

Abstract
Phenotypic plasticity––the capacity of a single genotype to produce different phenotypes in response to varying environmental conditions––is widespread. Yet, whether, and how, plasticity impacts evolutionary diversification is unclear. According to a widely discussed hypothesis, plasticity promotes rapid evolution because genes expressed differentially across different environments (i.e., genes with “biased” expression) experience relaxed genetic constraint and thereby accumulate variation faster than do genes with unbiased expression. Indeed, empirical studies confirm that biased genes evolve faster than unbiased genes in the same genome. An alternative hypothesis holds, however, that the relaxed constraint and faster evolutionary rates of biased genes may be a precondition for, rather than a consequence of, plasticity’s evolution. Here, we evaluated these alternative hypotheses by characterizing evolutionary rates of biased and unbiased genes in two species of frogs that exhibit a striking form of phenotypic plasticity. We also characterized orthologs of these genes in four species of frogs that had diverged from the two plastic species before the plasticity evolved. We found that the faster evolutionary rates of biased genes predated the evolution of the plasticity. Furthermore, biased genes showed greater expression variance than did unbiased genes, suggesting that they may be more dispensable. Phenotypic plasticity may therefore evolve when dispensable genes are co-opted for novel function in environmentally induced phenotypes. Thus, relaxed genetic constraint may be a cause––not a consequence––of the evolution of phenotypic plasticity, and thereby contribute to the evolution of novel traits.
Introduction
Phenotypic plasticity’s role in evolutionary diversification remains controversial (West-Eberhard 1989, 2003; Pfennig et al. 2010; Moczek et al. 2011). On the one hand, phenotypic plasticity has long been viewed as an impediment to evolutionary change (reviewed by Schlichting 2004). On the other hand, increasing evidence suggests that plasticity may facilitate evolutionary diversification (Pfennig et al. 2010; Moczek et al. 2011). Yet, the specific mechanisms by which phenotypic plasticity actually facilitates––or impedes––evolution remains unclear, particularly at the molecular level.
One way in which phenotypic plasticity may enhance diversification is by causing differences in gene expression between environmentally induced phenotypes (Aubin-Horth and Renn 2009). In particular, recent theory suggests that differentially expressed genes (“biased genes”) should be less constrained––and therefore free to evolve faster––than are genes that do not differ in expression between environmentally induced phenotypes (“unbiased genes”). Such diminished constraint can arise because biased genes evolve reduced pleiotropy [Fisher 1930 (1999); Pal et al. 2006; Snell-Rood et al. 2011]. Specifically, when alternative traits that are produced by genes with pleiotropic effects are under antagonistic selection, differential expression is thought to reduce this constraint and thereby enable rapid adaptive evolution in biased genes (Snell-Rood et al. 2011). Moreover, genetic constraints might be alleviated when biased genes experience relaxed selection in noninducing environments (Lahti et al. 2009; Snell-Rood et al. 2010; Van Dyken and Wade 2010). In particular, when compared to genes that are expressed constitutively, genes that are expressed facultatively should evolve more rapidly, because selection is less effective at removing deleterious alleles in genes that are expressed occasionally and/or in a subset of a population (Kawecki 1994; Kawecki et al. 1997; Van Dyken and Wade 2010). Indeed, recent empirical studies have confirmed that biased genes amass variation more rapidly and therefore evolve faster than do unbiased genes in the same genome (Hunt et al. 2010; Van Dyken and Wade 2010; Snell-Rood et al. 2011).
Finding that biased genes evolve faster than unbiased genes is also consistent with an alternative hypothesis, however. Indeed, rather than arising as a consequence of plasticity, enhanced evolutionary rates of biased genes might actually be a precondition for plasticity’s evolution (Hunt et al. 2011). Specifically, rapidly evolving genes may be more likely than slowly evolving genes to become co-opted for biased expression if they tend to be more “dispensable” (i.e., less critical to fitness and/or already less constrained by pleiotropy). Such genes should experience reduced purifying selection and therefore evolve faster (Hirsh and Fraser 2008).
Consistent with this hypothesis, in Hymenoptera, genes that are differentially expressed between castes evolve faster and appear to be more dispensable than are unbiased genes in the same genome that are not differentially expressed between castes (Hunt et al. 2010). Moreover, putative orthologs of caste-biased genes in a eusocial ant and a eusocial bee evolve more rapidly than do unbiased genes in a wasp lacking castes (Hunt et al. 2011), suggesting that rapid evolutionary rates may have preceded caste-biased gene expression. However, additional studies are needed to test these ideas.
Here, we evaluated the above two alternative hypotheses by asking two questions. First, does the evolution of phenotypic plasticity precede or follow relaxed genetic constraint? Second, if plasticity does follow relaxed genetic constraint, are biased genes more dispensable?
We addressed these two questions by focusing on a conspicuous example of phenotypic plasticity in spadefoot toads (genus Spea). Spea tadpoles develop either as an omnivore morph, or as a morphologically, behaviorally, and ecologically distinctive carnivore morph (Fig. 1a and b), which is triggered, in part, by diet; specifically, the ingestion of shrimp (Pfennig 1990; Ledón-Rettig et al. 2008; Ledón-Rettig and Pfennig 2011). Using this system, we identified a set of genes that display differential expression between these alternative carnivore and omnivore morphs (henceforth, “morph-biased” genes). We then compared the rate at which these genes evolve relative to a set of genes in the same genome that we determined did not exhibit morph-biased expression; i.e., unbiased genes.
Fig. 1.

Tadpoles of spadefoot toads (genus Spea) exhibit striking phenotypic plasticity. Depending on their environment, these tadpoles develop into either (a) omnivores that eat detritus, algae, and small invertebrates or (b) carnivores that specialize on fairy shrimp. (c) This plasticity is restricted to Spea (filled circles: taxa in which both morphs are present; open circles: taxa in which only omnivores are present). Generic names: Sp.: Spea; Sc.: Scaphiopus; Pd.: Pelodytes; Pb.: Pelobates; Br.: Brachytarsophrys; M.: Megophrys; L.:Leptolalax; X.: Xenopus; gray boxes indicate those species that were used in the present study. Panel (c) re-drawn from Ledón-Rettig and Pfennig (2011).
We also identified putative orthologs of these genes in four species that lack similar plasticity and compared the rate at which morph-biased genes evolved relative to the rate at which unbiased genes evolved in the latter four species. Reconstruction of ancestral character states reveals that carnivore-omnivore plasticity evolved in Spea after these four species had diverged from Spea’s ancestor (Fig. 1c). Thus, finding that morph-biased genes evolve faster than unbiased genes in Spea, but not in species that do not display carnivore–omnivore plasticity, would suggest that the faster evolutionary rates of morph-biased genes was likely an evolutionary consequence of the plasticity. In contrast, finding that morph-biased genes evolve faster than unbiased genes in all six species, but that morph-biased genes do not evolve faster in Spea, would instead suggest that the faster evolutionary rate of morph-biased genes is ancestral, and that this faster rate thus preceded the evolution of this plasticity, consistent with the second hypothesis above.
Materials and methods
Focal animals and the generation of alternative morphs
Two sibships of Spea bombifrons were produced from adult toads previously collected from the San Simon Valley of Cochise County, Arizona, and currently housed at the University of North Carolina Chapel Hill for the past 1–2 years. At 2 days posthatching, groups of 8 tadpoles per family were randomly selected and placed into 20 replicate (28 × 18 × 10 cm2) tanks filled with 6 L of dechlorinated tap water. Tadpoles were fed brine shrimp nauplii ad libitum until 3 days posthatching after which they were fed live brine shrimp ad libitum (ingestion of shrimp induces expression of the carnivore morph; Pfennig 1990; Ledón-Rettig et al. 2008). At 8 days posthatching (Gosner stage 29–32), each tadpole was scored as either a carnivore or an omnivore based on visual inspection of keratinized mouthparts and morphology of the jaw muscles (Pfennig 1990). Out of all tadpoles, we selected the eight individuals that expressed the most extreme carnivore-like morphology along with eight randomly selected omnivores. These 16 tadpoles were immediately flash frozen in liquid nitrogen.
Morph-biased gene discovery with heterologous microarrays
Tadpoles were extracted for total RNA using TRIzol Reagent and PureLink spin columns (Invitrogen) with on-column DNase I digestion following the manufacture’s protocol. RNA extracts were spiked with SuperaseIn RNase inhibitor (Ambion). DNA integrity was checked using gel electrophoresis and sample concentrations were quantified using a NanoDrop spectrophotometer (Thermo Scientific).
Microarray hybridization was conducted at the University of North Carolina Functional Genomics Core using Affymetrix GeneChip Xenopus tropicalis genome arrays according to the manufacture’s protocol. The resulting probe-level data were mas background corrected, normalized with quantiles, and pm corrected and summarized using a custom method with AFFY (Irizarry et al. 2003) in R v2.12.1 (R Development Core Team 2010). Our custom method evaluated all PM and MM probes within a probeset for the probe with the highest average expression across all samples (available upon request). The value of this single probe was then used as the final measure of expression for each sample. A fixed effect model was fit to the summarized log2 values (∼Morph + Family) and analyzed for significance using F-tests with permutation (1000 sample permutations) R/MAANOVA (Wu et al. 2003). The resulting P-values were corrected for multiple hypothesis testing using R/QVALUE (Storey and Tibshirani 2003).
Significant probesets were annotated using BLASTx with an e-value cutoff of 1 E−15. Probesets with no X. tropicalis best hit meeting the search criteria were removed from the candidate list. For each candidate, we attempted to sequence portions of coding region (250–900 bp) from a single Sp. bombifrons using primers designed from alignments of the X. tropicalis best hit for a given probeset and at least one other vertebrate ortholog (BLAST best hit). Genes with no orthologs were removed from the analysis.
Morph-biased gene discovery with 454 sequencing
To increase the likelihood of identifying genes with morph-biased expression, we developed qRT–PCR primers (see “Assessment of differential expression” section) from a library of expressed sequences. The library was created by Roche 454 transcriptome sequencing of a pool of tadpoles of Sp. multiplicata at various larval stages. mRNA was isolated from total RNA using the Poly(A)Purist mRNA purification kit (Ambion) and converted to cDNA (SuperScript II, Invitrogen). To provide a better representation of both 5′- and 3′-ends of the transcripts, the sample was partially digested with EcoRI and BamHI (New England Biolabs). This sample was prepared for 454-long read sequencing following standard protocols (Roche). For 454 sequencing, one-quarter plate of long reads was loaded and sequenced. This produced 135,797 filtered reads for a total of 47,844,572 bp (mean read length = 352 bp). Estimated per base error rate was 1.09%.
Read data were assembled using Newbler (Roche), with lenient parameters (25 bp overlap, 95% identity). Seventy-two percent of the reads were assembled into 4415 contigs, and 94.2% of bases had a Q score >39. These transcripts encompassed 1,661,519 bp of sequence. There were 32,750 reads that were not used for contig assembly (singletons). To identify putative homologs of known genes, we used tBLASTx (1 E−4) to align all proteins from the sequenced strain of X. tropicalis (RefSeq v4.2) to the Sp. multiplicata contigs. We limited primer development to contigs at least 600 bp in length and ribosomal RNA genes were removed from the candidate list.
Assessment of differential expression
As stated previously, candidate genes from the microarray analysis were those genes showing differential expression between morphs and for which we were able to obtain sequence data from Sp. bombifrons. Candidate genes from the 454 sequencings were from contigs of at least 600 bp in length from Sp. multiplicata and having an X. tropicalis ortholog (to the exclusion of ribosomal RNA genes). For each of these candidate genes, qRT–PCR was used to measure the degree of differential expression between omnivores and carnivores for the samples used in the microarray experiment (Supplementary Table S1). Primers were designed using PrimerQuest (Integrated DNA Technologies) and the following design parameters: product size = 90–160 bp, primer length = 22–28 bp, TM = 58–61, GC = 50%. Over half of these primer pairs were designed to span exon–intron boundaries for postPCR confirmation of no DNA contamination (Supplementary Table S1). Total RNA samples were tested with a subset of primer pairs for possible DNA contamination. cDNA was then generated from 2 µg of total RNA using a High-Capacity RNA to cDNA kit with both random hexamer and Oligo-dT primers (Applied Biosystems). All samples were run on a Bio-Rad CFX96 using FAST SYBR Green Master Mix (Applied Biosystems) for 40 cycles at 60°C.
The resulting threshold values (CT) were averaged across replicates and measures of relative expression were calculated using the ΔΔCT method (Livak and Schmittgen 2001). All samples were normalized relative to actg1 and actb, which were empirically determined to be the best endogenous controls for these samples (Leichty 2011). Expression level was relative to a calibrator sample, which was an arbitrary pool of tadpole cDNA. The mean expression level for omnivores and carnivores was subtracted and log2 transformed to produce a fold difference (FD) value for each gene. Mann–Whitney tests (R/wilcox.test) with FDR correction (R/p.adjust)(Benjamini and Hochberg 1995) were used to test significance of differential expression.
Based on magnitude of FD and degree of statistical significance (P-value), each gene was classified into one of three groups. Genes with an FD ≥ 1.0 and a P ≤ 0.05 were classified as morph biased. Genes with an FD < 0.2 and a P-value > 0.05 were classified as unbiased. All other genes were considered ambiguous.
Assessment of expression variance
For Sp. bombifrons, Scaphiopus couchii and Scaphiopus holbrookii, we measured the level of expression variation in morph-biased and -unbiased genes. We used the following nonrelative measure to calculate coefficient of variation within groups: 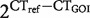 , where CTref is the mean cycle threshold of the endogenous control/s and CTGOI is the cycle threshold of the gene of interest. Expression levels were measured in Sc. couchii and Sc. holbrookii as previously described for Sp. bombifrons (see “Assessment of differential expression” section) with a few minor variations. Scaphiopus holbrookii samples were collected as eggs from the wild and reared under standard conditions at the University of North Carolina at Chapel Hill (UNC-CH) (n = 5). Scaphiopus couchii samples were from two clutches created at UNC-CH (n = 8). qPCR primers were developed from consensus alignments of Sc. couchii and Sc. holbrookii sequences (Supplementary Table S2). actg1, actb, 18S and 28S were used as endogenous controls for both Sc. couchii and Sc. holbrookii.
, where CTref is the mean cycle threshold of the endogenous control/s and CTGOI is the cycle threshold of the gene of interest. Expression levels were measured in Sc. couchii and Sc. holbrookii as previously described for Sp. bombifrons (see “Assessment of differential expression” section) with a few minor variations. Scaphiopus holbrookii samples were collected as eggs from the wild and reared under standard conditions at the University of North Carolina at Chapel Hill (UNC-CH) (n = 5). Scaphiopus couchii samples were from two clutches created at UNC-CH (n = 8). qPCR primers were developed from consensus alignments of Sc. couchii and Sc. holbrookii sequences (Supplementary Table S2). actg1, actb, 18S and 28S were used as endogenous controls for both Sc. couchii and Sc. holbrookii.
Sequencing and estimates of evolutionary rates
For genes classified as either morph-biased or unbiased, we sequenced orthologous portions of coding sequence from single individuals of Sp. multiplicata, Sc. couchii, Sc. holbrookii, and in some cases for X. laevis. In particular, primers were designed from Xenopus–Spea alignments. In some cases, further sequencing using standard PCR and 3′- and/or 5′- RACE (Invitrogen) were used to obtain additional sequences. PCR products were directly sequenced at the UNC-CH Genome Analysis Facility on an Applied Biosystems 3730XL Genetic Analyzer and sequences were assembled manually in Sequencer v4.10.1 (Gene Codes) at a minimum of 2X coverage. Polymorphic sites were coded using IUPAC nomenclature. For Xenopus tropicalis and X. laevis, coding sequences were extracted from Genbank (X. tropicalis RefSeq v4.2). Sequences for each gene were translated and aligned using MAFFT v6.483b (Katoh et al. 2002). We used PAL2NAL to convert protein alignments to codon alignments with the removal of gaps (Suyama et al. 2006).
Evolutionary rates were estimated with PAML v4.2 (Yang 2007) using the CODEML program. Branch models (Yang 1998) were fit for each gene class on an individual gene basis using the following unrooted phylogeny: [(Sp. bombifrons, Sp. multiplicata), (Sc. couchii, Sc. holbrookii), (X. tropicalis, X. laevis)]. The first model assumed a single dN/dS ratio across all branches of the phylogeny (the “one-ratio” model). A second model estimated two ratios, one for the clade formed by Sp. bombifrons and Sp. multiplicata (Spea), and a second for all other branches (the “two-ratio” model). The third model assumed a unique dN/dS ratio for each two-species clade (Spea, Scaphiopus, and Xenopus; the “three ratio” model). A fourth model assumed a unique ratio for each branch on the phylogeny (the “free-ratio” model). We then used likelihood ratio tests to compare models for a given gene class by summing the log-likelihoods of each model for each gene class (Yang and Swanson 2002). For the free-ratio model, we calculated mean dN/dS ratios for each branch on the phylogeny by pooling the estimated number of nonsynonymous and synonymous substitutions for each branch across genes within a gene class (i.e., morph-biased and unbiased).
We also sequenced homologous portions of seven genes from two closely related frog species, Pelodytes ibericus and Pelobates cultripes. For these seven genes, we used the three-ratio model to estimate clade-specific dN/dS ratios on the following tree: {[(Sp. bombifrons, Sp. multiplicata), (Sc. couchii, Sc. holbrookii)], (Pelodlytes ibericus, Pelobates cultripes), (X. tropicalis, X. laevis)}. We then compared these estimates for Spea, Scaphiopus, and Xenopus with those from the smaller tree (Supplementary Fig. S1). Additionally, we compared the difference between the likelihoods of the one- and two-ratio models for the small and large trees (Supplementary Fig. S2).
Results
Together, the heterologous microarrays and 454 transcriptome data identified 315 candidate genes for morph-biased expression. Of these candidates, we were able to sequence portions of the coding region and measure gene expression in Sp. bombifrons for 133 genes (Supplementary Table S1). Using quantitative PCR and Mann–Whitney U-tests, we classified each gene’s expression pattern as morph-biased or unbiased by comparing expression levels in carnivores and omnivores (Fig. 2).
Fig. 2.

Volcano plot of qPCR data. The horizontal line corresponds to a P-value of 0.05, such that all genes at, or above, this line are statistically significant in a test for differences between group medians (omnivores versus carnivores; see “Materials and methods section” for details). Open circles: unbiased genes; gray-filled circles: ambiguous genes; black-filled circles: morph-biased genes. See “Discussion” section for a description of the labeled genes.
As shown in Table 1, of these 133 genes, 25 were unambiguously morph-biased (of these, 19 genes were carnivore-biased, for which expression levels were significantly higher in carnivores than in omnivores, and 6 were omnivore-biased, for which expression levels were significantly higher in omnivores than in carnivores); 28 genes were unambiguously unbiased in their expression patterns; and 80 genes were ambiguous in their expression differences (this last group was therefore not included in subsequent analyses). We sequenced putative orthologs for 47 of these 53 morph-biased and unbiased genes in an additional plastic species, Sp. multiplicata, and in two nonplastic, spadefoot species, Sc. couchii and Sc. holbrookii. Additionally, we sequenced orthologs for X. laevis, for which sequences were not publicly available. Using these sequence data, and those from X. tropicalis (publicly available), we then compared the evolutionary rates of morph-biased and unbiased genes by fitting branch models for each gene separately for the two plastic species and the four nonplastic species. The average number of sites analyzed per gene was 522.6 with a range from 279 to 702.
Table 1.
Results of quantitative PCR for all candidate genes tested
| Mann–Whitney U-test |
Mean expression level |
|||||
|---|---|---|---|---|---|---|
| Gene ID | Gene annotation | P-value | FDR P-value | Carnivores | Omnivores | FD |
| Carnivore-biased genes (higher expression in carnivores) | ||||||
| btf3 | Basic transcription factor 3 | 0.0002 | 0.0021 | −0.069 | −1.594 | 1.525 |
| c2orf82 | Chromosome 2 open reading frame 82 | 0.0002 | 0.0021 | 1.58 | −0.12 | 1.7 |
| col2a1 | Collagen, type II, α 1 | 0.0003 | 0.0025 | −0.119 | −1.985 | 1.866 |
| col9a1 | Collagen α-1(IX) chain | 0.0003 | 0.0025 | 2.188 | 0.393 | 1.795 |
| csda | Cold shock domain protein A | 0.0006 | 0.0037 | 1.288 | −0.136 | 1.423 |
| des1 | Degenerative spermatocyte homolog 1, lipid desaturase | 0.0006 | 0.0037 | 2.117 | 0.865 | 1.251 |
| eif3c | Eukaryotic translation initiation factor 3, subunit C | 0.0003 | 0.0025 | 2.374 | 1.153 | 1.221 |
| gnb2l1 | Guanine nucleotide-binding protein (G protein), β polypeptide 2-like 1 | 0.0006 | 0.0037 | 2.338 | 1.252 | 1.086 |
| got2 | Glutamic-oxaloacetic transaminase 2, mitochondrial (aspartate aminotransferase 2) | 0.0002 | 0.0021 | 0.743 | −0.75 | 1.493 |
| krt5.2 | Keratin 5 gene 2 | 0.0006 | 0.0037 | −0.449 | −1.462 | 1.013 |
| krt19 | Keratin 19 | 0.0002 | 0.0021 | 0.204 | −1.121 | 1.325 |
| mrf1 | AT-rich interactive domain 5B (MRF1-like) | 0.0002 | 0.0021 | 1.539 | 0.233 | 1.305 |
| myl1 | Myosin, light chain 1, alkali; skeletal, fast | 0.0104 | 0.0287 | 0.999 | −0.028 | 1.027 |
| pm20d2 | Peptidase M20 domain containing | 0.0011 | 0.0049 | 0.851 | −1.416 | 2.267 |
| reep5 | Receptor accessory protein 5 | 0.0003 | 0.0025 | 1.026 | −0.073 | 1.1 |
| shisa7 | Shisa-7-like | 0.0019 | 0.0068 | 0.091 | −1.101 | 1.191 |
| tbx15 | T-box transcription factor TBX15-like | 0.0002 | 0.0021 | 1.981 | 0.89 | 1.091 |
| tnni2 | Troponin I type 2 (skeletal, fast) | 0.0047 | 0.014 | 1.176 | 0.071 | 1.105 |
| tnnt3 | Troponin T type 3 (skeletal, fast) | 0.0011 | 0.0049 | 0.654 | −0.368 | 1.022 |
| Omnivore-biased genes (higher expression in omnivores) | ||||||
| amy2a | Amylase, α 2A (pancreatic) | 0.003 | 0.0093 | −5.904 | −1.698 | 4.206 |
| c3 | Complement component 3 | 0.0011 | 0.0049 | −1.72 | −0.091 | 1.628 |
| mug1 | Murinoglobulin 1 | 0.0003 | 0.0025 | −3.838 | −1.373 | 2.465 |
| pglyrp1 | Peptidoglycan recognition protein 1 | 0.0003 | 0.0025 | −2.949 | −0.229 | 2.72 |
| pnlip | Pancreatic triacylglycerol lipase-like | 0.0002 | 0.0021 | −5.804 | 1.197 | 7.002 |
| tf | Transferrin | 0.0006 | 0.0037 | −3.302 | −1.63 | 1.671 |
| Unbiased genes | ||||||
| atp5g3 | ATP synthase, H+ transporting, mitochondrial Fo complex, subunit C3 (subunit 9) | 0.7984 | 0.8487 | −2.824 | −2.905 | 0.081 |
| bicd1 | Bicaudal D homolog 1-like | 1 | 1 | 1.701 | 1.62 | 0.081 |
| cab39l | Calcium-binding protein 39-like | 0.9591 | 0.9735 | −1.216 | −1.201 | 0.016 |
| ccnd2 | Cyclin D2 | 1 | 1 | 1.361 | 1.394 | 0.034 |
| ccni | Cyclin I | 0.5054 | 0.626 | 3.672 | 3.605 | 0.067 |
| col5a1 | Collagen, type V, α 1 | 0.6454 | 0.7322 | 3.879 | 3.795 | 0.085 |
| cox5a | Cytochrome c oxidase subunit Va | 0.8785 | 0.9194 | 0.371 | 0.357 | 0.014 |
| dach1 | Dachshund homolog 1 | 0.1949 | 0.277 | 0.217 | 0.385 | 0.168 |
| ercc3 | Excision repair cross-complementing rodent repair deficiency, complementation group 3 | 0.5737 | 0.6735 | 1.247 | 1.285 | 0.037 |
| faf1 | Fas (TNFRSF6) associated factor 1 | 0.7984 | 0.8487 | 2.257 | 2.262 | 0.006 |
| gas8 | Growth arrest-specific 8 | 0.3823 | 0.4869 | −0.826 | −0.997 | 0.171 |
| gtdc1 | Glycosyltransferase-like domain containing 1 | 0.4418 | 0.5574 | −0.19 | −0.361 | 0.171 |
| krt8 | Keratin 8 | 0.3282 | 0.4302 | 1.818 | 1.657 | 0.161 |
| lyst | Lysosomal trafficking regulator | 0.5737 | 0.6735 | 0.055 | 0.034 | 0.021 |
| mmaa | Methylmalonic aciduria type A protein, mitochondrial-like | 0.1304 | 0.2173 | 0.728 | 0.553 | 0.175 |
| npffr2 | Neuropeptide FF receptor 2-like | 0.5737 | 0.6735 | −1.614 | −1.523 | 0.09 |
| pitx2 | Paired-like homeodomain 2 | 0.3823 | 0.4869 | 1.39 | 1.286 | 0.104 |
| prss8 | Prostasin-like | 0.7209 | 0.7912 | 2.901 | 2.979 | 0.079 |
| psmd1 | Proteasome (prosome, macropain) 26S subunit non-ATPase 1 | 0.7209 | 0.7912 | 1.996 | 1.923 | 0.073 |
| ptafr | Platelet-activating factor receptor | 0.5737 | 0.6735 | −1.115 | −1.254 | 0.139 |
| rasd1 | RAS, dexamethasone-induced 1 | 0.7209 | 0.7912 | 1.626 | 1.557 | 0.068 |
| rbp1 | Retinol-binding protein 1, cellular | 0.9591 | 0.9735 | −1.223 | −1.196 | 0.027 |
| ric8a | Resistance to inhibitors of cholinesterase 8 homolog A | 0.7984 | 0.8487 | 0.73 | 0.68 | 0.051 |
| serbp1 | SERPINE1 mRNA-binding protein 1 | 0.8785 | 0.9194 | 3.021 | 3.073 | 0.052 |
| sgpp1 | Sphingosine-1-phosphate phosphatase 1 | 0.7984 | 0.8487 | 1.203 | 1.191 | 0.012 |
| thrap3 | Thyroid hormone receptor-associated protein 3 | 0.7209 | 0.7912 | 1.754 | 1.73 | 0.024 |
| wapal | Wings apart-like homolog | 0.9591 | 0.9735 | 0.706 | 0.674 | 0.033 |
| zfr | Zinc-finger RNA-binding protein | 0.9591 | 0.9735 | 2.904 | 2.946 | 0.042 |
| Ambiguous genes | ||||||
| a2m | α-2-macroglobulin | 0.007 | 0.0205 | −0.989 | −0.275 | 0.713 |
| abcc4 | ATP-binding cassette, sub-family C member 4 | 0.003 | 0.0093 | 1.242 | 2.051 | 0.809 |
| acp6 | Acid phosphatase 6, lysophosphatidic | 0.0281 | 0.0633 | 1.55 | 1.931 | 0.381 |
| actc1 | Actin, α, cardiac muscle 1 | 0.0499 | 0.1005 | −2.351 | −3.208 | 0.857 |
| aff2 | AF4/FMR2 family member 2-like | 0.065 | 0.1185 | −1.129 | −1.603 | 0.474 |
| aldoa | Aldolase A, fructose-bisphosphate | 0.1949 | 0.277 | 1.335 | 0.936 | 0.4 |
| arhgdig | Rho GDP dissociation inhibitor (GDI) gamma | 0.0379 | 0.0826 | −0.217 | −0.81 | 0.593 |
| arx | Homeobox protein ARX-like | 0.0006 | 0.0037 | 0.721 | −0.17 | 0.892 |
| atp2a1 | ATPase, Ca++ transporting, cardiac muscle, fast twitch 1 | 0.065 | 0.1185 | −1.207 | −2.023 | 0.816 |
| barx2 | Homeobox protein BarH-like 2-like | 0.0019 | 0.0068 | 2.38 | 1.504 | 0.875 |
| BC157756.1 | Novel protein similar to prothymosin, α | 0.1605 | 0.2408 | −0.647 | −0.904 | 0.257 |
| cbfb | Core-binding factor, β subunit, transcript variant 2 | 0.0011 | 0.0049 | 1.546 | 1.032 | 0.514 |
| cct8 | Chaperonin containing TCP1, subunit 8 (θ) | 0.0104 | 0.0287 | −0.023 | −0.55 | 0.527 |
| chchd1 | Coiled–coil–helix–coiled–coil–helix | 0.065 | 0.1185 | −0.485 | −0.181 | 0.303 |
| cirbp | Cold inducible RNA-binding protein | 0.083 | 0.1494 | −0.71 | −0.51 | 0.2 |
| clptm1 | Cleft lip and palate transmembrane protein 1 | 0.0148 | 0.0383 | 0.109 | 0.599 | 0.49 |
| cnn1 | Calponin 1, basic, smooth muscle | 0.0499 | 0.1005 | −0.147 | 0.169 | 0.316 |
| cryba4 | Crystallin, β A4 | 0.2786 | 0.3761 | −0.759 | −1.229 | 0.469 |
| crybb2 | Crystallin, β B2 | 0.065 | 0.1185 | −1.322 | −0.334 | 0.988 |
| crybb3 | Crystallin, β B3 | 0.1605 | 0.2408 | 0.435 | −0.299 | 0.733 |
| ddx1 | DEAD (Asp−Glu−Ala−Asp) box polypeptide 1 | 0.0281 | 0.0633 | 1.434 | 0.875 | 0.559 |
| ddx21 | DEAD (Asp−Glu−Ala−Asp) box polypeptide 21 | 0.0011 | 0.0049 | 1.089 | 0.556 | 0.533 |
| det1 | De-etiolated homolog 1 | 0.003 | 0.0093 | 0.272 | 0.822 | 0.55 |
| eef1b2 | Eukaryotic translation elongation factor 1 β 2 | 0.1605 | 0.2408 | −0.132 | −0.541 | 0.409 |
| eif3b | Eukaryotic translation initiation factor 3, subunit B | 0.003 | 0.0093 | −0.675 | −1.184 | 0.509 |
| eif3l | Eukaryotic translation initiation factor 3, subunit L | 0.0207 | 0.049 | −2.033 | −2.534 | 0.501 |
| elmod1 | ELMO/CED-12 domain containing 1 | 0.0499 | 0.1005 | 3.628 | 3.281 | 0.347 |
| eno3 | Enolase 3 β muscle | 0.1049 | 0.1816 | 2.258 | 2.662 | 0.404 |
| fam49a | Family with sequence similarity 49, member A | 0.2345 | 0.323 | −0.424 | −0.71 | 0.286 |
| fam73a | Family with sequence similarity 73, member A | 0.1949 | 0.277 | 2.814 | 2.453 | 0.361 |
| foxn3 | Forkhead box N3 | 0.0011 | 0.0049 | 1.791 | 2.652 | 0.86 |
| gak | Cyclin G-associated kinase | 0.1605 | 0.2408 | 1.489 | 1.729 | 0.24 |
| galntl6 | Polypeptide N-acetylgalactosaminyltransferase 6-like | 0.1605 | 0.2408 | −0.78 | −0.527 | 0.253 |
| gpm6a | Glycoprotein M6A | 0.0148 | 0.0383 | −1.423 | −1.902 | 0.479 |
| hoxa1 | Homeobox A1 | 0.0379 | 0.0826 | −0.087 | −0.543 | 0.455 |
| hpcal1 | Hippocalcin-like 1 | 0.0019 | 0.0068 | −1.25 | −0.706 | 0.544 |
| hsp90ab1 | Heat shock protein 90 kDa α (cytosolic), class B member 1 | 0.6454 | 0.7322 | 0.749 | 0.511 | 0.238 |
| htatsf1 | HIV-1 Tat specific factor 1 | 0.3282 | 0.4302 | −0.067 | −0.312 | 0.245 |
| itgb1bp3 | Integrin β 1-binding protein 3 | 0.0019 | 0.0068 | 0.171 | −0.703 | 0.874 |
| krt12 | Keratin 12 | 0.1605 | 0.2408 | −1.214 | −0.926 | 0.288 |
| kti12 | KTI12 homolog, chromatin associated | 0.0104 | 0.0287 | −0.735 | −0.464 | 0.271 |
| lyzc | Lysozyme C-like | 0.5054 | 0.626 | −1.851 | −1.573 | 0.278 |
| mdh2 | Malate dehydrogenase 2, NAD (mitochondrial) | 0.0003 | 0.0025 | 0.685 | 0.075 | 0.61 |
| myh2 | Myosin, heavy chain 2, skeletal muscle, adult | 0.065 | 0.1185 | 0.189 | −0.517 | 0.706 |
| myh8 | Myosin, heavy chain 8, skeletal muscle, perinatal | 0.6454 | 0.7322 | 1.361 | 1.028 | 0.332 |
| myl3 | Myosin, light chain 3, alkali; ventricular, skeletal, slow | 0.0011 | 0.0049 | −0.07 | −1.05 | 0.98 |
| mylpf | Myosin, light chain, phosphorylatable, fast skeletal muscle | 0.1605 | 0.2408 | 0.388 | 0.629 | 0.241 |
| nbeal2 | Neurobeachin-like protein 2-like | 0.0002 | 0.0021 | 1.499 | 2.252 | 0.753 |
| ndufab1 | NADH dehydrogenase (ubiquinone) 1, α/β subcomplex, 1, 8 kDa | 0.0019 | 0.0068 | −1.46 | −1.911 | 0.451 |
| neb | Nebulin-like | 0.6454 | 0.7322 | 1.449 | 1.171 | 0.278 |
| ngly1 | Peptide-N(4)-(N-acetyl-β-glucosaminyl)asparagine amidase-like | 0.1949 | 0.277 | −0.644 | −0.903 | 0.259 |
| nono | Non-POU domain containing, octamer-binding | 0.0207 | 0.049 | 2.179 | 2.653 | 0.474 |
| nsa2 | Ribosome biogenesis homolog | 0.3282 | 0.4302 | −1.369 | −1.065 | 0.304 |
| peli1 | Pellino homolog 1 | 0.1304 | 0.2173 | 1.219 | 1.519 | 0.3 |
| pgc | Progastricsin (pepsinogen C) | 0.0499 | 0.1005 | −0.252 | 0.17 | 0.422 |
| pif1 | PIF1 5′-to-3′ DNA helicase homolog | 0.3823 | 0.4869 | −1.224 | −0.742 | 0.482 |
| pou6f1 | POU class 6 homeobox 1 | 0.065 | 0.1185 | 2.109 | 2.352 | 0.243 |
| psmc6 | Proteasome (prosome, macropain) 26S subunit ATPase 6 | 0.0002 | 0.0021 | −0.864 | −1.676 | 0.813 |
| ptprs | Protein tyrosine phosphatase, receptor type, S | 0.003 | 0.0093 | −0.292 | −1.007 | 0.715 |
| rho | Rhodopsin | 0.0207 | 0.049 | −1.563 | −2.307 | 0.744 |
| sfxn5 | Sideroflexin 5 | 0.0207 | 0.049 | 1.652 | 0.914 | 0.738 |
| slc3a2 | Solute carrier family 3 (activators of dibasic and neutral amino acid transport), member 2 | 0.1605 | 0.2408 | −1.286 | −1.658 | 0.372 |
| slc25a3 | Solute carrier family 25 (mitochondrial carrier; adenine nucleotide translocator), member 3 | 0.2786 | 0.3761 | −1.278 | −1.644 | 0.365 |
| sox21 | SRY (sex determining region Y)-box 21 | 0.1304 | 0.2173 | 2.188 | 1.858 | 0.33 |
| ssg1 | Steroid sensitive gene 1 | 0.003 | 0.0093 | 0.963 | 0.042 | 0.921 |
| taf15 | TAF15 RNA polymerase II, TATA box-binding protein (TBP)-associated factor, 68 kDa | 0.0207 | 0.049 | −0.299 | −0.56 | 0.262 |
| tbc1d13 | TBC1 domain family, member 13 | 0.0002 | 0.0021 | 3.606 | 2.747 | 0.86 |
| tmem147 | Transmembrane protein 147 | 0.0047 | 0.014 | −1.153 | −0.717 | 0.436 |
| tmsb4 | Thymosin β 4 | 0.1605 | 0.2408 | −0.683 | −0.995 | 0.312 |
| tnnc2 | Troponin C type 2 (fast) | 0.1049 | 0.1816 | 0.076 | −0.301 | 0.377 |
| tpm1 | Tropomyosin 1 (α) | 0.1049 | 0.1816 | 0.48 | 0.136 | 0.343 |
| tpt1 | Tumor protein, translationally-controlled 1 | 0.2345 | 0.323 | −1.09 | −1.407 | 0.317 |
| trim63 | Tripartite motif-containing 63 | 0.0281 | 0.0633 | 2.713 | 1.249 | 1.464 |
| ttn | Titin | 0.0499 | 0.1005 | 1.248 | 0.326 | 0.922 |
| ubn2 | Ubinuclein 2 | 0.0148 | 0.0383 | 1.903 | 2.178 | 0.274 |
| ush2a | Usherin-like, Usher syndrome 2A | 0.0019 | 0.0068 | 2.072 | 1.127 | 0.945 |
| ybx1 | Y box-binding protein 1 | 0.0019 | 0.0068 | −0.98 | −0.254 | 0.726 |
| zbtb46 | Zinc-finger and BTB domain containing 46 | 0.1949 | 0.277 | 3.319 | 3.549 | 0.23 |
| znf326 | Zinc-finger protein 326 | 0.2345 | 0.323 | −0.204 | −0.462 | 0.258 |
| znf451 | Zinc-finger protein 451-like | 0.065 | 0.1185 | 2.313 | 2.731 | 0.419 |
We found that across all branches, except for the Sp. bombifrons branch, morph-biased genes, on average, evolve more quickly than do unbiased genes (Fig. 3; we provide the PAML estimates of dN/dS and related parameters for each gene individually in Supplementary Table S3). Indeed, the best model for both morph-biased and unbiased genes was a free-ratio model (Table 2). Since both plastic and nonplastic species are included in this analysis, the free-ratio model therefore suggests that the advent of plasticity did not increase evolutionary rates. Although this pattern did not hold for the Sp. bombifrons branch––in which morph-biased genes evolved more slowly than did unbiased genes––the rate difference in this branch is opposite to that predicted by the first hypothesis above.
Fig. 3.

Morph-biased genes have higher ancestral rates of molecular evolution than do unbiased genes. Comparison of unbiased genes and morph-biased genes in average rate of molecular evolution (dN/dS) for each branch on the phylogeny; gray boxes indicate the two species in which alternative, environmentally induced tadpoles have evolved. The tree is unrooted and dN/dS values were calculated by summing estimates of nonsynonymous and synonymous substitutions across genes within a gene class.
Table 2.
Tests for variation in rate of molecular evolution between lineages
| Alternative evolutionary models for unbiased and morph-biased gene classesa | |||||
|---|---|---|---|---|---|
| Median dN/dS |
|||||
| Model | npb | Log likelihood (lnL) | Spea | Scaphiopus | Xenopus |
| Unbiased genes (n = 23) | |||||
| One ratio: Spea = Scaphiopus = Xenopus | 253 | −28,230.24 | 0.050 | 0.050 | 0.050 |
| Two ratio: Spea, Scaphiopus = Xenopus | 276 | −28,214.41 | 0.046 | 0.052 | 0.052 |
| Three ratio: Spea, Scaphiopus, Xenopus | 299 | −28,204.44 | 0.038 | 0.056 | 0.051 |
| Free ratio: nine unique branches | 456 | −29,048.09 | – | – | – |
| Morph-biased genes (n = 24) | |||||
| One ratio: Spea = Scaphiopus = Xenopus | 264 | −33,416.98 | 0.093 | 0.093 | 0.093 |
| Two ratio: Spea, Scaphiopus = Xenopus | 288 | −33,410.82 | 0.078 | 0.093 | 0.093 |
| Three ratio: Spea, Scaphiopus, Xenopus | 312 | −33,388.72 | 0.074 | 0.161 | 0.089 |
| Free ratio: nine unique branches | 437 | −32,558.86 | – | – | – |
| Comparisons of models using likelihood ratio testsc | |||||
|---|---|---|---|---|---|
| Models compared | npb | 2ΔlnL | P-value† | ||
| Unbiased genes | |||||
| One- versus two-ratio model | 23 | 31.66 | 0.11 | ||
| One- versus three-ratio model | 46 | 51.61 | 0.26 | ||
| Two- versus three-ratio model | 23 | 19.94 | 0.65 | ||
| Morph-biased genes | |||||
| One- versus two-ratio model | 24 | 12.31 | 0.98 | ||
| One- versus three-ratio model | 48 | 56.51 | 0.19 | ||
| Two- versus three-ratio model | 24 | 44.20 | 0.007 | ||
aThree-nested models were fit for the estimation of the nonsynonymous/synonymous rate ratio (dN/dS) for each gene independently and then grouped based on a gene’s classification.
bTotal number of parameters in the model (sum of separate analyses);
cLikelihood ratio tests were used to determine which model of evolution best described each gene class.
†Based on a chi-square distribution with d.f. = Δnp.
Although the free-ratio model was statistically the best model tested, we compared the more simplistic models (ratio models one, two, and three) to increase the likelihood of detecting differences among lineages (Table 2). For unbiased genes, we predicted that they evolve no differently between lineages, and our results confirmed this prediction (one-ratio model versus two-ratio model, 2ΔlnL = 32.50, P = 0.11; one-ratio model versus three-ratio model, 2ΔlnL = 52.82, P = 0.29). For biased genes, we predicted that the two-ratio or three-ratio model would be a better fit than the one-ratio model if morph-biased gene expression (i.e., plasticity) affects evolutionary rates. We found no support for this prediction (one-ratio model versus two ratio model, 2ΔlnL = 12.30, P = 0.97; one-ratio model versus three-ratio model, 2ΔlnL = 55.74, P = 0.15). Additionally, the one-ratio model estimates of the dN/dS ratio for each gene class revealed that morph-biased genes do indeed evolve more quickly than do unbiased genes (one-tailed Mann–Whitney U-test, P = 0.008). Thus, the higher evolutionary rate of morph-biased genes is an ancestral characteristic shared by all the species that we examined, regardless of whether these species did, or did not, express the carnivore–omnivore plasticity.
One might argue that the saturation of substitutions on the branches between Xenopus and Spea/Scaphiopus decrease the accuracy of our dN/dS estimates, or, that inadequate sampling of species reduces our ability to detect differences between, evolutionary models. To test these possibilities, we sequenced orthologs in a subset of seven of the above 47 genes from two distantly related spadefoot species, Pelodytes ibericus, and Pelobates cultripes. Doing so provided a fuller phylogeny with shorter branch lengths between species (Fig. 1c). We then compared rate estimates from the smaller tree to that of the larger tree. We found a strong positive relationship between estimates of dN/dS ratios for Spea, Scaphiopus, and Xenopus (R2 = 0.94, F1,19 = 294.79, P < 0.0001; Supplementary Fig. S1). Furthermore, twice the difference in likelihoods between the one- and two-ratio models (2ΔlnL) for the small and large trees were highly correlated (R2 = 0.90, F1,5 = 45.45, P = 0.001; Supplementary Fig. S2). Since 2ΔlnL is the test statistic for the likelihood ratio test, this high correlation suggests that the smaller phylogeny is equivalent in detecting differences among evolutionary models. Thus, overly large evolutionary distances and small sample size cannot account for our findings.
An analysis of expression variance within Sp. bombifrons revealed that morph-biased genes have higher variance than do unbiased genes, both when compared within a morph class (two-tailed Mann–Whitney U-tests, omnivores, P = 0.0072; carnivores, P = 0.000064) or the average of both morph classes (P = 0.000033; Fig. 4). This trend was also observed in Sc. couchii and Sc. holbrookii when we analyzed expression variance for 47 of the putative orthologs to Sp. bombifrons morph-biased and unbiased genes (Sc. holbrookii, P = 0.0077; Sc. couchii, P = 0.02665; Fig. 4). Thus, biased genes appear more variable in their expression.
Fig. 4.

Morph-biased genes have higher levels of expression variance than do unbiased genes. Comparison of unbiased genes and morph-biased genes in coefficient of variation in levels of gene expression in Sp. bombifrons (separately for carnivore-biased genes, omnivore-biased genes, and both types of morph-biased genes combined), Sc. couchii, and Sc. holbrookii. Notches represent confidence intervals around the medians (black horizontal line); outliers were included in the analysis but are not displayed here.
Discussion
Our results revealed that enhanced evolutionary rates of morph-biased genes relative to unbiased genes preceded the evolution of morph-biased gene expression (Fig. 3). This finding is consistent with the hypothesis that the faster evolutionary rates of morph-biased genes is a consequence, rather than a cause, of morph-biased expression and, thus, of the evolution of phenotypic plasticity. Although the notion that relaxed constraint precedes the evolution of phenotypic plasticity does not preclude the possibility that plasticity also increases relaxation of constraint, our results provide no support for this hypothesis. Indeed, morph-biased genes did not evolve any faster in Spea (the clade in which the plasticity has evolved) than in the other species (Table 2 and Fig. 3).
One might contend that an evolutionary precursor of the carnivore–omnivore plasticity exists in all the species we examined, in which case, the evolution of plasticity may have preceded the evolution of relaxed genetic constraint on these genes. Consistent with this hypothesis, preexisting plasticity exists in at least one of the non-Spea species that we tested; Sc. couchii facultatively change the length of their gut in response to alternative diets of detritus and shrimp (Ledón-Rettig et al. 2008). This finding is suggestive, because, in Spea, the carnivore morph can be induced by ingestion of shrimp, and this morph develops a much shorter gut (Ledón-Rettig et al. 2008). Yet, even if such preexisting plasticity is present in all the non-Spea species––and whether or not this is the case is unclear––the carnivore–omnivore plasticity is expressed much more frequently, and to a much greater degree, in Spea than in the other species (Ledón-Rettig et al. 2008; see also Ledón-Rettig and Pfennig, this volume). Consequently, if plasticity were a cause of relaxed constraint, then one should find that: (1) morph-biased genes evolve faster than unbiased genes, which we did find in all species, except for Sp. bombifrons (Fig. 3) and (2) morph-biased genes evolve faster in Spea than in the other four species, which we did not find (Table 2 and Fig. 3). Thus, our results are more consistent with the alternative hypothesis that the faster evolutionary rates of morph-biased genes are ancestral to the evolution of the carnivore–omnivore plasticity.
The prediction that plasticity enhances evolutionary rates stems from theory that was developed for genes with morph-specific expression (i.e., genes that are either “on” or “off” in their expression in each morph). In contrast, the genes analyzed in this study have relatively modest differences in expression (although analysis of wild-caught samples tended to produce larger differences between morphs; Leichty 2011), and none is expressed in a single morph only. Indeed, the degree to which such graded variation in levels of expression influence the strength of selection acting on a particular gene remains an open question (Snell-Rood et al. 2010; Connallon and Clark 2011).
Finding that enhanced evolutionary rates predate plasticity is important because such a pattern suggests that plasticity may arise when dispensable genes are co-opted for novel function in environmentally induced phenotypes. Indeed, morph-biased genes may have initially become morph-biased in their expression precisely because they were capable of more rapid evolution. Although we were unable to test this prediction directly using genetic techniques, a gene’s dispensability appears to be positively correlated not only with its rate of evolution (Hirsh and Fraser 2008), but also with its expression variance (Fraser et al. 2004). Therefore, if morph-biased genes are more dispensable (Hunt et al. 2010), they should have higher levels of expression variance, as has been suggested for sex-biased genes (Mank and Ellegran 2009). Consistent with this prediction, we found that morph-biased genes did indeed show higher levels of expression variance than did unbiased genes, both within a plastic species (Sp. bombifrons), and two nonplastic species (Sc. couchii and Sc. holbrookii) (Fig. 4). Thus, higher evolutionary rates in biased genes may reflect greater dispensability of these genes.
Yet, following the acquisition of morph-biased expression, a gene’s importance to fitness, and possibly its level of dispensability, should change. The relationship between fitness and gene expression will depend on whether and how any changes in gene expression affect the performance of the alternative phenotypes. Genes whose expression is critical to the novel, derived phenotype will be more constrained and should begin evolving more slowly. This could explain why morph-biased genes actually evolved more slowly in Sp. bombifrons (Fig. 3). Of all species in the genus Spea, Sp. bombifrons have evolved the highest propensity to produce alternative carnivore and omnivore tadpoles (D. Pfennig, unpublished data). Therefore, genes whose expression is critical to the proper functioning of these morphs may have come under stronger selection in Sp. bombifrons, which could explain the slower evolutionary rate of morph-biased genes in this species. In other words, the slower evolutionary rate of morph-biased genes in Sp. bombifrons is consistent with the alternative hypothesis that relaxed genetic constraint is a cause––and not a consequence––of the evolution of phenotypic plasticity.
The small sample size of biased genes makes it difficult to reach any general conclusions regarding the function(s) of genes with morph-biased expression. However, the genes showing the highest levels of differences in expression between morphs are known to be involved in dietary (pnlip, amy2a, and pm20d2), immune (pglyrp1 and mug1) and structural (col2a1 and col9a1) functions in other species (Table 1 and Fig. 2). Although none of these genes likely determines which morph an individual expresses (i.e., none likely regulate morph development), most (if not all) may be crucial in determining the functionality of morphs. If this is the case, future work in this system could seek to compare the fitness consequences of reduced expression for these genes in embryos and early tadpoles of species with (Sp. bombifrons and Sp. multiplicata), and without, alternative morphs (i.e., Sc. couchii and Sc. holbrookii). If genes with greater dispensability tend to be co-opted into specifying alternative, environmentally induced phenotypes, then knock-down of morph-biased genes should be more deleterious in Sp. bombifrons or Sp. multiplicata than in Sc. couchii or Sc. holbrookii.
In summary, relaxed genetic constraint may be a cause––and not a consequence—of the evolution of phenotypic plasticity. Generally, genes that experience relaxed genetic constraint may provide the raw material that enables phenotypic plasticity to evolve. Indeed, because plasticity may be critical in the origins of novel phenotypes (West-Eberhard 2003; Moczek et al. 2011), such genes may even be crucial for evolutionary innovation to arise.
Accession numbers
Data from arrays have been submitted to ArrayExpress, accession E-MEXP-3324. Assembled 454 data are available from NCBI under BioProject 72529 and all sequences used in the evolutionary rate analysis have been submitted to GenBank, accessions JN639633–JN639840 (Supplementary Table S4).
Funding
This research was supported by a grant from the National Institutes of Health (NIH) Director's New Innovator Award Program to K.P. (1DP2OD004436-01). D.P. is supported by the National Science Foundation (NSF DEB-1019479), and C. J. is supported by the NIH (A10-0915-004) and NSF (MCB-0920196).
Supplementary Data
Supplementary Data are available at ICB online.
Acknowledgments
We thank T. Cruickshank, S. Dhole, D. Kikuchi, C. Ledon-Rettig, A. Moczek, M. Noor, A. Rice, J. Santos, E. Snell-Rood, two anonymous referees, and Harold Heatwole for comments on the manuscript and D. Buchholz and S. Kulkarni for supplying samples of Pelodytes ibericus and Pelobates cultripes. We also thank I. Dworkin, A. Moczek, F. Nijhout, and M. Wund for organizing the symposium and inviting us to participate and SICB and the NSF for funding the symposium.
References
- Aubin-Horth N, Renn SCP. Genomic reaction norms: using integrative biology to understand molecular mechanisms of phenotypic plasticity. Mol Ecol. 2009;18:3763–80. doi: 10.1111/j.1365-294X.2009.04313.x. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc Ser B Meth. 1995;57:289–300. [Google Scholar]
- Connallon T, Clark AG. Association between sex-biased gene expression and mutations with sex-specific phenotypic consequences in Drosophila. Genome Biol Evolut. 2011;3:151–5. doi: 10.1093/gbe/evr004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA. A complete variorum edition. New York: Oxford University Press; 1930 (1999). The genetical theory of natural selection. [Google Scholar]
- Fraser HB, Hirsh AE, Giaever G, Kumm J, Eisen MB. Noise minimization in eukaryotic gene expression. Public Libr Sci Biol. 2004;2:e137. doi: 10.1371/journal.pbio.0020137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsh AE, Fraser HB. Protein dispensability and rate of evolution. Nature. 2008;411:1046–9. doi: 10.1038/35082561. [DOI] [PubMed] [Google Scholar]
- Hunt BG, Ometto L, Wurmb Y, Shoemaker D, Yi SV, Keller L, Goodisman MAD. Relaxed selection is a precursor to the evolution of phenotypic plasticity. Proc Natl Acad Sci USA. 2011;108:15936–41. doi: 10.1073/pnas.1104825108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt BG, Wyder S, Elango N, Werren JH, Zdobnov EM, Yi SV, Goodisman MAD. Sociality is linked to rates of protein evolution in a highly social insect. Mol Biol Evol. 2010;27:497–500. doi: 10.1093/molbev/msp225. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–66. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawecki TJ. Accumulation of deleterious mutations and the evolutionary cost of being a generalist. Am Nat. 1994;144:833–8. [Google Scholar]
- Kawecki TJ, Barton NH, Fry JD. Mutational collapse of fitness in marginal habitats and the evolution of ecological specialisation. J Evol Biol. 1997;10:407–29. [Google Scholar]
- Lahti DC, Johnson NA, Ajie BC, Otto SP, Hendry AP, Blumstein DT, Coss RG, Donohue K, Foster SA. Relaxed selection in the wild. Trends Ecol Evol. 2009;24:487–96. doi: 10.1016/j.tree.2009.03.010. [DOI] [PubMed] [Google Scholar]
- Ledón-Rettig CC, Pfennig DW. Emerging model systems in eco-evo-devo: the environmentally responsive spadefoot toad. Evol Dev. 2011;13:391–400. doi: 10.1111/j.1525-142X.2011.00494.x. [DOI] [PubMed] [Google Scholar]
- Ledón-Rettig CC, Pfennig DW, Nascone-Yoder N. Ancestral variation and the potential for genetic accommodation in larval amphibians: implications for the evolution of novel feeding strategies. Evol Dev. 2008;10:316–25. doi: 10.1111/j.1525-142X.2008.00240.x. [DOI] [PubMed] [Google Scholar]
- Leichty AR. [Chapel Hill (NC)]: University of North Carolina; 2011. The evolution of morph-biased genes in spadefoot toads [Master's thesis] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Mank JE, Ellegran H. Are sex-biased genes more dispensable? Biol Lett. 2009;5:409–12. doi: 10.1098/rsbl.2008.0732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moczek AP, Sultan SE, Foster S, Ledon-Rettig C, Dworkin I, Nijhout HF, Abouheif E, Pfennig DW. The role of developmental plasticity in evolutionary innovation. Proc Roy Soc Ser B. 2011;278:2705–13. doi: 10.1098/rspb.2011.0971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal C, Papp B, Lercher MJ. An intergrated view of protein evolution. Nat Rev Genet. 2006;7:337–48. doi: 10.1038/nrg1838. [DOI] [PubMed] [Google Scholar]
- Pfennig DW. The adaptive significance of an environmentally-cued developmental switch in an anuran tadpole. Oecologia. 1990;85:101–7. doi: 10.1007/BF00317349. [DOI] [PubMed] [Google Scholar]
- Pfennig DW, Wund MA, Snell-Rood EC, Cruickshank T, Schlichting CD, Moczek AP. Phenotypic plasticity’s impacts on diversification and speciation. Trends Ecol Evol. 2010;25:459–67. doi: 10.1016/j.tree.2010.05.006. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. 2010. R. Vienna, Austria: R Foundation for Statistical Computing.
- Schlichting CD. The role of phenotypic plasticity in diversification. In: DeWitt TJ, Scheiner SM, editors. Phenotypic plasticity: functional and conceptual approaches. Oxford, UK: Oxford University Press; 2004. pp. 191–200. [Google Scholar]
- Snell-Rood EC, Cash A, Han MV, Kijimoto T, Andrews A, Moczek AP. Developmental decoupling of alternative phenotypes: insights from the transcriptomes of horn-polyphenic beetles. Evolution. 2011;65:231–45. doi: 10.1111/j.1558-5646.2010.01106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell-Rood EC, Van Dyken JD, Cruickshank T, Wade MJ, Moczek AP. Toward a population genetic framework of developmental evolution: the costs, limits, and consequences of phenotypic plasticity. Bioessays. 2010;32:71–81. doi: 10.1002/bies.200900132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–5. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–12. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyken JD, Wade MJ. The genetic signature of conditional expression. Genetics. 2010;184:557–70. doi: 10.1534/genetics.109.110163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West-Eberhard MJ. Phenotypic plasticity and the origins of diversity. Ann Rev Ecol Sys. 1989;20:249–78. [Google Scholar]
- West-Eberhard MJ. New York: Oxford University Press; 2003. Developmental plasticity and evolution. [Google Scholar]
- Wu H, Kerr M, Cui X, Churchill G. MAANOVA: a software package for the analysis of spotted cDNA microarray experiments. In: Parmigiani G, Garrett E, Irizarry R, Zeger S, editors. The analysis of gene expression data. New York: Springer; 2003. pp. 313–41. [Google Scholar]
- Yang Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol. 1998;15:568–73. doi: 10.1093/oxfordjournals.molbev.a025957. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: a program package for phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yang Z, Swanson WJ. Codon-substitution models to detect adaptive evolution that account for heterogeneous selective pressures among site classes. Mol Biol Evol. 2002;19:49–57. doi: 10.1093/oxfordjournals.molbev.a003981. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.