

Abstract
The synthesis of a carboxyamidopyridine-pyrazolate dinucleating ligand (H5bppap) is described. Bimetallic iron and cobalt complexes of H5bppap ([MII2H2bppap]+) showed structural differences in both their primary and secondary coordination spheres. The binding of small molecules into the pre-organized ligand cavity is verified by the hydration of [FeII2H2bppap]+ and [CoII2H2bppap]+, leading to the formation of the complexes [{CoII(OH)}CoIIH3bppap]+ and [{FeII(OH)}-FeIIH3bppap]+, in which one of the metal centers has a terminal hydroxo ligand.
Hydrogen bonds (H-bonds) within the secondary coordination sphere of metalloprotein active sites are effective at regulating metal-mediated processes.1 In many cases these intramolecular H-bonds are formed between amino residues and external ligands, which are often derived from water molecules. A key feature of biological H-bonding networks is their ability to change structure during a chemical transformation to aid in the transfer of protons and electrons, or in the orientation of substrates.2 There have been numerous efforts to incorporate these properties in synthetic systems and notable advancements have been made.3 One approach is to place basic functional groups proximal to metal centers that upon protonation can serve as H-bond donors.4 Following this concept, we have developed a new dinucleating ligand 3,5-bis[bis(N-6-pivalamido-2-pyridylmethyl)aminomethyl]-1H-pyrazole (H5bppap), that when bonded to metal ions, adopted a variety of structures depending on its protonation state.5 This report illustrates that H5bppap can produce dinuclear species, in which each metal ion can have a different primary coordination sphere. Furthermore, we show that the ligand can aid in forming varied intramolecular H-bonding networks.
Nearly all of the binucleating ligands that have appeared in the literature provide symmetrical primary coordination spheres around each metal ion.6 However, there are several situations in which differing metal coordination geometries would be beneficial for function.6b, 7 For example, the reduced form of the respiratory protein hemerthyrin contains one six- and one five-coordinate ferrous center: dioxygen binding occurs at the coordinately unsaturated site 8 Our group 9 and others 10 have been utilizing carboxyamidopyridyl units in the design of multidentate ligands because it potentially coordinates to metal ions in two distinct modes. In one case, the carboxyamido group is deprotonated and functions as a bidentate ligand through coordination of the pyridyl nitrogen and carbonyl oxygen atoms (Figure 1A). Alternatively, the protonated form of the carboxyamido group assumes a conformation such that it is an intramolecular H-bond donor (Figure 1B). We have found that coupling these units to a pyrazolate ring led to H5bppap, a dinucleating ligand whose complexes can have metal centers with differing primary and secondary coordination spheres.
Figure 1.
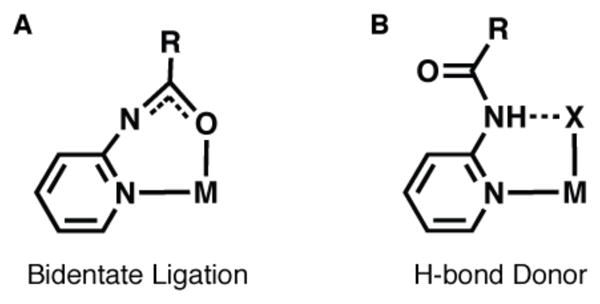
Possible binding modes of the carboxyamido unit.
H5bppap was synthesized in multi-gram quantities via a convergent synthetic route as outlined in Scheme 1. The protected bridging unit, 3,5-bis(chloromethyl)-(tetrahydropyran-2-yl)-1H-pyrazole5a,11,12 and the bis[N-(6-pivalamido-2-pyridylmethyl)]amine1 were coupled to afford a protected-form of the ligand, which was subsequently deprotected to afford H5bppap as a white powder in good yield (84 %).
Scheme 1.

Synthesis of H5bppap.a
a Conditions: (a) Na2CO3, MeCN, Δ, 24 h; then HCl(aq)/EtOH, rt, 21 h, 80%.
To evaluate the structural properties of metal complexes with H5bppap, [FeII2H2bppap]+ and [CoII2H2bppap]+ were prepared (Scheme 2) and crystallized.12 Treating H5bppap with 3 equiv of KH in anhydrous THF gave the [H2bppap]3− anion, which was allowed to react with 2 equiv of Fe(OTf)2. The salt [FeII2H2bppap](OTf) was isolated in almost quantitative yields via separation of the product from insoluble KCl. [CoII2H2bppap](BPh4) was prepared by treating a methanolic solution of H5bppap with NaOMe, Co(OAc)2 and NaBPh4; the salt was isolated as a lime green solid in high yield (86 %). Both complexes were stable in a dry, anaerobic environment for several weeks.
Scheme 2.
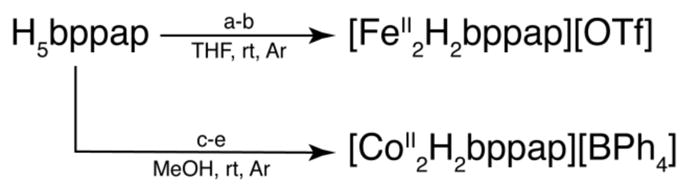
Synthesis of bimetallic complexes of H5bappap.a
aConditions: (a) 3 equiv KH; (b) 2 equiv Fe(OTf)2; (c) 3 equiv NaOMe; (d) 2 equiv Co(OAc)2; (e) 1 equiv NaBPh4.
The molecular structures for [FeII2H2bppap]+ and [CoII2H2bppap]+ were determined by X-ray diffraction methods (Figure 2) and illustrate the versatility of the [H2bppap]3− ligand in regulating the primary and secondary coordination spheres. The structure [CoII2H2bppap]+ shows that both CoII centers have nearly identical distorted trigonal bipyramidal coordination geometries (τCo1 = 0.74; τCo2 = 0.76).13 The pyridyl and pyrazolate nitrogen atoms define the trigonal planes: for pyrazolate nitrogen atoms, the Co1–N4 and Co2–N10 bond distances are equivalent within experimental error. However, each cobalt center has two distinct Co–Npy bond lengths: for example, the Co1–N2 and Co1–N3 bond distances are 2.028(2) and 2.135(2) Å, respectively. This difference in bond lengths reflects the fact that N2 is contained within two chelating rings, while N3 is only involved in a single chelating ring. The apical nitrogen atoms N1 and N9 bind to one of the axial coordination sites on each metal ion (avg. Co–Naxial = 2.171(2) Å). The other axial positions around each cobalt center are occupied by atoms O1 and O3 from the deprotonated carboxyamido groups, which coordinate in an identical manner (Table S2). The remaining carboxyamido groups are protonated, and form intramolecular H-bonds with O1 and O3, in which the N6···O1 and N12···O3 distances of less than 3 Å are observed. Co1···Co2 separation of 4.513 (1) Å was observed in [CoII2H2bppap]+.
Figure 2.

Thermal ellipsoid plots and schematic drawings for [CoII2H2bppap]+ (A) and [FeII2H2bppap]+ (B). Only carboxyamido hydrogen atoms are shown for clarity. Selected metrical parameters are in supporting information.12
The structural analysis of [FeII2H2bppap]+ revealed that each iron center has different primary and secondary coordination spheres (Figure 2B). Fe1 has a distorted trigonal bipyramidal geometry (τFe1 = 0.68), containing an N4O donor set that is similar to what was observed for the cobalt centers in [CoII2H2bppap]+. In contrast, Fe2 is six-coordinate, having an N4O2 arrangement of donor atoms, in which both oxygen atoms of the carboxyamido groups are bonded to the iron center. The significant difference in the Fe2–O3 and Fe2–O4 bond lengths of 1.957(2) and 2.267(2) Å reflects the protonation states of the carboxyamido groups. The shorter Fe2–O3 bond length agrees with the carboxyamido group containing O3 being deprotonating and serving as an anionic ligand. The carboxyamido group that includes O4 is protonated and thus is a neutral donor. Differences in the ligand N–C(O) bond lengths support this assignment: for the neutral group the N12–C45 is 1.358(3) Å, whereas for the anionic group the N11–C34 is 1.307(4) Å, indicating increased double bond character. Further- more, this type of coordination prevents the formation of intramolecular H-bonds around Fe2. It is not clear why there is a difference in coordination spheres for the two iron centers in [FeII2H2bppap]+.
The synthetic and structural results with the [MII2H2bppap]+ (MII = Fe, Co) complexes suggested that the appended carboxyamido groups of [H2bppap]3− are flexible and could readily adopt different conformations that may be useful in controlling the binding of exogenous ligands. To examine this possibility and evaluate the accessibility of the metal centers, we study the binding of water molecules to [MII2H2bppap]+ complexes (Scheme 3). Treating either complex with 1 equiv of water produced the mono-hydroxide complexes, [{MII(OH)}MIIH3bppap]+.12,14 Our analytic and spectroscopic studies suggested that water addition caused protonation of the dinucleating ligand with concomitant binding of a hydroxide ion to one of the metal centers. Note that additional amounts of water, up to 40 equiv, afford only the mono-hydroxo complexes.
Scheme 3.
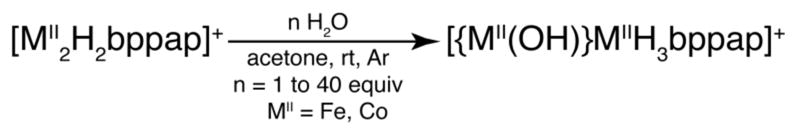
Synthesis of dimetallic [{MII(OH)}MIIH3bppap]+ complexes.12
X-ray diffraction studies revealed that the cobalt ions in [{CoII(OH)}CoIIH3bppap]+ (Figure 3A) have dissimilar trigonal bipyramidal coordination geometry (τCo1 = 0.88; τCo2 = 0.74). For Co2, the metrical parameters and overall structures of the primary and secondary coordination spheres are nearly identical to those found for the cobalt centers in [CoII2H2bppap]+. For Co1, the hydroxo ligand is terminally bonded at one of the axial sites with a Co1–O5 bond length of 1.931(2) Å and N1–Co1–O5 bond angle of 176.154°. This binding caused a major structural change in the dinucleating ligand: O1 is not bonded to the Co1 (as in [CoII2H2bppap]+) but is positioned outside the cavity surrounding the metal ions. Moreover, the appended carboxyamido group containing O1 is now protonated, with the N5–H5B vector placed within the cavity. The structural rearrangement resulted in two intramolecular H-bonds forming between the hydroxo ligand and the carboxyamido groups (N5···O5, 2.773 and N3···O5, 2.773 Å). The Fourier transform IR spectrum supports this premise by having a single ν(O–H) peak at 3613 cm−1 and several broad ν(N–H) signals between 3400 and 3200 cm−1, consistent with H-bonds involving the carboxyamido units.14e
Figure 3.

Thermal ellipsoid plots and schematic drawings for [{CoII(OH)}CoIIH3bppap]+ (A) and [{FeII(OH)}FeIIH3bppap]+. (B). Only carboxyamido hydrogen atoms are shown for clarity. Selected metrical parameters are in supporting information.12
The structural analysis on [{FeII(OH)}FeIIH3bppap]+ showed that again one five- and six-coordinate iron center exists, yet now an endogenous hydroxo ligand is terminally bonded to one of the iron sites. The hydroxo ligand is bonded to the five-coordinate iron site (τFe1 = 0.68), with an Fe1–O5 bond length of 1.938(2) Å and N1–Fe1–O5 bond angle of 172.62(9)° (Figure 3B). Both carboxyamido groups are protonated around Fe1 and form intramolecular H-bonds to the Fe1–O5 unit in a similar manner to that observed in [{CoII(OH)}CoIIH3bppap]+. The Fe2 site remained six-co ordinate, with both O3 and O4 of the appended carboxyamido groups bonded to the iron center.
In summary, we have developed a new dinucleating ligand that utilizes a pyrazolate bridge and carboxyamidopyridyl groups. The structures of [MII2H2bppap]+ and their hydrated products [{MII(OH)}MIIH3bppap]+ showed the versatility of this design to produce complexes with varied coordination chemistry. The formation of a single metal ion site with a terminal hydroxo ligand is rare because of the propensity of this ligand to bridge between metal ions. The formation of only one M–OH site in each complex is attributed to several factors, including the spacing provided by the pyrazolate unit,5b the intramolecular H-bonds, and the bidentate binding-mode of the carboxyamido groups. Our findings illustrate the utility of this design in preparing new classes of unsymmetrical dinuclear complexes.
Supplementary Material
Acknowledgments
Acknowledgement is made to the NIH (GM50781 to A.S.B) for financial support.
Footnotes
Supporting Information Available: Details for all experiments, spectra, and crystallographic details (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Shook RL, Borovik AS. Inorg Chem. 2010;49:3646–3660. doi: 10.1021/ic901550k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Marshall NM, Garner DK, Wilson TD, Gao Y-G, Robinson H, Nilges MJ, Lu Yi. Nature. 2009;462:113–116. doi: 10.1038/nature08551. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Miller AF. Acc Chem Res. 2007;41:501–510. doi: 10.1021/ar700237u. [DOI] [PubMed] [Google Scholar]; (b) Jackson TA, Brunold TC. Acc Chem Res. 2004;37:461–470. doi: 10.1021/ar030272h. [DOI] [PubMed] [Google Scholar]
- 3.(a) Collman JP, Zhang XM, Wong K, Brauman JI. J Am Chem Soc. 1994;116:6245–6251. [Google Scholar]; (b) Harata M, Jitsukawa K, Masuda H, Einaga H. J Am Chem Soc. 1994;116:10817–10818. [Google Scholar]; (c) Wada A, Harata M, Hasegawa K, Jitsukawa K, Masuda H, Mukai M, Kitagawa T, Einaga H. Angew Chem Int Ed. 1998;37:798–799. doi: 10.1002/(SICI)1521-3773(19980403)37:6<798::AID-ANIE798>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]; (d) Wada A, Yamaguchi S, Jitsukawa K, Masuda H. Angew Chem Int Ed. 2005;44:5698–5701. doi: 10.1002/anie.200501157. [DOI] [PubMed] [Google Scholar]; (e) Natale D, Mareque-Rivas JC. Chem Commun. 2008:425–437. doi: 10.1039/b709650j. [DOI] [PubMed] [Google Scholar]; (f) Shook RL, Borovik AS. Chem Commun. 2008:6095–6107. doi: 10.1039/b810957e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Kendall AJ, Zakharov LN, Gilbertson JD. Inorg Chem. 2010;49:8656–8658. doi: 10.1021/ic101408e. [DOI] [PubMed] [Google Scholar]; (b) Kovari E, Kramer R. J Am Chem Soc. 1996;118:12704–12709. [Google Scholar]
- 5.(a) Zinn PJ, Powell DR, Day VW, Hendrich MP, Sorrell TN, Borovik AS. Inorg Chem. 2006;45:3484–3486. doi: 10.1021/ic060009n. [DOI] [PubMed] [Google Scholar]; (b) Klingele J, Dechert S, Meyer F. Coord Chem Rev. 2009;253:2698–2741. [Google Scholar]
- 6.Gavrilova AL, Bosnich B. Chem Rev. 2004;104:349–383. doi: 10.1021/cr020604g.A recent review in unsymmetric ligand designs: Jarenmark M, Carlsson H, Nordlander E. C R Chimie. 2007;10:433–462.
- 7.Du Bois J, Mizoguchi TJ, Lippard SJ. Coord Chem Rev. 2000;200:443–485. [Google Scholar]
- 8.(a) Stenkamp RE. Chem Rev. 1994;94(3):715–726. [Google Scholar]; (b) Holmes MA, Letrong I, Turley S, Sieker LC, Stenkamp RE. J of Mol Biol. 1991;218:583–593. doi: 10.1016/0022-2836(91)90703-9. [DOI] [PubMed] [Google Scholar]
- 9.(a) Shook RL, Gunderson WA, Greaves J, Ziller JW, Hendrich MP, Borovik AS. J Am Chem Soc. 2008;130:8888–8889. doi: 10.1021/ja802775e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Powell-Jia DA, Pham MTN, Ziller JW, Borovik AS. Inorg Chim Acta. 2010;363:2728–2733. doi: 10.1016/j.ica.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Ingle GK, Makowska-Grzyska MM, Szajna-Fuller E, Sen I, Price JC, Arif AM, Berreau LM. Inorg Chem. 2007;46:1471–1480. doi: 10.1021/ic062020t. [DOI] [PubMed] [Google Scholar]; (b) Rudzka K, Arif AM, Berreau LM. J Am Chem Soc. 2006;128:17018–7023. doi: 10.1021/ja0601336. [DOI] [PubMed] [Google Scholar]; (c) Mareque-Rivas JC, Salvagni E, Parsons S. Dalton Trans. 2004:4185–4192. doi: 10.1039/b414223c. [DOI] [PubMed] [Google Scholar]; (d) Mareques-Rivas JC, de Rosales RTM, Parsons S. Dalton Trans. 2003:2156–2163. [Google Scholar]; (e) Mareques-Rivas JC, Salvagni E, de Rosales RTM, Parsons S. Dalton Trans. 2003:3339–3349. [Google Scholar]
- 11.Roder JC, Meyer F, Pritzkow H. Organometallics. 2001;20:811–817. [Google Scholar]
- 12.See supporting information for details.
- 13.The parameter τ is the index of trigonality for 5-coordinate complexes: Addison AW, Rao TN, Reedijk J, Van Rijn J, Verschoor GCJCS. Dalton Trans. 1984:1349–1356.
- 14.(a) Rutsch P, Meyer F. Chem Commun. 1998:1037–1038. [Google Scholar]; (b) Bauer-Siebenlist B, Meyer F, Farkas E, Vidovic D, Dechert S. Chem Eur J. 2005;11:4349–4360. doi: 10.1002/chem.200400932. [DOI] [PubMed] [Google Scholar]; (c) Bozoglian F, Romain S, Ertem MZ, Todorova TK, Sens C, Mola J, Rodriguez M, Romero I, Benet-Buchholz J, Fontrodona X, Cramer CJ, Gagliardi L, Llobet A. J Am Chem Soc. 2009;131:15176–15187. doi: 10.1021/ja9036127. [DOI] [PubMed] [Google Scholar]; (d) Strautmann JBH, Walleck S, Bogge H, Stammler A, Glaser T. Chem Commun. 2010;47:695–697. doi: 10.1039/c0cc03098h. [DOI] [PubMed] [Google Scholar]; (e) Macbeth CE, Hammes BS, Young VG, Jr, Borovik AS. Inorg Chem. 2001;40:4733–4741. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.