

Abstract
Human T-cell leukemia virus type I (HTLV-I)-associated myelopathy/tropical spastic paraparesis (HAM/TSP) is a neurodegenerative disease characterized by selective loss of axons and myelin in the corticospinal tracts. This central axonopathy may originate from the impairment of anterograde axoplasmic transport. Previous work showed tau hyperphosphorylation at T181 in cerebrospinal fluid of HAM/TSP patients. Similar hyperphosphorylation occurs in SH-SY5Y cells incubated with supernatant from MT-2 cells (HTLV-I-infected lymphocytes secreting viral proteins, including Tax) that produce neurite shortening. Tau phosphorylation at T181 is attributable to glycogen synthase kinase 3-β (GSK3-β) and cyclin-dependent kinase 5 (CDK5) activation. Here we investigate whether neurite retraction in the SH-SY5Y model associates with concurrent changes in other tau hyperphosphorylable residues. Threonine 181 turned out to be the only tau hyperphosphorylated residue. We also evaluate the role of GSK3-β and CDK5 in this process by using specific kinase inhibitors (LiCl, TDZD-8, and roscovitine). Changes in both GSK3-β active and inactive forms were followed by measuring the regulatory phosphorylable sites (S9 and Y216, inactivating and activating phosphorylation, respectively) together with changes in β-catenin protein levels. Our results showed that LiCl and TDZD-8 were unable to prevent MT-2 supernatant-mediated neurite retraction and also that neither Y216 nor S9 phosphorylations were changed in GSK3-β. Thus, GSK3-β seems not to play a role in T181 hyperphosphorylation. On the other hand, the CDK5 involvement in tau phosphorylation was confirmed by both the increase in its enzymatic activity and the absence of MT-2 neurite retraction in the presence of roscovitine or CDK5 siRNA transfection.
Keywords: HAM/TSP, MT-2, SH-SY5Y, CDK5-siRNA, Tax
Human T-cell leukemia virus type I (HTLV-I)-associated myelopathy/tropical spastic paraparesis (HAM/TSP) is a central axonopathy characterized by distal axonal degeneration of the corticospinal tracts (Osame, 1986; Cartier et al., 1997). This disease could originate from the impairment of axoplasmic transport in these long axons, followed by accumulation of amyloid precursor protein (Coleman, 2005; Cartier et al., 2007). We previously found a significant increase in tau phosphorylation only at T181 in cerebrospinal fluid (CSF) from HAM/TSP patients (Maldonado et al., 2008). T181 lies outside the tau-tubulin interaction site, but within the projection domain. The implications of this phosphorylation are not clear; however, tau phosphorylation in the region containing residues 172–251 causes a slightly lower activity in stimulating microtubule assembly (Liu et al., 2007). Although this hyperphosphorylation is probably not associated with axonal microtubule dysfunction, an effect on fast axonal transport cannot be ruled out (Shahani and Brandt, 2002; Avila et al., 2004).
CD4+ cells are the main HTLV-I reservoir, but both CD8+ cells and astrocytes may be infected, but not neurons (Matsuura et al., 2010). Axonal degeneration may be the result of the extracellular action of the secreted viral protein Tax (Boxus et al., 2008). Tax increases the TNF-α mRNA expression in neuronal cellular lines (Cowan et al., 1997). Although 40% of patients with HAM/TSP are seronegative for virus, some of them exhibit a truncated provirus form including the Tax gene (Ramirez et al., 2003). To elucidate the extracellular action of viral proteins, we have initiated studies on the human neuroblastoma SH-SY5Y cell line, phenotypically similar to CNS neurons (Sayas et al., 1999; Encinas et al., 2000). We found that treatment of this neuronal cells with the secreted products from HTLV-I-infected MT-2 cells (including Tax protein) produced neurite retraction and also an increase in tau phosphorylation at T181, similar to that observed in patients with HAM/TSP (Maldonado et al., 2008). Glycogen synthase kinase 3-β (GSK3-β) and cyclin-dependent kinase 5 (CDK5) are the most important kinases involved in the phosphorylation of this residue (Hanger et al., 2009).
CDK5 and GSK3-β have extensive regulatory mechanisms (Cruz and Tsai, 2004; Dhariwala and Rajadhyaksha, 2008). Monomeric CDK5 has no enzymatic activity and requires association with a regulatory binding partner, p35 or p39. These partners confer discrete substrate specificities; for example, the specificity of p39-CDK5 is higher than that of the p35-CDK5 complex for tau as a substrate. Neurotoxic insults can deregulate CDK5 activity, increasing intercellular calcium and activating calpain; p35 and p39 are cleaved by calpain, generating p25 and p29 fragments, which can bind to CDK5, forming p25-CDK5 and p29-CDK5 complexes (Kusakawa et al., 2000; Dhariwala and Rajadhyaksha, 2008). These complexes have higher activities and longer half-lives than p35-CDK5 and p39-CDK5 forms.
GSK3-β is regulated mainly by phosphorylation of specific sites, Y216 (activation) and S9 (inactivation; Bhat et al., 2000; Liang and Chuang, 2007). In addition, GSK3-β activity is also dependent on its cellular location, the extent of phosphorylation of its substrates, and cross-talk with CDK5 kinase (Cho and Johnson, 2003; Meares and Jope, 2007; Engmann and Giese, 2009). Both CDK5 and GSK3-β may be implicated in axonal degeneration, as described for Alzheimer’s disease (Hanger et al., 2009). The main aim of this study was to evaluate the participation of these kinases in both neurite retraction and tau hyperphosphorylation caused by secreted products from HTLV-I-infected MT-2 cells containing Tax protein (Alefantis et al., 2007).
MATERIALS AND METHODS
Human SH-SY5Y Cell Cultures and Neurite Retraction Studies
Human SH-SY5Y neuroblastoma were maintained in a mixture of Dulbecco’s modified Eagle’s medium nutrient and F12-Ham in a proportion of 1:1 (Sigma-Aldrich, St. Louis, MO), supplemented with 6% heat-inactivated fetal bovine serum (FBS; Hyclone, Thermo Fischer Scientific, South Logan, UT). Cells were seeded at an initial density of 2.2 × 103 cells/cm2. The differentiation protocol was based on the report of Encinas et al. (2000), with some modifications. Briefly, after 24 hr of seeding, the differentiation to a neuronal-type was induced by addition of 10 μM all-trans-retinoic acid (Sigma-Aldrich) with daily gradual reduction of the FBS concentration (from 6 to 0%) over 5 days. Then, 50 ng/ml of brain-derived neurotrophic factor (BDNF; Alomone Laboratories, Jerusalem, Israel) was added over 2 days. BDNF was removed from culture medium and replaced by a mixture of DMEM-F12 Ham’s without serum, 4 hr before addition of supernatants of MT-2 and K562 cultures. In all experiments, differentiated cells were incubated for 1 hr with MT-2 and K562 supernatants. MT-2 and K562 cells (5–10 million in 10 ml) were cultivated in RPMI as reported by Ramirez et al. (2003), except for reduction in FBS from 10% to 0.2% for 7 days.
The participation of GSK3-β and CDK5 in retraction and tau hyperphosphorylation at T181 was evaluated using SH-SY5Y differentiated cells pretreated for 1 hr with two inhibitors of GSK3-β [5 mM and 20 mM LiCl or 10 μM TDZD-8(4-benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione)] and an inhibitor for CDK5 (10 μM roscovitine, prepared in DMSO), before the incubation with MT-2 (with HTLV-I provirus) supernatant and K562 supernatant (control). The final concentration of DMSO in cultures was 0.1%, an innocuous concentration for SH-SY5Y cells (Garrofe-Ochoa et al., 2008).
Tax neutralization from MT-2 supernatant was performed by adding 10 μl/ml of Tax antibody (HTLV-I Tax Hyb 168A51-2, obtained through the NIHAIDS Research and Reference Reagent Program Division of AIDS, NIAID, NIH) 1 hr before the incubation of SH-SY5Y differentiated cells. Anti-rabbit (Pierce, Rockford, IL) was used as control antibody.
Neurite Length Measurement
Cells were examined under a phase-contrast microscope and directly captured as digital micrographs in black and white. Images acquired using a ×20 objective were photographed from seven different fields with Z configuration and further analyzed. Neurite length was measured with the NIH ImageJ 1.38d plugin NeuronJ; the neurite length corresponds to the net extension away from the cell body, i.e., from the margin of the cell body to its terminus. The measurement was made using the program’s handheld cursor. Data collected from 200–300 neurites were used for each condition, and the program gives the population average neurite length in micrometers. All neurite measured were included in the statistical analysis.
Transfection of CDK5 siRNA Duplexes
The siRNA duplex targeting CDK5 (Smartpool) and scramble control of siRNA were purchased from Thermo Fisher Scientific (Dharmacon, Lafayette, CO). SH-SY5Y cells were differentiated for 5 days with retinoic acid (according to a previously described protocol) and then transfected for 3 hr with 2 μg of each siRNA using the TransMessenger transfection reagent, following the manufacturer’s protocol (Qiagen, Valencia, CA). Transfected cells were washed three times with DMEM-F12 Ham and then treated with 50 ng/ml BDNF for 2 days. Neurite retraction studies were performed as previously described.
Western Blot Analysis
Cell extracts were prepared as follows. Cells were washed with phosphate-buffered saline and then resuspended in RIPA buffer (25 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% Nonidet P40, 1% sodium deoxycholate, 0.1% SDS), with 2 μl/ml protease inhibitor cocktail (catalog P8340; Sigma-Aldrich) and 1 mM sodium-o-vanadate, followed of mechanical disruption. The soluble fraction was obtained by centrifugation at 14,000g for 15 min at 4°C. Tax protein was inmunoprecipitated from MT-2 and K562 supernatants using an AminoLink Plus inmobilization kit and anti-Tax antibody (CVL-MAB0022; Covalab, Lyon, France), following the manufacturer’s protocol (Pierce). Protein determination was done using the BCA Protein Assay kit from Pierce, according to the manufacturer’s instructions.
SDS-PAGE was performed with 12% polyacrylamide gels, and portions of 25 μg protein of cell lysate were used. The buffer for electrotransfer to nitrocellulose membranes (Bio-Rad, Hercules, CA) contained 25 mM Tris-HCl, 192 mM glycine, and 20% (v/v) methanol, and electrotransfer was done at a total of 600 mA at 4°C. After electrotransfer, membranes were blocked for 1 hr at room temperature with 6% Quick-Blocker (Chemicon, Temecula, CA) dissolved in TBS-T [20 mM Tris-HCl, 137 mM NaCl, 0.1% (v/v) Tween-20, pH 7.6], then incubated overnight at 4°C with the different primary antibodies at the appropriate dilution in TBS-T buffer. The following monoclonal antibodies were used: antibodies against tau (dilution 1:1,000; catalog AHB0042; BioSource-Invitrogen, Carlsbad, CA), GSK3-β-phospho-S9 (dilution 1:2,000; catalog 05–643; Upstate Biotechnology, Waltham, MA), and GSK3-β-phospho-Y216 (dilution 1:2,000; catalog 05–413; Upstate Biotechnology). The following polyclonal antibodies were used: antibodies against tau-phospho-T181 (1:2,000; catalog ab38505; Abcam, Cambridge, United Kingdom) and six different phosphorylated residues of tau (dilution 1:2,000; catalog 4477G; BioSource-Invitrogen), GSK3-β (dilution 1:2,000; catalog AB8687; Upstate Biotechnology), CDK5 (dilution 1:1,000; catalog sc-173), and β-catenin (dilution 1:1,000; catalog sc-7199) from Santa Cruz Biotechnology (Santa Cruz, CA). After washing three times (10 min each wash) with TBS-T (without milk), membranes were incubated with the corresponding secondary antibody. As a secondary antibody we used anti-rabbit conjugated with peroxidase diluted 1:20,000 (catalog 1858415; Pierce) or anti-mouse conjugated with peroxidase diluted 1:10,000 (catalog 1858413; Pierce). Blots were incubated for 1 hr with peroxidase-conjugated secondary antibodies. After rinsing three times (10 min each rinse) with TBS-T (without milk), positive reactions were identified by using enhanced chemiluminiscence SuperSignal West Femto Chemiluminiscent substrate (Pierce) in all the other analyses. X-ray films (CL-Xposure film; Pierce) were exposed for varying times. Control experiments (without primary antibodies) with only secondary antibodies did not give any chemiluminescent signal. For consecutive analyses with various antibodies, stripping was performed using the ReBlot plus mild antibody solution (Chemicon) according to the manufacturer’s instructions. Blots were then blocked, and probing was performed as described above. Quantification of blots was carried out by scanning films using the Un-Scan-It program (Silk Scientific, Orem, UT).
CDK5 Activity Assay
Cell lysates containing 200 μg protein were diluted in T-PER (Thermo Fisher Scientific, Pierce Protein Research Products, Rockford, IL) to a volume of 500 μl and precleared with 30 μl protein A-agarose beads (50% slurry in lysis buffer; Santa Cruz Biotechnology) at 4°C for 1 hr. CDK5 was immunoprecipitated using 4 μg anti-CDK5 IgG from pre-cleared lysates by overnight incubation at 4°C, followed by 2 hr of incubation at 4°C with 25 μl protein A-agarose beads. Immunoprecipitates were washed three times with cold PBS and twice with kinase buffer (20 mM Tris-HCl pH 7.4, 10 mM MgCl2, 1 mM EDTA) and resuspended in 20 μl of 1× buffer kinase. Five microliters of 5× kinase assay mixture (100 mM Tris-HCl, pH 7.4, 50 mM MgCl2, 5 mM EDTA, 50 μM NaF, 5 mM DTT and 2.5 mM dNTPs) and 5 μl histone H1 (1 mg/ml; Sigma-Aldrich) were added to 15 μl of the immunoprecipitates. Kinase assays were carried out at 30 °C for 60 min by addition of 5 μCi [γ32P] ATP (0.5 mM). The reaction was stopped by adding 5× SDS sample buffer and boiling for 10 min at 100°C. Then, 25-μl aliquots were run on 4–20% polyacrylamide gel, which was exposed for 1–3 hr at −80°C to a film, and the scanned bands were quantified in Scion Image.
Cell Viability Assay
Trypan blue exclusion analysis was performed as previously described (Vasko et al., 2005). Briefly, cells were detached from the plate using a 0.05% trypsin-EDTA solution, and medium was added. Equal volumes of the cell suspension and 0.4% (w/v) trypan blue in PBS were mixed, and the cells were scored under a phase-contrast microscope using a Neubauer camera counting the four external squares. Percentage survival was calculated as percentage of live cells divided by total cell number (including dead and live cells) and then normalized by the initial condition.
Statistical Analysis
All statistical analysis in these studies were performed with the Statistical Package of the Social Sciences (SPSS). Statistical analysis on neurite length between studied groups and viability of cells was carried out using the Student’s t-test for two independent samples. ANOVA was used for the Western blots analysis. Statistical significance was assumed at P < 0.05 in all cases.
RESULTS
Effect of Tax Neutralization from MT-2 Supernatant on Neurite Retraction
Differentiated human SH-SY5Y neuroblastoma cells treated with MT-2 (HTLV-I-infected) cell supernatant exhibited neurite retraction, but not the control K562 supernatant (Fig. 1A,B). Tax protein was detected in MT-2 cell supernatant (Fig. 1C). Tax neutralization with anti-Tax antibody prevented the neurite shortening in differentiated cells (Fig. 1A,B). No change in neurite length was observed when adding control antibody.
Fig. 1.
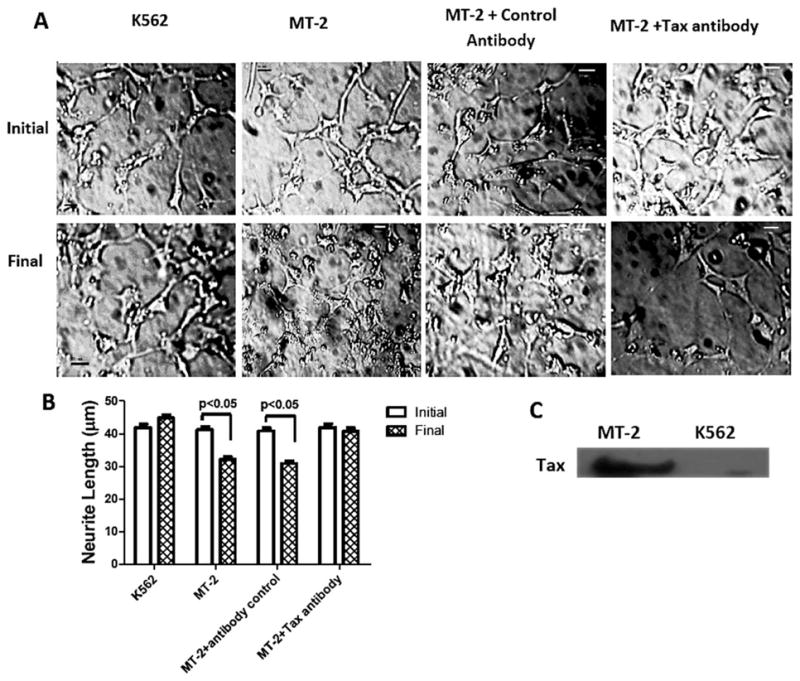
A: Effect of MT-2 cell supernatant on neurite length in differentiated SH-SY5Y cells. Representative microphotographs initially and after 1-hr treatments of differentiated SH-SY5Y cells with MT-2 and K562 supernatants in the presence or absence of anti-Tax. Neurite length is expressed as the population average ± SEM collected from 200–300 neurites for each condition measured with the ImageJ 1.38d plugin NeuronJ (B), and the presence of Tax protein in MT-2 cell supernatant was analyzed by Western blot; K562 cells supernatant was used as negative control (C). Bars represent mean ± SEM of triplicate cultures. Significant differences was assumed at P < 0.05. Scale bars = 20 μm.
Effect of MT-2 Supernatant on Tau Phosphorylation
Differentiated SH-SY5Y cells treated with MT-2 cell supernatant exhibit tau hyperphosphorylation at T181, as previously reported by Maldonado et al. (2008). The current study extends to other putative tau phosphorylation sites (S199, T205, S231, S262, S356, and S396; Hanger et al., 2009). We detected no significant differences in phosphorylation after incubating differentiated SH-SY5Y cells with MT-2 or K562 supernatants (Table I). Some representative Western Blots for tau phosphorylation are shown in Figure 2.
TABLE I.
Effect of Secretable Products of MT-2 on Tau Phosphorylation in SH-SY5Y Cells Measured by Western Blot†
| Residual number | Group | Mean value p-tau/tau | SD | P |
|---|---|---|---|---|
| pT181 | Initial | 1.0 | 0.3 | 0.0004 |
| K562 | 1.1 | 0.4 | ||
| MT-2★ | 3.3 | 0.4 | ||
| pS199 | Initial | 0.8 | 0.3 | 0.43 |
| K562 | 0.6 | 0.1 | ||
| MT-2 | 0.58 | 0.06 | ||
| pT205 | Initial | 1.3 | 0.4 | 0.42 |
| K562 | 1.0 | 0.3 | ||
| MT-2 | 0.9 | 0.3 | ||
| pS231 | Initial | 1.1 | 0.6 | 0.64 |
| K562 | 1.1 | 0.2 | ||
| MT-2 | 0.8 | 0.2 | ||
| pS262 | Initial | 0.8 | 0.1 | 0.23 |
| K562 | 1.1 | 0.2 | ||
| MT-2 | 0.9 | 0.1 | ||
| pS356 | Initial | 1.8 | 0.4 | 0.90 |
| K562 | 1.7 | 0.3 | ||
| MT-2 | 1.7 | 0.1 | ||
| pS396 | Initial | 1.9 | 0.5 | 0.56 |
| K562 | 1.6 | 0.2 | ||
| MT-2 | 1.8 | 0.3 |
The phosphorylation degree of tau at different residues, in SH-SY5Y cells incubated with MT-2 supernatant, was evaluated by using specific phospho-tau antibodies. The densitometric results were normalized to total tau and expressed as mean ± SD of triplicate experiments.
P < 0.05.
Fig. 2.
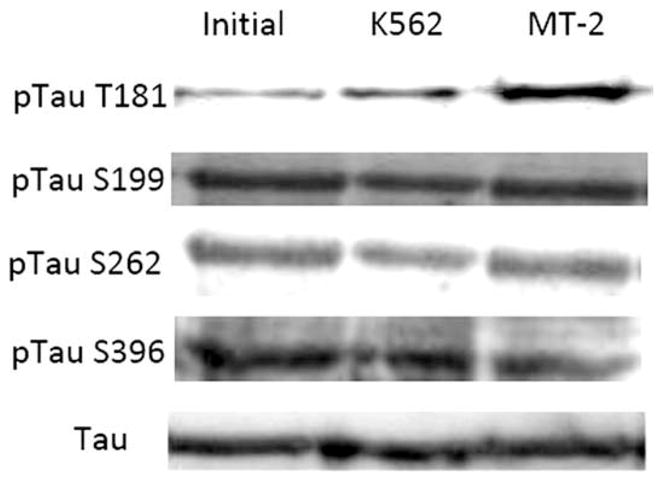
Effect of MT-2 supernatant on various phosphorylable tau residues. Some examples of representative Western blots of cellular lysates from differentiated SH-SY5Y cell treated with MT-2 or K562 supernatants for 1 hr compared with the initial condition. Quantitative analyses of all residues are shown in Table I.
Effect of GSK3-β Inhibition on Neurite Retraction
Neither LiCl nor TDZ-8 preincubations prevented the neurite retraction in SH-SY5Y cells treated with MT-2 cell supernatant (Fig. 3A,B). Control cell preincubation with LiCl led to a significant decrease in T181 phosphorylation, indicating that GSK3-β has a role in maintaining basal phosphorylation (Fig. 3C). A fivefold increase in pT181 was detected after incubation with MT-2 supernatant. LiCl preincubation was significantly attenuated (twofold increase). Cell viability was unaffected by the various treatments (Fig. 3D).
Fig. 3.
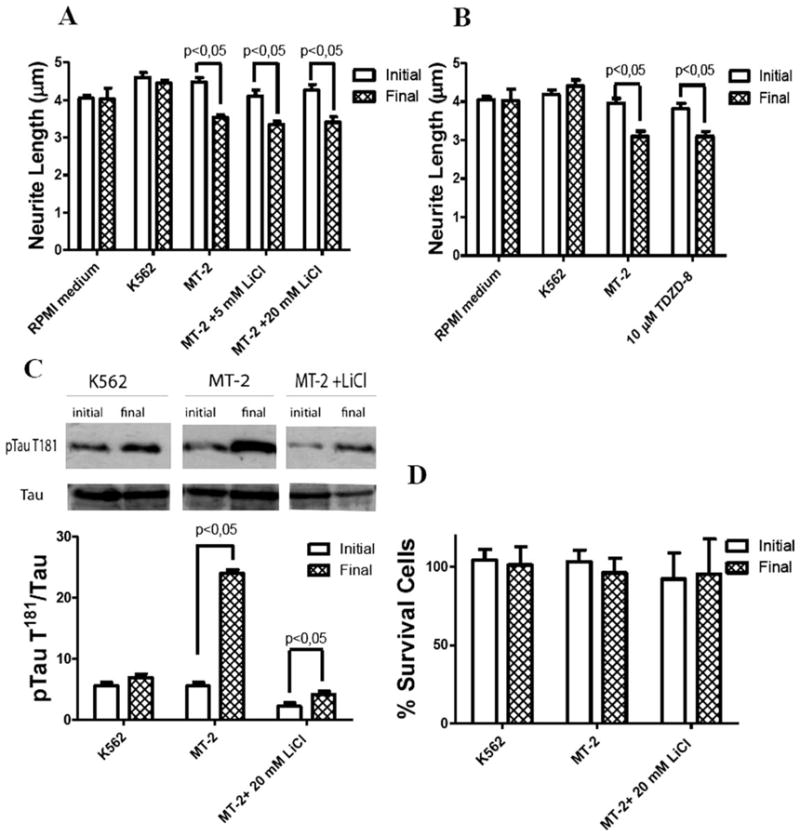
Effect of GSK3-β inhibitors on neurite retraction and tau hyperphosphorylation by MT-2 supernatant. Differentiated SH-SY5Y cells were preincubated with 5 and 20 mM of LiCl or 10 μM of TDZD-8 for 1 hr and then treated for 1 hr with MT-2 or K562 supernatant. Effect of inhibitors, LiCl (A) and TDZD-8 (B), on neurite length, collecting data from 200–300 neurites for each condition; results were expressed as the population average ± SEM. C: Effect of LiCl in tau phosphorylation at T181; representative Western blot of p-tau T181 and data standardized by total tau and expressed as mean of three different experiments ± SEM; a representative Western blot is shown. D: Effect of LiCl 20 mM on cell viability of SH-SY5Y differentiated cells; the ordinate represents percent of cells surviving after exposure to MT-2 and K562 supernatants for 1 hr measured by trypan blue exclusion; each column represents mean ± SEM of independent triplicates assays. Statistical significance was assumed at P < 0.05.
Effect of MT-2 Supernatant on GSK3-β Phosphorylation
We evaluated two site-specific phosphorylations of GSK3-β, one activating (Y216) and another inactivating (S9) site. GSK3-β regulates the proteosomal breakdown of β-catenin so that an increased kinase activity stimulates β-catenin degradation (Grimes and Jope, 2001). We observed no change in either phosphorylation or β-catenin (Fig. 4), which implies a lack of increase in the kinase activity.
Fig. 4.
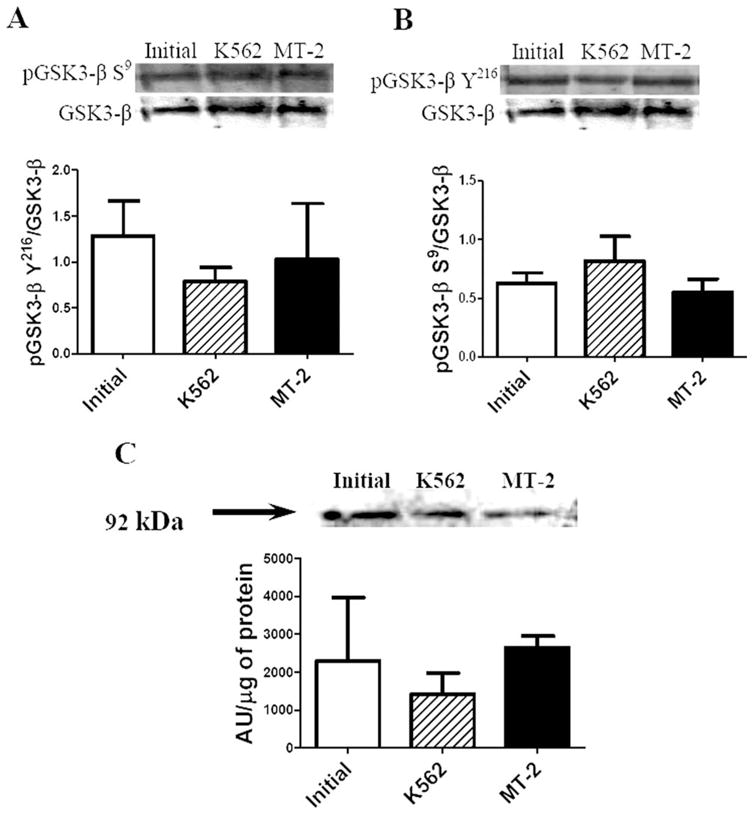
Effect of MT-2 supernatant on GSK3-β phosphorylation and β-catenin levels on SH-SY5Y differentiated cells. Twenty-five micrograms of cell lysates were analyzed by Western blot with the following primary antibodies: pGSK3-β S9 (A), pGSK3-β Y216 (B), and β-catenin (C). Data on phosphorylated GSK3-β forms were standardized by total GSK3-β; β-catenin was expressed as the ratio of pixels, arbitrary units (AU), to total protein. All results are presented as mean ± SEM of three independent experiments. Statistical significance was assumed at P < 0.05.
Effect of CDK5 Inhibition and CDK5 Knockdown on Neurite Retraction
Roscovitine prevented neurite shortening caused by MT-2 supernatant (Fig. 5A) and also attenuated both T181 basal phosphorylation and hyperphosphorylation by MT-2 supernatant (Fig. 5B). There was no change in cellular viability in any of the groups under study (Fig. 5C). The CDK5 knockdown (56%) using siRNA allowed us to prevent neurite shortening by MT-2 treatment (Fig. 6A,B). An siRNA scramble was used as a control.
Fig. 5.
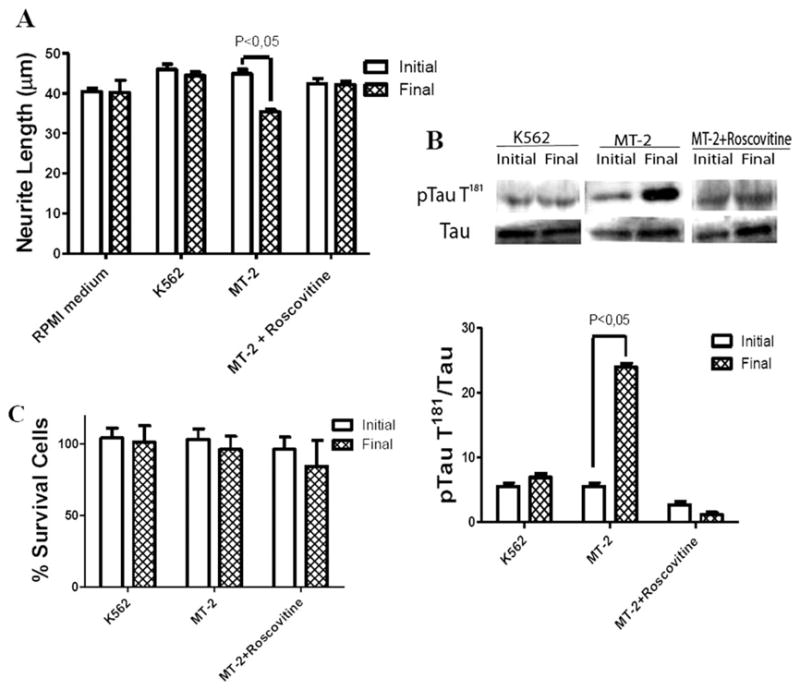
Effect of CDK5 inhibitor on neurite retraction and tau hyperphosphorylation caused by MT-2 supernatant. Differentiated SH-SY5Y cells were preincubated with 10 μM roscovitine for 1 hr and then treated for 1 hr with MT-2 or K562 supernatant. A: Effect of inhibitor on neurite length was followed, collecting data from 200–300 neurites for each condition; results were expressed as the population average ± SEM. B: Effect of roscovitine on tau phosphorylation at T181; the values were standardized by total tau and expressed as the mean of three different experiments ± SEM; a representative Western blot is shown. C: Effect of roscovitine on cell viability of SH-SY5Y differentiated cells; the ordinate represents percentage of cells surviving after exposure to MT-2 and K562 supernatants for 1 hr as measured by trypan blue exclusion; values represent mean ± SEM of independent triplicates. Statistical significance was assumed at P < 0.05.
Fig. 6.
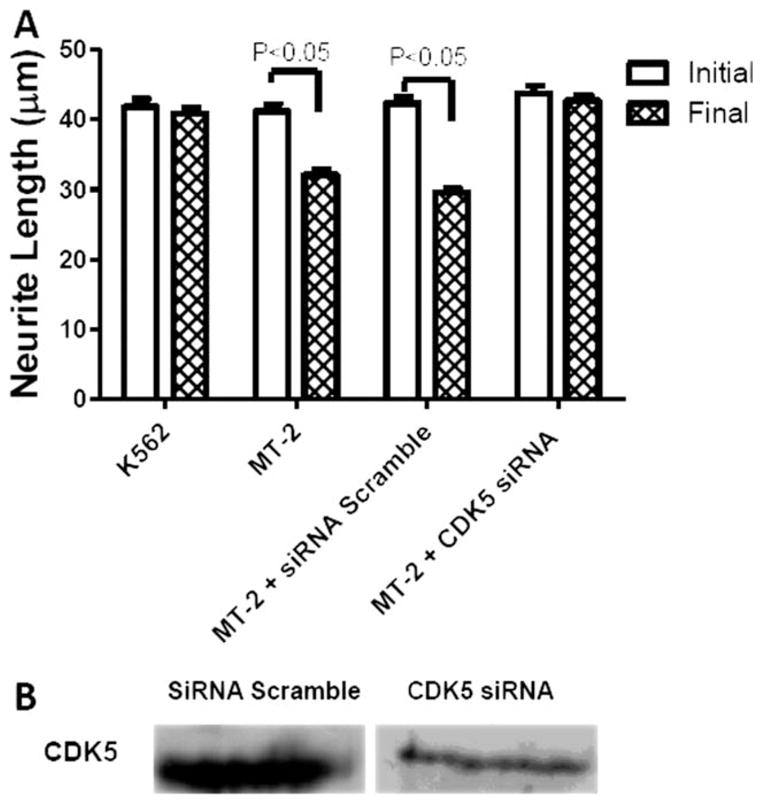
Effect of CDK5 siRNA on neurite retraction caused by MT-2 supernatant. SH-SY5Y differentiated cells were transfected with CDK5 siRNA Smartpool or Scramble control siRNA and then treated for 1 hr with MT-2 or K562 supernatant. A: Effect of CDK5 siRNA on neurite length was followed collecting data from 200–300 neurites for each condition. B: CDK5 knock down was tested by Western blot analysis. Measurements were expressed as average ± SEM from three independent experiments. Statistical significance was assumed at P < 0.05.
Effect of MT-2 Supernatant on CDK5 Activity
A significant increase in CDK5 activity, but not in CDK5 total protein, was found in the neuroblastoma differentiated cells treated with MT-2 supernatant (Fig. 7).
Fig. 7.
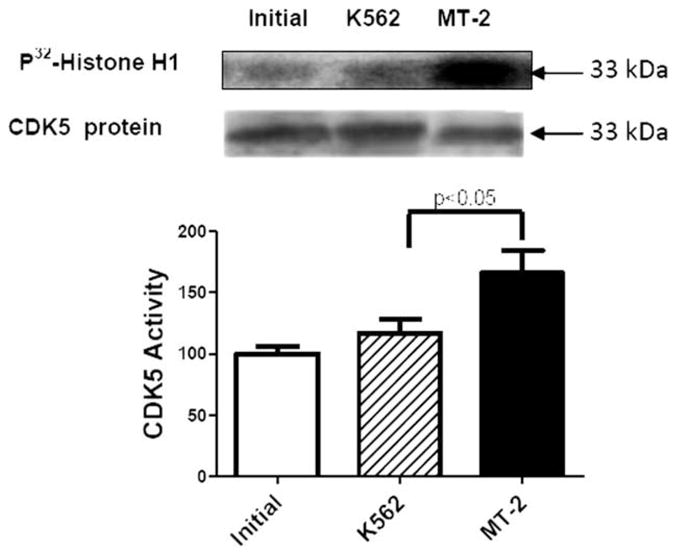
Effect of secretable products from MT-2 cells on CDK5 activity. CDK5 was inmunoprecipitated from SH-SY5Y cell lysate samples and subjected to a histone H1 kinase assay. A representative autoradiogram of the phosphorylated histone H1 band is shown (upper lane). CDK5 protein was determined by Western blot (lower lane). Activity measurements were expressed as average ± SEM from six independent samples for each condition. Statistical significance was assumed at P < 0.05.
DISCUSSION
Our ultimate objective is a better understanding of the action of extracellular viral proteins in HAM/TSP, with a view to pharmacological intervention. In this study, we have attempted to assess the extent of the participation of two kinases (GSK3-β and CDK5) in neurite retraction and tau hyperphosphorylation in SH-SY5Y treated cells with HTLV-I-infected MT-2 cells.
Tau plays an essential role not only in neurite outgrowth but also in elongation and maintenance of axon morphology. We have previously reported increased tau phosphorylation at T181 in CSF of HAM/TSP patients (Maldonado et al., 2008). This increase has also been observed in SH-SY5Y cells treated with MT-2 cell supernatant, which contains the viral Tax protein, exhibiting neurite retraction reminiscent of the axonal degeneration in vivo. Here we have investigated whether other tau phosphorylation sites (pS199, pT205, pS231, pS262, pS356, and pS396) are hyperphosphorylated in the presence of MT-2 supernatant. Such hyperphosphorylations occur in several neurodegenerative, diseases such as Creutzfeldt-Jakob disease, amyotrophic lateral sclerosis, Alzheimer’s disease, and relapsing-remitting multiple sclerosis (Olsson et al., 2005; Buerger et al., 2006; Otto et al., 2008; Skinningsrud et al., 2008). However, no similar hyper-phosphorylations occurred in our experiments. The similarity and extent of tau phosphorylation both in our model and in HAM/TSP in vivo suggest that the kinases involved might be the same (Maldonado et al., 2008). The two kinases GSK3-β and CDK5 phosphorylate tau at T181 (Hanger et al., 2009).
Our experiments with kinase inhibitors suggest that the MT-2 secreted products can induce neurite retraction by activation of CDK5, but not of GSK3-β, leading to an increase in tau pT181. This cell model could mimic the effect of HTLV-I-infected T-lymphocytes on cortical spinal neurons from patients, being a novel model for the possible participation of CDK5 in HAM/TSP.
Neurite retraction involves extracellular cues that mediate rapid remodeling of the cytoskeleton, including rearrangements of microtubules, actin microfilaments, and neurofilaments (Luo and O’Leary, 2005; Fukushima and Morita, 2006). Axonal stability depends on the capacity of the cytoskeleton to assemble and disassemble in response to extracellular signals (Sayas et al., 2002). Blockage of anterograde axonal transport in the CNS might trigger axon degeneration (Coleman and Perry, 2002). APP accumulation in axons precedes axolemmal disruption (e.g., in HAM/TSP and traumatic brain injury), which suggests that axonal transport failure is an early event in axonal degeneration (Coleman, 2005; De Vos et al., 2008). Increased tau phosphorylation at T181 suggests that microtubules are likely implicated in the neurite shortening in SH-SY5Y cells. However, we cannot discard the additional effects of CDK5 on other cytokeleton proteins as CRMP and actin (Hou et al., 2008).
Although the LiCl treatment only attenuated tau hyperphosphorylation, the roscovitine treatment of SH-SY5Y thoroughly blocked hyperphosphorylation at T181 induced by MT-2 supernatant. In addition, CDK5 may be involved in this hyperphosphorylation when GSK3-β is inhibited. Lithium inhibits GSK3-β directly by competing with Mg2+ and, indirectly, through phosphatase inhibition, PP2A, then increasing phosphorylation at S9 (Tajes et al., 2009). In addition, lithium reduces intracellular Ca2+ that promotes calpain-mediated proteolysis of p35 into p25, thus reducing CDK5 hyperactivation (Jorda et al., 2005; Crespo-Biel et al., 2009; Crews et al., 2009; Tajes et al., 2009). While studying the effect of gp120 from HIV on SH-SY5Y cells, Crews et al. (2009) observed a lower CDK5 activity in cells preincubated with LiCl in the absence of the neurotoxic agent. Thus, the reduction in tau phosphorylation at T181 observed under our experimental conditions with lithium treatment could be attributed to a lower amount of p25-CDK5.
Abnormal tau phosphorylation can originate from an imbalance between kinase and phosphatase activities. Studies in SH-SY5Y cells have shown that inhibition of PP-2A and PP-1 leads to tau hyperphosphorylation at specific sites and up-regulation of the activities of cdc2 kinase, CDK5, and MAPK, but not of GSK3-β (Tanaka et al., 1998). We believe that tau hyperphosphorylation is attributable at least in part to CDK5, but we cannot discard a possible decreased dephosphorylation. CDK5 regulates phosphorylation of several downstream targets in cytoskeletal function (Kanungo et al., 2009), so CDK5 signaling pathways could be involved in mediating retraction by MT-2 secreted viral products that contain Tax (Alefantis et al., 2005).
The reduced level of hyperphosphorylated tau in the presence of lithium does not allow us to attribute this alteration to neurite shortening. However, a higher level of pT181 might be required to observe this damage under our experimental conditions (1 hr of treatment). MT-2 secreted products increased CDK5 activity in differentiated SH-SY5Y cells. The kinase inhibition by either roscovitine or CDK5 siRNA prevented neurite retraction. All these results support the idea that CDK5 is involved in neurite shortening in our model. How could CDK5 be involved?
Deregulation of CDK5 activity by increasing the neurotoxic activator p25 might contribute to the pathogenesis of various neurodegenerative pathologies (Cruz and Tsai, 2004). Activation of calpains cleaves p35 and p39, generating the p25 and p29 fragments, respectively, then forming p25-CDK5 or p29-CDK5 with higher activity and longer half-life than those noncleaved complexes. In addition, tau phosphorylation by p39-CDK5 during brain development reduces tau binding to microtubules (Takahashi et al., 2003). The p35-CDK5 activity can additionally be up-regulated by CDK5 phosphorylation at Y15 mediated by Abl-Cables and Fyn-CDK5 pathways, which leads to an increase in cytoskeleton phosphorylation, including tau, CRMPs, and actin (Zukerberg et al., 2000; Hou et al., 2008).
We have demonstrated that Tax neutralization with anti-Tax serum reduced the ability of MT-2 supernatant to cause neurite shortening. This is the first evidence of direct participation of Tax protein in our model. Tax has been considered an important protein in HAM/TSP progression (Ramirez et al., 2003; Boxus et al., 2008; Matsuura et al., 2010). This viral protein in lymphocytes interacts with transcriptional factors, cell signaling proteins, and PDZ protein domains. We think that extracellular Tax could activate CDK5 in HAM/TSP patients through changes in some CDK5 activators.
One of the current strategies targeting tau in neurodegenerative diseases consists of reduction of tau phosphorylation by specific protein kinase inhibition (Hanger et al., 2009). In HIV infection, a pharmacological development has been reported using a selective blockage of the viral proteins Tat or Rev by means of their neutralization with either antibody or apten (Rusnati and Presta, 2002; Zhou et al., 2008). The results of this study lead us to conclude that CDK5 or Tax inhibitors may be therapeutically useful in HAM/TSP.
Acknowledgments
Contract grant sponsor: FONDECYT; Contract grant number: 108-0396.
We thank Dr. Marcelo Kogan for providing SH-SY5Y cells, Dr. Jenny Fiedler for supplying β-catenin antibody, Dr. Pablo Caviedes for his advice, and Dr. Chris Pogson and Mr. Claudio Telha for their critical reading of the manuscript.
References
- Alefantis T, Mostoller K, Jain P, Harhaj E, Grant C, Wigdahl B. Secretion of the human T cell leukemia virus type I transactivator protein tax. J Biol Chem. 2005;280:17353–17362. doi: 10.1074/jbc.M409851200. [DOI] [PubMed] [Google Scholar]
- Alefantis T, Flaig KE, Wigdahl B, Jain P. Interaction of HTLV-1 Tax protein with calreticulin: implications for Tax nuclear export and secretion. Biomed Pharmacother. 2007;61:194–200. doi: 10.1016/j.biopha.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila J, Lucas JJ, Perez J, Hernandez Role of tau protein in both physiological and pathological conditions. Physiol Rev. 2004;84:361–384. doi: 10.1152/physrev.00024.2003. [DOI] [PubMed] [Google Scholar]
- Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, Scott CW, Lee CM. Regulation and localization of tyrosine 216 phosphorylation of glycogen synthase kinase-3β in cellular and animal models of neuronal degeneration. Proc Natl Acad Sci U S A. 2000;97:11074–11079. doi: 10.1073/pnas.190297597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxus M, Twizere J-C, Legros S, Dewulf J-F, Kettmann R, Willems L. The HTLV-1 Tax interactome. Retrovirology. 2008;5:76. doi: 10.1186/1742-4690-5-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buerger K, Ewers M, Pirttila T, Zinkowski R, Alafuzoff I, Teipel SJ, DeBernardis J, Kerkman D, McCulloch C, Soininen H, Hampel H. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain. 2006;129:3035–3041. doi: 10.1093/brain/awl269. [DOI] [PubMed] [Google Scholar]
- Cartier LM, Cea JG, Vergara C, Araya F, Born P. Clinical and neuropathological study of six patients with spastic paraparesis associated with HTLV-I: an axomyelinic degeneration of the central nervous system. J Neuropathol Exp Neurol. 1997;56:403–413. doi: 10.1097/00005072-199704000-00009. [DOI] [PubMed] [Google Scholar]
- Cartier L, Vergara C, Valenzuela M. Immunohistochemistry of degenerative changes in the central nervous system in spastic paraparesis associate to human T lymphotropic virus type I. Rev Med Chil. 2007;135:1139–1146. doi: 10.4067/s0034-98872007000900007. [DOI] [PubMed] [Google Scholar]
- Cho JH, Johnson GV. Glycogen synthase kinase 3β phosphorylates tau at both primed and unprimed sites. Differential impact on microtubule binding. J Biol Chem. 2003;278:187–193. doi: 10.1074/jbc.M206236200. [DOI] [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Coleman MP, Perry VH. Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci. 2002;25:532–537. doi: 10.1016/s0166-2236(02)02255-5. [DOI] [PubMed] [Google Scholar]
- Cowan EP, Alexander RK, Daniel S, Kashanchi F, Brady JN. Induction of tumor necrosis factor alpha in human neuronal cells by extracellular human T-cell lymphotropic virus type 1 Tax. J Virol. 1997;71:6982–6989. doi: 10.1128/jvi.71.9.6982-6989.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo-Biel N, Camins A, Pallas M, Canudas AM. Evidence of calpain/cdk5 pathway inhibition by lithium in 3-nitropropionic acid toxicity in vivo and in vitro. Neuropharmacology. 2009;56:422–428. doi: 10.1016/j.neuropharm.2008.09.012. [DOI] [PubMed] [Google Scholar]
- Crews L, Patrick C, Achim CL, Everall IP, Masliah E. Molecular pathology of neuro-AIDS (CNS-HIV) Int J Mol Sci. 2009;10:1045–1063. doi: 10.3390/ijms10031045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JC, Tsai L-H. Cdk5 deregulation in the pathogenesis of Alzheimer’s disease. Trends Mol Med. 2004;10:452–458. doi: 10.1016/j.molmed.2004.07.001. [DOI] [PubMed] [Google Scholar]
- De Vos KJ, Grierson AJ, Ackerley S, Miller CCJ. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- Dhariwala FA, Rajadhyaksha MS. An unusual member of the Cdk family: Cdk5. Cell Mol Neurobiol. 2008;28:351–369. doi: 10.1007/s10571-007-9242-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas M, Iglesias M, Liu Y, Wang H, Muhaisen A. Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J Neurochem. 2000;75:991–1003. doi: 10.1046/j.1471-4159.2000.0750991.x. [DOI] [PubMed] [Google Scholar]
- Engmann O, Giese KP. Crosstalk between Cdk5 and GSK3β: implications for Alzheimer’s disease. Front Mol Neurosci. 2009;2:2. doi: 10.3389/neuro.02.002.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima N, Morita Y. Actomyosin-dependent microtubule rearrangement in lysophosphatidic acid-induced neurite remodeling of young cortical neurons. Brain Res. 2006;1094:65–75. doi: 10.1016/j.brainres.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Garrofe-Ochoa X, Melero-Fernandez de Mera RM, Fernandez-Gomez FJ, Ribas J, Jordan J, Boix J. BAX and BAK proteins are required for cyclin-dependent kinase inhibitory drugs to cause apoptosis. Mol Cancer Ther. 2008;7:3800–3806. doi: 10.1158/1535-7163.MCT-08-0655. [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15:112–119. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Hou ST, Jiang SX, Smith RA, Kwang WJ. Permissive and repulsive cues and signalling pathways of axonal outgrowth and regeneration. Rev Cell Mol Biol. 2008:125–181. doi: 10.1016/S1937-6448(08)00603-5. [DOI] [PubMed] [Google Scholar]
- Jorda EG, Verdaguer E, Canudas AM, Jimenez A, Garcia de Arriba S, Allgaier C, Pallas M, Camins A. Implication of cyclin-dependent kinase 5 in the neuroprotective properties of lithium. Neuroscience. 2005;134:1001–1011. doi: 10.1016/j.neuroscience.2005.04.061. [DOI] [PubMed] [Google Scholar]
- Kanungo J, Zheng YL, Amin ND, Pant HC. Targeting Cdk5 activity in neuronal degeneration and regeneration. Cell Mol Neurobiol. 2009;29:1073–1080. doi: 10.1007/s10571-009-9410-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakawa G-I, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga SI. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem. 2000;275:17166–17172. doi: 10.1074/jbc.M907757199. [DOI] [PubMed] [Google Scholar]
- Liang M-H, Chuang D-M. Regulation and function of glycogen synthase kinase-3 isoforms in neuronal survival. J Biol Chem. 2007;282:3904–3917. doi: 10.1074/jbc.M605178200. [DOI] [PubMed] [Google Scholar]
- Liu F, Li B, Tung EJ, Grundke-Iqbal I, Iqbal K, Gong CX. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur J Neurosci. 2007;26:3429–3436. doi: 10.1111/j.1460-9568.2007.05955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, O’Leary DD. Axon retraction and degeneration in development and disease. Annu Rev Neurosci. 2005;28:127–156. doi: 10.1146/annurev.neuro.28.061604.135632. [DOI] [PubMed] [Google Scholar]
- Maldonado H, Ortiz-Riano E, Krause B, Barriga A, Medina F, Pando ME, Alberti C, Kettlun AM, Collados L, Garcia L, Cartier L, Valenzuela MA. Microtubule proteins and their post-translational forms in the cerebrospinal fluid of patients with paraparesis associated with HTLV-I infection and in SH-SY5Y cells: an in vitro model of HTLV-I-induced disease. Biol Res. 2008;41:239–259. [PubMed] [Google Scholar]
- Matsuura E, Yamano Y, Jacobson S. Neuroimmunity of HTLV-I infection. J Neuroimmun Pharmacol. 2010;5:310–325. doi: 10.1007/s11481-010-9216-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meares GP, Jope RS. Resolution of the nuclear localization mechanism of glycogen synthase kinase-3: functional effects in apoptosis. J Biol Chem. 2007;282:16989–17001. doi: 10.1074/jbc.M700610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A, Vanderstichele H, Andreasen N, De Meyer G, Wallin A, Holmberg B, Rosengren L, Vanmechelen E, Blennow K. Simultaneous measurement of β-amyloid(1–42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin Chem. 2005;51:336–345. doi: 10.1373/clinchem.2004.039347. [DOI] [PubMed] [Google Scholar]
- Osame M. HTLV-I associated myelopathy, a new clinical entity. Lancet. 1986;1:1031–1032. doi: 10.1016/s0140-6736(86)91298-5. [DOI] [PubMed] [Google Scholar]
- Otto M, Lewczuk P, Wiltfang J. Neurochemical approaches of cerebrospinal fluid diagnostics in neurodegenerative diseases. Methods. 2008;44:289–298. doi: 10.1016/j.ymeth.2007.06.012. [DOI] [PubMed] [Google Scholar]
- Ramirez E, Fernandez J, Cartier L, Villota C, Rios M. Defective human T-cell lymphotropic virus type I (HTLV-I) provirus in seronegative tropical spastic paraparesis/HTLV-I-associated myelopathy (TSP/HAM) patients. Virus Res. 2003;91:231–239. doi: 10.1016/s0168-1702(02)00276-9. [DOI] [PubMed] [Google Scholar]
- Rusnati M, Presta M. HIV-1 Tat protein: a target for the development of anti-AIDS therapies. Drugs Future. 2002;27:481–493. [Google Scholar]
- Sayas CL, Moreno-Flores MT, Avila J, Wandosell F. The neurite retraction induced by lysophosphatidic acid increases Alzheimer’s disease-like tau phosphorylation. J Biol Chem. 1999;274:37046–37052. doi: 10.1074/jbc.274.52.37046. [DOI] [PubMed] [Google Scholar]
- Sayas CL, Avila J, Wandosell F. Regulation of neuronal cytoskeleton by lysophosphatidic acid: role of GSK-3. Biochim Biophys Acta. 2002;1582:144–153. doi: 10.1016/s1388-1981(02)00149-x. [DOI] [PubMed] [Google Scholar]
- Shahani N, Brandt R. Functions and malfunctions of the tau proteins. Cell Mol Life Sci. 2002;59:1668–1680. doi: 10.1007/PL00012495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinningsrud A, Stenset V, Gundersen AS, Fladby T. Cerebrospinal fluid markers in Creutzfeldt-Jakob disease. Cerebrospinal Fluid Res. 2008;5:14. doi: 10.1186/1743-8454-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajes M, Yeste-Velasco M, Zhu X, Chou SP, Smith MA, Pallas M, Camins A, Casadesus G. Activation of Akt by lithium: pro-survival pathways in aging. Mech Ageing Dev. 2009;130:253–261. doi: 10.1016/j.mad.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Saito T, Hisanaga S, Pant HC, Kulkarni AB. Tau phosphorylation by cyclin-dependent kinase 5/p39 during brain development reduces its affinity for microtubules. J Biol Chem. 2003;278:10506–10515. doi: 10.1074/jbc.M211964200. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Zhong J, Iqbal K, Trenkner E, Grundke-Iqbal I. The regulation of phosphorylation of tau in SY5Y neuroblastoma cells: the role of protein phosphatases. FEBS Lett. 1998;426:248–254. doi: 10.1016/s0014-5793(98)00346-9. [DOI] [PubMed] [Google Scholar]
- Vasko MR, Guo C, Kelley MR. The multifunctional DNA repair/redox enzyme Ape1/Ref-1 promotes survival of neurons after oxidative stress. DNA Repair. 2005;4:367–379. doi: 10.1016/j.dnarep.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Zhou J, Li H, Li S, Zaia J, Rossi JJ. Novel dual inhibitory function aptamer-siRNA delivery system for HIV-1 therapy. Mol Ther. 2008;16:1481–1489. doi: 10.1038/mt.2008.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zukerberg LR, Patrick GN, Nikolic M, Humbert S, Wu CL, Lanier LM, Gertler FB, Vidal M, Van Etten RA, Tsai LH. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron. 2000;26:633–646. doi: 10.1016/s0896-6273(00)81200-3. [DOI] [PubMed] [Google Scholar]