embryonic days 15–16 (Fuzhou Animal Center, Fuzhou, China) were prepared as described previously [7, 28]. All procedures followed the ethical guidelines for investigations of experimental pain in conscious animals and were approved by the Institutional Animal Care Committee of Fujian Medical University. Following the above procedures, cultures typically contained more than 90% neurons as assessed by staining with an antibody against phosphorylated neurofilament (data not shown). After 3 days in vitro, the cells were preincubated with Rg1 or other drugs for 24 h, and then co-incubated with Aβ1-42 (5 μM final concentration) for 48 h or other indicated lengths of time.
Mitochondria Membrane Potential
Loss of mitochondrial membrane potential was determined using the JC-1 mitochondrial transmembrane potential detection kit (Cell Technology, Inc.) according to the manufacturer instructions. Briefly, neurons were treated with ginsenoside Rg1 and/or oligomeric Aβ1-42 for different time points. Neurons were then collected, suspended in 0.5mL 1× JC-1 reagent, and incubated at 37°C in a 5% CO2 incubator for 15 minutes. The cells were then collected by centrifugation and the pellet was washed with 2 ml assay buffer (1×) and resuspended in 0.3 ml Assay Buffer. One hundred μl cell suspension was transferred into a black 96-well plate (triplicate for each sample). Red fluorescence (excitation 550 nm, emission 600 nm) and green fluorescence (excitation 485 nm, emission 535 nm) were measured with a fluorescence plate reader (BioTek Senergy HT, Vermont, USA). The ratio of red to green fluorescence, an indicator for membrane potential, was determined. The ratio was decreased in dead cells and in cells undergoing apoptosis in comparison with healthy cells.
ROS Measurement
The dichlorofluorescein (DCF) assay was used to measure the levels of ROS. The cell-permeable dichlorofluorescein diacetate (DCFH-DA) crosses into the cytoplasm, where it is de-esterified by cellular esterase resulting in DCFH. DCFH in turn is converted upon oxidation to the highly fluorescent DCF. By measuring the fluorescence, we are able to quantify the levels of ROS. After incubation with Aβ1-42 for varying lengths of time, neurons were washed with PBS and incubated with 10 μM of nonfluorescent DCFH-DA for 30 min at 37 ° in the incubator with 5 % CO2. The measurements were performed on a microplate fluorometer (BioTek Senergy HT, Vermont, USA) with λex 480 nm and λem 530 nm.
Intracellular production of H2O2 was fluorometrically determined according to the manufacturer's instructions (Amplex Red, Molecular Probes). Cells plated on 96-well plates were incubated with the fresh growth medium containing agents to be tested. Cultures were exposed to a working solution containing 50 μM Amplex Red reagent and 0.1 U/ml horseradish peroxidase (HRP) for 60 min. Fluorescence was measured with the excitation at 540 nm and the emission at 590 nm using a microplate fluorometer (BioTek Senergy HT, Vermont, USA).
Measurement of Cytochrome c Oxidase (CcO) Activity
Complex IV activity was measured as described previously [7]. Briefly, neuronal cultures in six-well plates were washed with ice-cold PBS, and cells were harvested, centrifuged, and suspended in 50 μl of isolation buffer containing 250 mM sucrose, 20 mM HEPES, pH 7.2, and 1 mM EDTA. Cell suspensions (containing ~3–4 mg of protein/ml) were added to a cuvette containing 0.95 ml of 1× assay buffer (10 mM Tris-HCl, pH 7.0, and 120 mM KCl), and the reaction volume was brought to 1.05 ml with 1× enzyme dilution buffer (10 mM Tris-HCl, pH 7.0). The reaction was then started by the addition of 50 μl of ferrocytochrome c substrate solution (0.22 mM) (from Sigma); the change in absorbance of cytochrome c at 550 nm was measured using a DU640 spectrophotometer (Beckman company, USA). The reading was recorded every 5 sec during the first 3 minutes. Background levels were measured without cell suspensions.
ATP Measurement
ATP levels were determined using a luciferin/luciferase-based ATP assay kit (from Roche). Briefly, neurons were treated with ginsenoside Rg1 and/or oligomeric Aβ1-42 for different lengths of time. Neurons were harvested, centrifuged and diluted at a concentration of 1 × 104 cells/ml. The same volume of cell lysis reagent was added to the samples and then incubated them for 5 min at 25 °C. An appropriate volume of luciferase reagents were added to the samples and the reading was recorded consecutively from 1 to 10 s with an interval of 1 s using a Microplate Luminometer MPL4, (Berthold, Pforzheim, Germany). The change between different groups was compared.
Isolation of Mitochondria and Cytochrome c Release
The Mitochondrial Fractionation kit (from Active Motif, Inc.) was used to isolate mitochondrial and cytosolic fractions from cells according to the manufacturer's instructions. Briefly, primary cortical neurons were treated with ginsenoside Rg1 and/or oligomeric Aβ1-42. The treated neurons were scraped and spun twice at 600 × g for 5 minutes. Ice-cold 1X cytosolic buffer was added and the cell pellet was resuspended and incubated on ice for 15 minutes. Cells were homogenized and the lysate was spun twice at 800 × g for 20 minutes. The resultant supernatant contained the cytosol, including mitochondria; the supernatant was spun at 10,000 × g for 20 minutes to pellet the mitochondria. The mitochondrial pellet was washed and spun with 1X cytosolic buffer at 10,000 × g for 10 minutes, and then lysed by adding Complete Mitochondria Buffer followed by incubation on ice for 15 minutes, the result of which was the Mitochondrial fraction. At the same time, the supernatant was centrifuged at 16,000 × g for 25 minutes. The centrifuged supernatant was the cytosolic fraction. The protein concentration was measured by using a Bio-Rad protein assay.
After isolation of mitochondria and cytosolic fraction, the ELISA Cytochrome C kit (from Active Motif, Inc.) was used to measure the level of cytochrome c according to the manufacturer's instructions.
Assessment of Apoptosis
Apoptosis in cultured neurons was assessed by TUNEL assay under light microscopy. Neurons grown on coverslips were washed and fixed in 3.7 % paraformaldehyde for 10 min at 18–24°C and then post-fixed with 100 % alcohol for 20 min and washed in PBS for 10 min. The previously treated neurons were first covered with 50 μl of NeuroPore, incubated for 25 minutes, and washed twice with PBS. Neurons were immersed in quenching solution (3% hydrogen peroxide in Methanol) for 5 minutes, washed with PBS, and incubated in 1X TdT Labeling Buffer for 5 minutes, and further covered with 50 μl of labeling reaction mix followed by incubation at 37 °C for 1 hour in a humidity chamber. The labeling reaction was stopped with 1X TdT Stop Buffer for 5 minutes. Finally, the treated neurons were carefully washed, covered with 50 μl of Strep-HRP Solution for 10 minutes, and immersed in DAB solution for 2 to 7 minutes. TUNEL-positive cells were counted in five fields per well and averaged.
Caspase 3 Activity
Caspase-3 activity was measured according to the manufacturer's instructions (Clonteck Laboratories). Briefly, primary cortical neurons were treated with ginsenoside Rg1 and/or oligomeric Aβ1-42 for different lengths of time. The neurons were scraped and centrifuged at 400 × g for 5 min and then re-suspended in 50 μl chilled cell lysis buffer per 2 × 105cells and incubated on ice for 10 min. Next, they were centrifuged again at maximum speed for 10 min at 4°C to precipitate cellular debris. The supernatants were transferred to new microcentrifuge tubes and 50 μl of 2X Reaction Buffer/ DTT Mix (10 μl of 1 M DTT stock per 1 ml of 2X Reaction Buffer) was added. One μl of caspase-3 inhibitor (DEVD-CHO) was added to 50 μl of one supernatant. Transferred supernatants were incubated at 37°C for 1 hr in a water bath. A parallel control reaction that did not contain conjugated substrate was set up. Caspase 3 activity was assessed by reading excitation at 380-nm and emission at 460-nm in a microplate fluorometer (BioTek Senergy HT, Vermont, USA).
Statistical Analysis
All numerical results were expressed as the mean± standard error of the mean (S.E.M.). Data were analyzed by analysis of variance (ANOVA) followed by analysis of significance (Tukey test) for multiple time points groups. The results from Rg1 or/and Abeta treated groups for indicated time were compared using t test. Statistical analysis was carried out using SPSS (version 11.0 for Windows).
RESULTS
Effect of Ginsenoside Rg1 on Oligomeric Aβ1-42-Induced Mitochondrial Function
To determine the effect of Rg1 on mitochondrial function and properties, we measured mitochondrial membrane potential, ATP level, mitochondrial enzyme activity associated with the respiratory chain, cytochrome c oxidase (CcO), and ROS levels in Aβ-treated neurons as compared with Rg1 – treated neurons. First, we evaluated the loss of mitochondrial membrane potential (ΔΨM) using JC-1 as an indicator. Upon Aβ exposure, cortical neurons showed a statistically significant time-dependent decrease in ΔΨM when compared to the control Fig. (2A). However, pre-treatment with varying doses of ginsenoside Rg1 (2.5–10 μM) showed a dose-dependent increase in ΔΨM as compared to neurons treated with Aβ alone for either 24h or 48h Fig. (2B). The ΔΨM in vehicle-treated control or ginsenoside Rg1 groups was higher than that of the Aβ1-42 treatment for 24h or 48h.
Fig. (2). The effects of ginsenoside Rg1 on Aβ-induced mitochondrial abnormalities.

Neurons were pretreated with ginsenoside Rg1 at 2.5–10 μM for 24 h, then exposed to Aβ1-42 at 5.0 μM for the indicated time points. Levels of mitochondrial membrane potential (Panels A and B), ATP (Panels C and D), and cyto-chrome c oxidase (CcO, Panels E and F) were measured. NS: non significance. N = 3–5 per group of cells. Studies were repeated four times and data were shown as mean ± SEM.
Second, we measured ATP levels. As shown in Fig. (2C), ATP generation in neurons treated with Aβ1-42 was time-dependent decreased when compared to the control group, particularly at 12h, 24h or 48h. However, in neurons pre-incubated with ginsenoside Rg1 and then co-incubated with A for 24h or 48h, the ATP levels were significantly increased compared with that of Aβ1-42 group for 24h or 48h Fig. (2D). Decreases in ATP levels correlated with the change in ΔΨM.
Third, we analyzed mitochondrial respiratory function by measuring CcO activity. The mitochondrial respiratory chain is composed of five enzyme complexes, and of these CcO (complex IV) is vital. Defects in CcO activity have been shown in early stage AD brains from AD mouse models and Aβ-treated neurons [29–34]. Neurons treated with 5μM oligomeric Aβ1-42 for 12h to 48h demonstrated significantly decreased CcO activity Fig. (2E). Notably, Rg1 pre-treatment significantly increased CcO activity in a dose-dependent manner as compared to cells treated with Aβ1-42 alone for 24h or 48h Fig. (2F).
Given that mitochondria are a major source for generation and accumulation of reactive oxygen species (ROS) and that mitochondrial dysfunction causes an increase in radical production, we next determined the effect of Rg1 on reactive oxygen species (ROS) production. ROS and H2O2, two predominant oxygen radicals, were detected simultaneously. ROS and H2O2 were significantly increased in neurons exposed to Aβ1-42 Fig. (3A, C), especially in the group incubated with Aβ1-42 for 24h or 48h as compared to the control group. Cells that were pre-incubated with Rg1 exhibited significantly reduced levels of ROS and H2O2 when insulted by Aβ Fig. (3B, D).
Fig. (3). The effect of ginsenoside Rg1 on ROS generation.
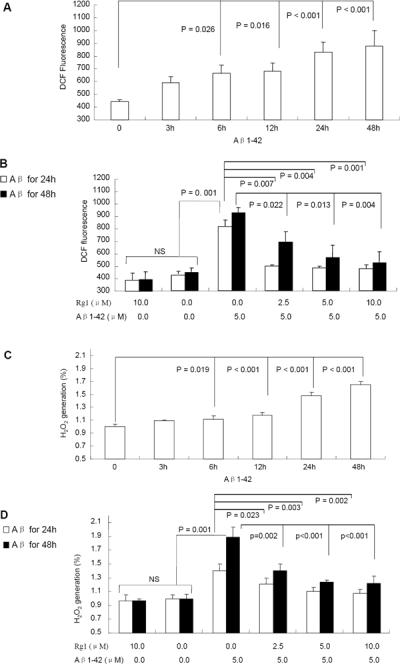
Neurons treated with Aβ1-42 (5.0 μM) at the indicated time points showed an increase in DCF fluorescence intensity (an indicator of ROS generation (Panel A) and H2O2 production (Panel C). Ginsenoside Rg1 treatment at the indicated concentration abolished oligomeric Aβ1-42-induced ROS generation and H2O2 production (Panels B and D). NS: non significance. N = 3–5 per group of cells. Data were expressed as mean ± SEM. Studies were repeated at least three times.
Cytochrome c Release: the Effect of Ginsenoside Rg1 and Oligomeric Aβ1-42
In most apoptosis pathways, mitochondrial cytochrome c release is a key event in initiating the cascade of reactions leading to apoptotic cell death [35]. Therefore, cytochrome c levels in cytosolic and mitochondrial fraction were detected separately after the neurons were treated with 5 μM oligomeric Aβ1-42 for different time points (3h, 6h, 12h, 24h or 48h). Levels of cytochrome c in cytosolic fraction were increased in a time-dependent manner in parallel with the Aβ1-42 incubation time Fig. (4A), especially at 24h or 48h, when compared to the control group. Similarly, cytochrome c levels in mitochondrial fraction were decreased. However, we found less cytochrome c protein in Rg1-treated neurons in the cytosolic fraction than vehicle-treated neuron in the presence of Aβ Fig. (4B). Similarly, cytochrome c levels in mitochondrial fractions were increased significantly in Rg1-treated group in comparison with that of the group treated only with Aβ1-42. These results suggest that ginsenoside Rg1 attenuates Aβ-induced cytochrome c release from mitochondria.
Fig. (4). The effect of ginsenoside Rg1 on Aβ-induced the release of mitochondrial cytochrome c.

Neurons were treated with oligomeric Aβ1-42 (5 μM) alone (Panel A) or plus ginsenoside Rg1 (Panel B). Levels of cytochrome c in the cytosolic fraction or mitochondrial fractions were determined by ELISA. Ginsenoside Rg1 attenuated mitochondrial cytochrome c releasing to cytosolic fraction in neurons induced by oligomeric Aβ1-42 at 5μM for 48 hr (Panel B). * and # P <0.001 vs. Aβ treatment in mitochondrial fraction and cytosolic fraction, respectively. Data were determined from four experiments.
The Effect of Ginsenoside Rg1 on Aβ1-42 -Induced Neurotoxicity
To determine whether the protective effect of ginsenoside Rg1 on Aβ-induced mitochondrial dysfunction correlates to neurotoxicity, we assessed changes in morphology, apoptosis and caspase-3 activity. Neurons were pre-treated with varying doses of ginsenoside Rg1 for 24h and co-incubated with or without oligomeric Aβ1-42 for 48h and then analyzed under light microscopy Fig. (5A). Our results showed a normal dense and consistent neurite network radiating from the cell bodies to the periphery Fig. (5A1). By contrast, Aβ1-42 –treated neurons showed peripheral fragmentation, partial loss of disordered neurites Fig. (5A2), while the deformed neurites showed improvement when cells were preincubated with 10μM ginsenoside Rg1 for 24h Fig. (5A4). Neurons treated with 10μM ginsenoside Rg1 grew well Fig. (5A3), suggesting no toxic effect of Rg1 on neuronal function and that ginsenoside Rg1 protects the neurons from Aβ1-42–-induced toxicity.
Fig. (5). The effect of ginsenoside Rg1 on Aβ1-42 -induced neurotoxicity.

Cortical neurons were incubated in the absence (Panel A1, A3) or in the presence of 5uM oligomeric Aβ1-42 for 48h (Panel A2 and A4) or in the presence of 10uM ginsenoside Rg1 (A3) alone, respectively. The cells (Panel A4) were preincubated with 10uM ginsenoside Rg1 for 24h and co-incubated with 5uM oligomeric Aβ1-42 for 48h. Panel B–D. The effect of ginsenoside Rg1 on apoptosis. The percentage of TUNEL-positive neurons was increased in neurons treated with oligomeric Aβ1-42 at 5μM for 48 hours in a dose-dependent manner (B). The ginsenoside Rg1 treatment decreased the Aβ-induced TUNEL-positive neurons in a reverse dose-dependent manner (C). N = 3–5 per group of cells. Studies were repeated five times and data were expressed as mean ± SEM. D. The representative images showed TUNEL-positive apoptotic cells.
To further test the protective effect of gisenoside Rg1 on Aβ1-42, the TUNEL method was used to detect apoptotic neurons treated with gisenoside Rg1 and/or Aβ1-42. Upon exposure to Aβ1-42, cortical neurons showed a dose-dependent increase in the number of apoptotic nuclei compared to vehicle-treated control neurons Fig. (5B). In agreement with morphologic observations Fig. (5A), ginsenoside Rg1 treatment significantly diminished oligomeric Aβ1-42-induced apoptosis Fig. (5C–D). Neurons treated with ginsenoside Rg1 alone grew similar to those in the control group, indicating that ginsenoside Rg1 at 10μM concentration is not harmful to neurons.
Given that caspase-3 plays a major role in the apoptosis process in response to stress, [36, 37], we next measured caspase-3 activity. Neurons treated with Aβ 1-42 showed significantly increased caspase-3 activity in a dose-dependent manner but not in Rg1-treated neurons Fig. (6 A–B). These data indicate that ginsenoside Rg1 treatment attenuates Aβ1-42-induced neuronal damage through decreased apoptosis and rescued caspase activity.
Fig. (6).

Effect of Rg1 on oligomeric Aβ1-42-induced neuronal caspase-3 activity. Oligomeric Aβ1-42 at the indicated doses increased caspase-3-like activity (A), whereas ginsenoside Rg1 largely attenuated that increase (Panel B). Data were determined from five experiments and expressed as mean ± SEM.
DISCUSSION
Mitochondrial oxidative stress has been well characterized as a hallmark of both AD and Aβ toxicity [38, 39]. In the present study, Aβ 1-42 treatment resulted in dose-dependent changes in caspase-3-like activity and TUNEL with the most-pronounced change at 5 μM in cortical neurons. Our results are consistent with the published observation that Aβ1-42 insult results in neurotoxicity.
Mitochondria function as “cellular power plants” because they generate most of the cellular ATP, which is used as a source of chemical energy. These organelles produce ATP through the coupling of oxidative phosphorylation with respiration. Mitochondrial dysfunction is present and decreases in several mitochondrial enzyme activities are present in AD, including pyruvate dehydrogenase complex, ketoglutarate dehydrogenase complex, and CcO Vmax activities [40]. The CcO defect, in particular, is central to mitochondrial dys-function in AD. Reduced enzyme activity, especially the CcO defect, results in elevated ROS and reduced ATP levels; such reduced activity may also encourage excessive mitochondrial membrane potential depolarization, increase cytoplasmic cytochrome c, and elevate caspase-3 activity, resulting in the apoptosis of neurons [41, 42].
Therefore, the inhibition of neuronal mitochondrial oxidative stress may hold potential benefits for the treatment of AD. In the present study, we found that oligomeric Aβ1-42 reduced CcO activity, decreased ATP levels, mitochondrial membrane potential and cytochrome c in mitochondria, in addition to increasing levels of ROS and H2O2 and cyto-chrome c in cytosolic fractions of cortical neurons. Our observations are consistent with the previous report on deleterious effects of Aβ on mitochondrial function. Intriguingly, the addition of Rg1 to neurons in the presence of Aβ significantly attenuates compromised mitochondrial function, resulting in rescued neuronal function.
Release of cytochrome c and loss of membrane potential are key events in mitochondrial apoptosis. In the current study, we showed that Rg1 protects neurons from Aβ1-42-induced apoptosis, and that ginsenoside Rg1 reduced cyto-chrome c release from mitochondria to cytosolic fraction that was induced by Aβ1-42. Ginsenoside Rg1 also attenuated the loss of membrane potential. These findings suggest that ginsenoside Rg1 attenuates Aβ-induced impairment of mitochondrial function.
Mitochondria are also major generators of ATP and reactive oxygen species (ROS) in cells and tissues. To confirm the protective effect of ginsenoside Rg1, we analyzed levels of ROS, H2O2 and ATP, and found that ginsenoside Rg1 attenuated the increase of ROS and H2O2, and also attenuated the decreased ATP induced by oligomeric Aβ1-42. Changes in ATP levels, accumulation of ROS, and impaired mitochondrial membrane potential are related to the mitochondrial electron transport chain (ETC).
The CcO defect is central in AD mitochondria among the several mitochondrial enzyme activities in the ETC. CcO reduction occurs at all stages of AD, including mild cognitive impairment (MCI) [33]. Interestingly, we found ginsenoside Rg1 partly protected CcO from the effects of oligomeric Aβ1-42 treatment. Therefore, we conclude that ginsenoside Rg1 can preserve the ETC by protecting CcO, thereby promoting ATP generation, decreasing ROS generation/accumulation, and eventually increasing mitochondria membrane potential and reducing cytochrome c release from mitochondria.
Given that the CcO enzyme complex contains 13 protein subunits and we know that ten are encoded by nuclear and three by mtDNA genes [43, 44], further investigation is needed to elucidate whether ginsenoside Rg1 improves CcO activity through its protein subunits or mtDNA [45]. Alternatively, CcO activity may be improved if ginsenoside Rg1 protects the mitochondrial function in other pathways, such as through anti-oxidant mechanisms. The detailed mechanisms underlying the ginsenoside Rg1-mediated pathways also require further investigation.
Currently, there are several drug candidates in the pipeline, directly targeting the underlying mechanisms of AD. Among the investigative therapies, mitochondria and anti-oxidant therapeutic strategies have shown promise [46]. Previous studies have demonstrated ginsenoside Rg1 has effective anti-amnesic, anti-aging and anti-oxidant effects [21, 47], but it is unclear whether Rg1 has a protective effect on Aβ-impaired mitochondrial dysfunction. The present studies provide the evidence of a beneficial effect of Rg1 on Aβ-induced mitochondrial toxicity.
In summary, our findings suggest that mitochondria are key players in Aβ-induced neuronal damage in the pathogenesis of AD. The present study demonstrates that ginsenoside Rg1, one of the main components of ginseng, has a protective effect in the maintenance of mitochondrial function in addition to neuronal protection. In view of the significance of mitochondria-mediated cell deaths including apoptosis and necrosis and the protective effect of Rg1 on Aβ-induced mitochondrial damage including cytochrome c release, which will trigger mitochondria-involved apoptosis, the present studies suggest that the protective effects of Rg1 on mitochondrial function contribute, at least in part, to Aβ-insulted neuronal damage. The detailed cause-effects of Rg1 on Aβ-induced mitochondrial and neuronal dysfunction require further investigation. Our results provide a piece of telling evidence that this ancient herb may indeed hold potential benefit for halting and treating age-related diseases including AD.
ACKNOWLEDGEMENTS
This work was supported by grants from National Natural Science Foundation of China (No. 30772555) to Dr. X. Chen and from NIH/NIA (R37AG037319) to Dr. SS Yan.
ABBREVIATIONS
- AD
Alzheimer's disease
- Aβ1-42
Amyloid-beta peptide1-42
- CcO
Cytochrome c oxidase
- ROS
Reactive oxygen species
- H2O2
Hydrogen peroxide
- ATP
Adenosine-triphosphate
Footnotes
CONFLICT OF INTEREST None
REFERENCES
- [1].Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–5. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- [2].Haass C, Steiner H. Protofibrils, the unifying toxic molecule of neurodegenerative disorders? Nat Neurosci. 2001;4:859–60. doi: 10.1038/nn0901-859. [DOI] [PubMed] [Google Scholar]
- [3].Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- [4].Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–7. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- [5].Shankar GM, Li S, Mehta TH, Shepardson NE, Smith I, Brett FM, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14(8):837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen JX, Yan SD. Amyloid-beta-Induced Mitochondrial Dysfunction. J Alzheimers Dis. 2007;12:177–84. doi: 10.3233/jad-2007-12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Takuma K, Yao J, Huang J, Xu H, Chen X, Luddy J, et al. ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. FASEB J. 2005;19:597–8. doi: 10.1096/fj.04-2582fje. [DOI] [PubMed] [Google Scholar]
- [8].Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, et al. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 2005;19:2040–1. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- [9].Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008;14:1097–105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004;304:448–52. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- [11].Yao J, Du H, Yan S, Fang F, Wang C, Lue LF, et al. Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer's disease. J Neurosci. 2011;31:2313–20. doi: 10.1523/JNEUROSCI.4717-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–49. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- [13].Dragicevic N, Mamcarz M, Zhu Y, Buzzeo R, Tan J, Arendash GW, et al. Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer's transgenic mice. J Alzheimers Dis. 2010;20(2):S535–50. doi: 10.3233/JAD-2010-100342. [DOI] [PubMed] [Google Scholar]
- [14].Chen JX, Yan SS. Role of mitochondrial amyloid-beta in Alzheimer's disease. J Alzheimers Dis. 2010;20(2):S569–78. doi: 10.3233/JAD-2010-100357. [DOI] [PubMed] [Google Scholar]
- [15].Du H, Guo L, Zhang W, Rydzewska M, Yan S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol Aging. 2011;32(3):398–406. doi: 10.1016/j.neurobiolaging.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2009;106:14670–5. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Moreira PI, Cardoso SM, Santos MS, Oliveira CR. The key role of mitochondria in Alzheimer's disease. J Alzheimers Dis. 2006;9:101–10. doi: 10.3233/jad-2006-9202. [DOI] [PubMed] [Google Scholar]
- [18].Moreira PI, Santos MS, Oliveira CR. Alzheimer's disease: a lesson from mitochondrial dysfunction. Antioxid Redox Signal. 2007;9:1621–30. doi: 10.1089/ars.2007.1703. [DOI] [PubMed] [Google Scholar]
- [19].Moreira PI, Duarte AI, Santos MS, Rego AC, Oliveira CR. An integrative view of the role of oxidative stress, mitochondria and insulin in Alzheimer's disease. J Alzheimers Dis. 2009;16:741–61. doi: 10.3233/JAD-2009-0972. [DOI] [PubMed] [Google Scholar]
- [20].Swerdlow RH, Khan SM. The Alzheimer's disease mitochondrial cascade hypothesis: an update. Exp Neurol. 2009;218:308–15. doi: 10.1016/j.expneurol.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cheng Y, Shen LH, Zhang JT. Anti-amnestic and anti-aging effects of ginsenoside Rg1 and Rb1 and its mechanism of action. Acta Pharmacol Sin. 2005;26:143–9. doi: 10.1111/j.1745-7254.2005.00034.x. [DOI] [PubMed] [Google Scholar]
- [22].Chen F, Eckman EA, Eckman CB. Reductions in levels of the Alzheimer's amyloid beta peptide after oral administration of ginsenosides. FASEB J. 2006;20:1269–71. doi: 10.1096/fj.05-5530fje. [DOI] [PubMed] [Google Scholar]
- [23].Shi YQ, Huang TW, Chen LM, Pan XD, Zhang J, Zhu YG, et al. Ginsenoside Rg1 attenuates amyloid-beta content, regulates PKA/CREB activity, and improves cognitive performance in SAMP8 mice. J Alzheimers Dis. 2010;19:977–89. doi: 10.3233/JAD-2010-1296. [DOI] [PubMed] [Google Scholar]
- [24].Wei C, Jia J, Liang P, Guan Y. Ginsenoside Rg1 attenuates beta-amyloid-induced apoptosis in mutant PS1 M146L cells. Neurosci Lett. 2008;443:145–9. doi: 10.1016/j.neulet.2008.07.089. [DOI] [PubMed] [Google Scholar]
- [25].Xu H, Jiang H, Wang J, Xie J. Rg1 protects iron-induced neurotoxicity through antioxidant and iron regulatory proteins in 6-OHDA-treated MES23.5 cells. J Cell Biochem. 2010;111:1537–45. doi: 10.1002/jcb.22885. [DOI] [PubMed] [Google Scholar]
- [26].Zhu D, Wu L, Li CR, Wang XW, Ma YJ, Zhong ZY, et al. Ginsenoside Rg1 protects rat cardiomyocyte from hypoxia/reoxygenation oxidative injury via antioxidant and intracellular calcium homeostasis. J Cell Biochem. 2009;108:117–24. doi: 10.1002/jcb.22233. [DOI] [PubMed] [Google Scholar]
- [27].Leung KW, Yung KK, Mak NK, Chan YS, Fan TP, Wong RN. Neuroprotective effects of ginsenoside-Rg1 in primary nigral neurons against rotenone toxicity. Neuropharmacology. 2007;52:827–35. doi: 10.1016/j.neuropharm.2006.10.001. [DOI] [PubMed] [Google Scholar]
- [28].Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–76. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- [29].Chen X, Stern D, Yan SD. Mitochondrial dysfunction and Alzheimer's disease. Curr Alzheimer Res. 2006;3:515–20. doi: 10.2174/156720506779025215. [DOI] [PubMed] [Google Scholar]
- [30].Atamna H, Frey WH., 2nd Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer's disease. Mitochondrion. 2007;7:297–310. doi: 10.1016/j.mito.2007.06.001. [DOI] [PubMed] [Google Scholar]
- [31].Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci USA. 2010;107:18670–5. doi: 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pickrell AM, Fukui H, Moraes CT. The role of cytochrome c oxidase deficiency in ROS and amyloid plaque formation. J Bioenerg Biomembr. 2009;41:453–6. doi: 10.1007/s10863-009-9245-3. [DOI] [PubMed] [Google Scholar]
- [33].Valla J, Schneider L, Niedzielko T, Coon KD, Caselli R, Sabbagh MN, et al. Impaired platelet mitochondrial activity in Alzheimer's disease and mild cognitive impairment. Mitochondrion. 2006;6:323–30. doi: 10.1016/j.mito.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chen X, Stern D, Yan SD. Cellular targets of amyloid beta peptide: potential roles in neuronal cell stress and toxicity. third ed. Oxford Publication; 2007. [Google Scholar]
- [35].Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–60. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- [36].DiPietrantonio AM, Hsieh T, Wu JM. Activation of caspase 3 in HL-60 cells exposed to hydrogen peroxide. Biochem Biophys Res Commun. 1999;255:477–82. doi: 10.1006/bbrc.1999.0208. [DOI] [PubMed] [Google Scholar]
- [37].Matsura T, Kai M, Fujii Y, Ito H, Yamada K. Hydrogen peroxide-induced apoptosis in HL-60 cells requires caspase-3 activation. Free Radic Res. 1999;30:73–83. doi: 10.1080/10715769900300081. [DOI] [PubMed] [Google Scholar]
- [38].Chauhan V, Chauhan A. Oxidative stress in Alzheimer's disease. Pathophysiology. 2006;13:195–208. doi: 10.1016/j.pathophys.2006.05.004. [DOI] [PubMed] [Google Scholar]
- [39].Chen JX, Yan SS. Role of mitochondrial amyloid-beta in Alzheimer's disease. J Alzheimers Dis. 2010;20(2):S569–78. doi: 10.3233/JAD-2010-100357. [DOI] [PubMed] [Google Scholar]
- [40].Swerdlow RH, Kish SJ. Mitochondria in Alzheimer's disease. Int Rev Neurobiol. 2002;53:341–85. doi: 10.1016/s0074-7742(02)53013-0. [DOI] [PubMed] [Google Scholar]
- [41].Cardoso SM, Santana I, Swerdlow RH, Oliveira CR. Mitochondria dysfunction of Alzheimer's disease cybrids enhances Abeta toxicity. J Neurochem. 2004;89:1417–26. doi: 10.1111/j.1471-4159.2004.02438.x. [DOI] [PubMed] [Google Scholar]
- [42].Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:29–44. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- [43].Swerdlow RH. Mitochondria in cybrids containing mtDNA from persons with mitochondriopathies. J Neurosci Res. 2007;85:3416–28. doi: 10.1002/jnr.21167. [DOI] [PubMed] [Google Scholar]
- [44].Swerdlow RH, Parks JK, Cassarino DS, Maguire DJ, Maguire RS, Bennett JP, Jr, et al. Cybrids in Alzheimer's disease: a cellular model of the disease? Neurology. 1997;49:918–25. doi: 10.1212/wnl.49.4.918. [DOI] [PubMed] [Google Scholar]
- [45].Tanaka N, Goto Y, Akanuma J, Kato M, Kinoshita T, Yamashita F, et al. Mitochondrial DNA variants in a Japanese population of patients with Alzheimer's disease. Mitochondrion. 2010;10:32–7. doi: 10.1016/j.mito.2009.08.008. [DOI] [PubMed] [Google Scholar]
- [46].Dumont M, Lin MT, Beal MF. Mitochondria and antioxidant targeted therapeutic strategies for Alzheimer's disease. J Alzheimers Dis. 2010;20(Suppl 2):S633–43. doi: 10.3233/JAD-2010-100507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chen XC, Zhou YC, Chen Y, Zhu YG, Fang F, Chen LM. Ginsenoside Rg1 reduces MPTP-induced substantia nigra neuron loss by suppressing oxidative stress. Acta Pharmacol Sin. 2005;26:56–62. doi: 10.1111/j.1745-7254.2005.00019.x. [DOI] [PubMed] [Google Scholar]