

Abstract
Motivation: Next-generation sequencing greatly increases the capacity to detect rare-variant complex-trait associations. However, it is still expensive to sequence a large number of samples and therefore often small datasets are used. Given cost constraints, a potentially more powerful two-step strategy is to sequence a subset of the sample to discover variants, and genotype the identified variants in the remaining sample. If only cases are sequenced, directly combining sequence and genotype data will lead to inflated type-I errors in rare-variant association analysis. Although several methods have been developed to correct for the bias, they are either underpowered or theoretically invalid. We proposed a new method SEQCHIP to integrate genotype and sequence data, which can be used with most existing rare-variant tests.
Results: It is demonstrated using both simulated and real datasets that the SEQCHIP method has controlled type-I errors, and is substantially more powerful than all other currently available methods.
Availability: SEQCHIP is implemented in an R-Package and is available at http://linkage.rockefeller.edu/suzanne/seqchip/Seqchip.htm
Contacts: dajiang@umich.edu or sleal@bcm.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
There is solid evidence that rare variants play an important role in the etiology of complex traits (Bodmer and Bonilla, 2008; Cohen et al., 2004; Cohen et al., 2006; Fearnhead et al., 2004; Ji et al., 2008; Kryukov et al., 2007; Romeo et al., 2007; Romeo et al., 2009). The development and implementation of next-generation sequencing in genetic studies of complex traits have made possible the detection of rare variant associations. However, it is still very expensive to sequence a large number of samples at high coverage depth, which is necessary to accurately detect rare variants for association studies. Instead of sequencing all samples, a two-stage strategy can be applied, where a subset of the sample is sequenced to discover variants, and the identified variants are then genotyped in the remaining sample. Genotyping currently is considerably less expensive than sequencing, therefore given a fixed budget, a two-stage design can be much more powerful than a one-stage design, where only sequencing is used. This is because for the same financial expenditure, genetic information from a much larger number of samples can be extracted and analyzed in a two-stage study. In fact, the two-stage study design has been widely applied. Many rare variant associations were identified for a number of clinically important traits, including colorectal adenomas (Fearnhead et al., 2004), age-related macular degeneration (Raychaudhuri et al., 2011), lipids level (Sanna et al., 2011) and inflammatory bowel disease (Rivas et al., 2011).
In a two-stage study, candidate genes that were previously implicated in the etiology of complex trait through genome-wide association studies or functional studies may be sequenced to identify rare variants. It was demonstrated that sequencing >500 cases can uncover variants that can explain over 80% of the locus population attributable risk (Liu and Leal, 2010b). When more sophisticated methods are used for selecting samples, e.g. calculating expected number of causal variants for each individual and sequencing the individuals with the maximal counts (Edwards et al., 2010), it is potentially possible to enrich for causal variants using a smaller sample size than if samples are randomly selected. To make the two-stage-design cost effective for large scale studies, the commercially available exome chip can also be customized and up to 30 000 variants can be added. Alternatively, custom genotyping arrays can also be developed.
For the two-stage study design integrating sequencing and genotyping, it was shown that for a fixed number samples sequenced, sequencing only cases can be more powerful than sequencing a balanced number of cases and controls, for detecting associations with causal variants that are enriched in cases (Longmate et al., 2010). This is because a larger portion of low frequency causal variants can be identified by sequencing only cases. However, it has also been shown (Li and Leal, 2009) that type-I errors will be inflated for two-stage studies where only cases are sequenced to discover variants and naïve analysis is implemented, which directly integrates sequence and genotype data and compares aggregated variant frequencies between cases and controls. A straightforward correction is to exclude the sequence data used for variant discovery and use only the genotyped samples in the downstream association analysis (GSO – genotype samples only). However, this approach does not make full use of the available data and is underpowered, especially when a large portion (e.g. >50%) of affected individuals are sequenced for variant discovery. In Longmate et al. (2010), the authors suggested removing one variant carrier from the sequenced sample per variant nucleotide site (ROPS). When data are integrated using the ROPS method, rare variant association tests have controlled type-I error. However, this method is not without problems: (i) when there are many variant sites within the analyzed region, the ROPS method may result in removing a large number of variant carriers from the analysis, which is highly inefficient; and (ii) When covariates are included in the analysis, variant carriers may not be interchangeable under the null hypothesis, which makes it statistically invalid to apply the ROPS method.
Given the limitations of existing methods, it is desirable to have a statistical method that can correct for the bias induced by the two-stage study design and can be integrated with existing rare variant association methods (Bhatia et al., 2010; Han and Pan, 2010; Ionita-Laza et al., 2011; Liu and Leal, 2010a; Madsen and Browning, 2009; Neale et al., 2011; Price et al., 2010; Wu et al., 2011). In this article, SEQCHIP, a likelihood-based method for integrating sequence and genotype data was developed to correct for the bias created by two-stage study design. The method corrects for the variant genotypes obtained from sequencing, such that the corrected genotypes approximately follow the same distribution as that of the genotyped samples. The method can be used with any existing rare variant association tests that can analyze uncertain genotypes, e.g. imputed genotypes. In particular, the weighted sum statistics (WSS) have been extended to incorporate imputed genotypes ()(Zawistowski et al., 2010). The extensions of the test of the aggregated number of rare variants (ANRV; Morris and Zeggini, 2010) is straightforward where the genotype coding at each variant site can be replaced by ‘dosage’ (Guan and Stephens, 2008; Li et al., 2010; Zheng et al., 2011). The variable threshold (VT) test (Price et al., 2010) computes the ANRV test statistics for each frequency threshold and then uses their maximum as the test statistic. To control for multiple testing, p-values of VT are obtained empirically. The three tests are used to evaluate the performance of different data integration methods, i.e. ROPS, SEQCHIP and GSO.
We evaluated the performance of SEQCHIP through extensive simulations. Genetic data were generated using a realistic population genetic model (Kryukov et al., 2009). Disease phenotypes were simulated based upon parameters estimated from complex trait studies. We showed that when data are integrated using SEQCHIP, type-I errors for rare variant association tests are well controlled and the power is consistently higher than integrating data through ROPS (Longmate et al., 2010) or analyzing genotyped samples only (GSO) in all the scenarios that were examined.
As an application of the SEQCHIP method, we re-analyzed samples from a case control study of colorectal adenomas (Fearnhead et al., 2004). Sequence data were generated on five genes, which were previously implicated in the etiology of colorectal adenomas (Frayling et al., 1998; Kim et al., 2000; Lamlum et al., 2000; Lipton et al., 2003). In the study, 124 cases were sequenced to discover variants, and the identified variants were followed up and genotyped in 483 controls. Association analysis was carried-out by directly comparing total variant frequencies between sequenced cases and genotyped controls, and therefore the estimated p-values could be inflated. We re-analyzed this dataset, where sequence and genotype data were integrated using SEQCHIP and ROPS methods. The ANRV, WSS and VT tests were implemented to detect associations with rare variants. The association signal was statistically significant when the correction for the two-stage design was made using SEQCHIP, but not when ROPS was used. It should be noted that the GSO method could not be applied because all cases were sequenced and only controls were genotyped. The results verified the association with colorectal adenomas using valid methods and established that the SEQCHIP method is essential for integrating sequence and genotype data in cost-effective two-stage association studies.
2 METHODS
2.1 SEQCHIP method
We assume that there are NA affected individuals and NU unaffected individuals in the sample. Among the affected individuals, NS (i.e. individuals 1,2,…, NS) are sequenced to discover variants. An additional NG=NA−NS cases (i.e. individuals NS+1,…, NA) and NU controls are genotyped at the variant nucleotide sites that were uncovered in the sequence sample. The multi-site genotype for an individual i at the candidate gene locus is denoted by a vector, i.e.
where each entry k represents a site with di-allelic single-nucleotide variations, and xik,j is an indicator of whether the jth variant at site k is the minor allele. The total number of minor alleles observed at site k in the sequenced sample follows a truncated binomial distribution, i.e.
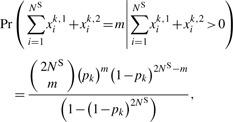 |
(1) |
where pk is the minor allele frequency (MAF) at site k.
It is clear from the above equation that the expected number of minor alleles at site k in the sequenced cases satisfies
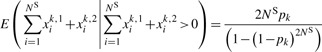 |
(2) |
Therefore, a naïve estimate of allele frequencies (i.e. the mean number of minor alleles per chromosome) will be inflated by a factor of 1/ck, where ck=1−(1−pk)2NS.
The idea behind the SEQCHIP method is to correct for the genotypes of the samples that are sequenced, such that corrected sequence genotypes approximately follow the same distribution as that of the genotyped samples. Specifically, auxiliary variables are defined for the samples that are sequenced, i.e.
where the marginal distribution for each entry element 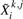 satisfies
satisfies
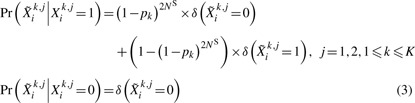 |
(3) |
It was shown in Supplementary Appendix SA and Supplementary Figure S1 that the modified genotype coding approximately follow the same distribution as that of the genotyped samples. The allele frequencies in (3) are generally unknown. In practice, they can be estimated from the data. Details for the MAF estimators are displayed in Supplementary Appendix SB.
Instead of analyzing the original dataset 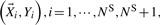
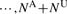 , the ‘new’ dataset with corrected sequence genotypes are analyzed, i.e.
, the ‘new’ dataset with corrected sequence genotypes are analyzed, i.e. 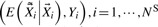 and
and 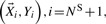
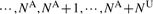
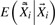 can be considered as the ‘dosage’ for the corrected genotypes conditional on the observed genotypes. In principle, it is also possible to use a mixture likelihood which integrates uncertainties in the modified genotype. However, when multiple rare variants are analyzed, calculating the mixture likelihood can be computationally intensive and numerically unstable. Therefore, the mixture likelihood approach is not pursued.
can be considered as the ‘dosage’ for the corrected genotypes conditional on the observed genotypes. In principle, it is also possible to use a mixture likelihood which integrates uncertainties in the modified genotype. However, when multiple rare variants are analyzed, calculating the mixture likelihood can be computationally intensive and numerically unstable. Therefore, the mixture likelihood approach is not pursued.
In principle, all rare variant association tests that can analyze uncertain genotypes (e.g. imputed genotypes) can be directly applied to analyze the SEQCHIP corrected genotypes. Score tests can be implemented, and standard permutation procedure can be used to obtain empirical p-values.
2.2 ROPS and GSO method
We compared the SEQCHIP method with the ROPS (Longmate et al., 2010) and GSO methods. Specifically, the ROPS method removes one randomly chosen variant carrier for each uncovered variant site. Therefore, if there are K variant sites uncovered in the sample, a total of K samples will be removed and NA+NU−K samples will be analyzed following the ROPS approach (Longmate et al., 2010). For the GSO method, all individuals that are sequenced are removed from subsequent association analyses.
2.3 Generation of genetic and phenotypic data
We simulated genetic data using a four-parameter population genetic model (Adams and Hudson, 2004; Kryukov et al., 2009). The model was estimated using sequence data from the Ottawa Obesity Study (Ahituv et al., 2007), and incorporates both demographic change (i.e. bottleneck and exponential expansion) and purifying selection, which are believed to affect rare variant site frequency spectrums. Details for the population genetics model parameters can be found in (Kryukov et al., 2009). Hardy–Weinberg equilibrium was assumed for the general population. Phenotype data were generated according to the following logistic regression model, i.e.
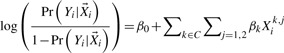 |
Under the alternative hypothesis, two types of phenotypic models were considered. In the first model, a certain proportion of rare variants sites C are randomly selected to be causal, and affect disease status. The power was investigated when 10, 70 and 90% of the variant sites are causal. In the second model, the causality of variants is determined by the selection coefficient. Power was evaluated, where variants with selection coefficients >10−4, 10−3 and 10−2 are causal and affect the disease risk.
Under both models, it is assumed that each causal variant has an odds ratio of 3 and non-causal variants have an odds ratio of 1, i.e.
as suggested by Bodmer and Bonilla (2008). Under the null hypothesis, all βk′s are set to be 0.
2.4 Evaluation of type-I errors and power
Type-I errors of rare variant association tests were evaluated under four different data integration strategies, i.e. (i) naïve method, which directly combines data without corrections,;ii) SEQCHIP, (iii) ROPS and (iv) GSO. Scenarios were considered where 500 cases/500 controls and 1500 cases/1500 controls were analyzed. For each case control dataset, we considered study designs where 10, 50 and 90% of the cases were sequenced to discover variants, and the identified rare variants (with MAF < 1%) were followed-up and genotyped in the remaining samples. For the data integration strategies under which the rare variant association tests have controlled type-I errors, (i.e. SEQCHIP, ROPS and GSO), the power was also compared. One sided tests were performed, i.e. the alternative hypothesis that there is an increased number of rare causal alleles in cases is tested. For the ANRV method, statistical significance was obtained analytically, whereas the p-values for WSS and VT were obtained empirically using 2000 permutations. The power and type-I errors for different tests were evaluated using 10 000 replicates.
2.5 Evaluation of study designs
We also evaluated different two-stage study designs that sequence a portion of the sample to discover variants and genotype identified variants in the remaining samples. Specifically, by applying the SEQCHIP method and performing rare variant association testing using ANRV, WSS and VT, we compared the study design of sequencing only cases with that of sequencing an equal number of cases and controls, and combining sequence and genotype data via meta-analysis methods. p-values for ANRV were evaluated analytically, whereas that for WSS and VT were calculated using 2000 permutations. For meta-analysis, p-values were transformed to Z-score statistics, which were then weighted by the square root of the sample size and combined (Munafo and Flint, 2004). Power at each sequence sample size was obtained based upon 10 000 replicates. A significance level of α=0.05 is used.
2.6 The analysis of sequence dataset of colorectal adenomas
In the colorectal adenoma dataset, five genes were first sequenced in cases for variant discovery and the identified variants were then genotyped in controls. In this article, we re-analyze the dataset using two valid data integration methods, i.e. ROPS and SEQCHIP. ANRV, WSS and VT tests were applied to detect associations with rare variants.
3 RESULTS
3.1 Evaluation of type-I errors
Type-I errors of the ANRV test were displayed in (Table 1) and that of the WSS and VT tests are shown in (Supplementary Table S1), for different strategies of integrating sequence and genotype data. In the naïve analyses where no corrections were made for the sequence data, type-I errors for all tests were highly inflated. In some scenarios, the type-I error can be >30% under a significance level of α = 0.05. For example, when 90% of the cases were sequenced in a sample of 1500 cases and 1500 controls, the type-I errors for the ANRV, WSS and VT tests are, respectively, 30.3, 29.9 and 60.4%.
Table 1.
Type-I error for rare variant association test ANRV
| Sample | Percentage of | Type-I errors for ANRVb |
|||
|---|---|---|---|---|---|
| sizea | cases sequenced | Naïve | SEQCHIP | ROPS | GSO |
| 1000 | 0.1 | 0.158 | 0.041 | 0.037 | 0.042 |
| 1000 | 0.5 | 0.217 | 0.035 | 0.022 | 0.048 |
| 1000 | 0.9 | 0.284 | 0.037 | 0.015 | 0.051 |
| 3000 | 0.1 | 0.149 | 0.043 | 0.037 | 0.043 |
| 3000 | 0.5 | 0.234 | 0.038 | 0 .021 | 0.049 |
| 3000 | 0.9 | 0.303 | 0.036 | 0.018 | 0.047 |
Scenarios were considered when sequence and genotype data were combined using the naïve method, SEQCHIP method and ROPS method, or when only genotype samples are analyzed.
aTotal sample size with equal number of cases and controls.
bType-I error was evaluated under a significance level of α=0.05. p-values for ANRV were obtained analytically.
When sequence and genotype data were combined using SEQCHIP and ROPS, the type-I errors for rare variant association tests were controlled. However, the correction by ROPS can be overly conservative. For example, the type-I error for ANRV, WSS and VT are, respectively, 0.022 (with 95% CI [0.019, 0.025]), 0.015 (with 95% CI [0.013, 0.017]) and 0.017 (with 95% CI [0.014, 0.020]) when 50% cases were sequenced in a sample of 500 cases and 500 controls. For the same scenario, if the SEQCHIP method is used, the type-I error for the three tests are also conservative, but to a lesser extent (i.e. ANRV: 0.035 with 95% CI [0.031, 0.039], WSS: 0.036 with 95% CI [0.032, 0.040] and VT: 0.043 with 95% CI [0.039, 0.047]).
3.2 Power comparisons
We compared the performance of different data integration methods, for which rare variant association tests have controlled type-I errors, i.e. SEQCHIP, ROPS and GSO. SEQCHIP method outperforms other methods in most scenarios. In some scenarios the power improvement can be as high as 30%. For example, when variant causality is determined by fitness, (i.e. variants with selection coefficients >10−4 are causal with effects βk=log(3), k∈C), if 90% of the cases were sequenced in a sample of 1500 cases and 1500 controls, the power for VT test is 76.5% (Fig. 1f). In the same scenario, if data are integrated by ROPS and GSO methods, the power for VT is only 55.6 and 34.1%, respectively.
Fig. 1.

Comparisons of SEQCHIP, ROPS and GSO. It is assumed that variants with selection coefficients >10−4 are causal. ANRV, WSS and VT are used to analyze data. p-values for ANRV were obtained analytically, whereas the p-values for the other methods were obtained using 5000 permutations. The power was evaluated under a significance level of α=0.05 using 10 000 replicates
ROPS methods can be conservative for correcting the genotypes of low frequencies variants. This is reflected by two observations: (i) when a small number of cases are sequenced and the data are integrated using ROPS, the power may be lower than when only genotype samples are analyzed (i.e. GSO method); (ii) when a larger portion cases are sequenced, the power for rare variant association tests may decrease. For example, when causality is assumed to be independent of fitness, (i.e.70% of the variants are causal with effect βk =log(3), k∈C), if 90% of the cases are sequenced for a cohort of 1500 cases and 1500 controls and ROPS is used to integrate the data, the power for WSS is 61.6%, which is lower than the power (67.4%) when 50% of the cases are sequenced (Fig. 2e). This is because for the ROPS method, by taking out one variant carrier from the sequenced cases, a greater proportion of the carriers are removed from the sequenced cases than from the entire sample, and the variant frequencies may be slightly underestimated. Specifically, when NS cases are sequenced to discover variants and NG cases and NU controls are genotyped, the variant frequencies in cases are estimated by 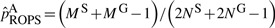 , where MS and MG are the number of minor alleles observed in sequenced and genotyped cases for a given site. On the other hand, the variant frequencies in genotyped cases are estimated by
, where MS and MG are the number of minor alleles observed in sequenced and genotyped cases for a given site. On the other hand, the variant frequencies in genotyped cases are estimated by 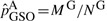 . It is easy to verify numerically that when variant frequencies are low,
. It is easy to verify numerically that when variant frequencies are low, 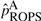 can be smaller in value than
can be smaller in value than 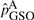 . For example, when only one variant is observed in the sequenced cases for a given site,
. For example, when only one variant is observed in the sequenced cases for a given site, 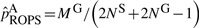 , which is smaller than
, which is smaller than 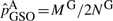 . We also proved rigorously using probability theory (Supplementary Appendix SA), that when SEQCHIP or ROPS are used to integrate data, the variant frequencies can be slightly underestimated, i.e.
. We also proved rigorously using probability theory (Supplementary Appendix SA), that when SEQCHIP or ROPS are used to integrate data, the variant frequencies can be slightly underestimated, i.e. 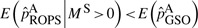 . Therefore, although more causal variants may be uncovered by sequencing additional samples and sample size can be increased by integrating sequence samples, the accumulated downward biases may mitigate the variant frequencies between cases and controls, and reduce the power for rare variant association tests.
. Therefore, although more causal variants may be uncovered by sequencing additional samples and sample size can be increased by integrating sequence samples, the accumulated downward biases may mitigate the variant frequencies between cases and controls, and reduce the power for rare variant association tests.
Fig. 2.

Comparisons of SEQCHIP, ROPS and GSO. It is assumed that 70% of variants are randomly chosen to be causal. ANRV, WSS and VT are used to analyze data. p-values for ANRV were obtained analytically, whereas the p-values for the other methods were obtained using 5000 permutations. The power was evaluated under a significance level of α=0.05 using 10 000 replicates
The GSO method only analyzes genotyped samples, and therefore can be vastly underpowered when a large portion of the cases are sequenced. For example, under the model assuming that variant causality is determined by fitness (i.e. variants with selection coefficients >10−4 are causal with effects βk=log(3), k∈C), for a sample of 1500 cases and 1500 controls, if 90% of the cases are sequenced for variant discoveries, the power of WSS is 38.2%. However, when sequence and genotype data are combined using SEQCHIP or ROPS, the power for WSS is, respectively, 73.5 and 58.2% (Fig. 1f). Due to the conservativeness in the estimation of allele frequencies, when a smaller proportion of cases are sequenced, e.g. 10 or 50%, the GSO method can have greater power than the ROPS method. For example, in a sample of 1500 cases and 1500 controls, when 50% of the cases are sequenced, the power for WSS is 61.7% if data are integrated using ROPS, and 66.2% if GSO method is used and only genotype data are analyzed (Fig. 1e).
The results of the power analyses for alternative selection coefficient cutoffs (i.e. 10−2 and 10−3) can be found in Supplementary Figures S2 and S3. Additionally Supplementary Figures S4 and S5 display the results of the power analysis when 10 and 90% of the variants are randomly chosen to be causal. For these additional power comparisons, the relative performances of different data integration methods remain unchanged. Among the three rare variant association methods that were examined, there is not a single method that is consistently the most powerful. The advantage of different methods over each other is small. This is concordant with other existing reviews on rare variant association analyses methods (Basu and Pan, 2011; Ladouceur et al., 2012).
3.3 Comparison of study designs
Figure 3 displays the power of the two study designs which sequence a portion of the samples to discover variants and genotype the identified variants in the remaining sample. It is demonstrated that when only a small number of samples are sequenced, sequencing only cases is more powerful than sequencing a balanced number of cases and controls. However, as the number of sequenced samples increases, the advantage of sequencing only cases diminishes because the data integration methods may be conservative for estimating rare variant frequencies. It is more powerful to sequence a balanced number of cases and controls to discover variants and then combine the sequence and genotype data by meta-analysis methods. The power comparisons remain similar when the ANRV (Supplementary Fig. S6) and WSS (Supplementary Fig. 7) tests are implemented.
Fig. 3.

Power comparison of two-stage study designs. Results are shown when only cases are sequenced for variant discovery (solid lines with circles) and when a balanced number of cases and controls (dashed lines with triangles) are sequenced. (a) displays results when 70% of the variants are randomly chosen to be causal, and (b) displays results where variants with selection coefficients >10−4 are causal. Scenarios are examined when different numbers of samples are sequenced from 1500 cases and 1500 controls. When only cases are sequenced, SEQCHIP is used to correct for the bias. When an equal number of cases and controls are sequenced, sequence and genotype data are analyzed separately and meta-analysis is used to combine the results. For all scenarios VT is used to detect rare variant associations
As a comparison, we also calculated the power when all samples are sequenced for the study (Supplementary Tables S2 and S3). The results can be viewed as the ‘maximal’ achievable power if there are no budgetary constraints and the entire sample can be sequenced.
3.4 Analysis of colorectal adenoma dataset
We jointly analyzed variants in five genes i.e. APC, AXIN1, CTNNB1, hMLH1 and hMSH2. A total of 12 missense mutations were identified through sequencing 124 cases with colorectal cancer, and the identified variants were then genotyped in 483 controls. Three analyses were performed for the dataset.
First we combined sequence and genotype data with SEQCHIP method, and analyzed the resulting dataset with the ANRV, WSS and VT. One-sided tests were performed, which tests for the enrichment of rare variant alleles in colorectal adenomas patients. The p-values are, respectively, given by pANRVSEQCHIP=0.034, pWSSSEQCHIP=0.023 and pVTSEQCHIP=0.106 (Table 2). The p-values for ANRV and WSS are significant. Second, we combined sequence and genotype dataset by the ROPS method. No significant results were observed (i.e. pANRVROPS=0.080, pWSSROPS=0.088 and pVTROPS=0.144).
Table 2.
The analysis of colorectal adenoma dataset
| Corrections |
p-values for rare variant association tests |
||
|---|---|---|---|
| ANRVa | WSSb | VTb | |
| SEQCHIP | 0.034 | 0.023 | 0.106 |
| ROPS | 0.080 | 0.088 | 0.144 |
| Naïve | 0.004 | 0.005 | 0.005 |
ap-values for ANRV test were obtained analytically.
bp-Values for WSS and VT tests were obtained by 10 000 permutations.
Finally, for comparison purposes, the dataset was also analyzed under the naïve strategy, where sequence and genotype data are directly combined without corrections. The p-values are clearly biased, for all tests (i.e. pANRVnaive=0.004, pWSSnaive=0.005 and pVTnaive=0.005). This is concordant with our theoretical expectations and observations from simulated dataset. We also analyzed each gene individually. However, each gene alone contains too few rare variants and the analyses were not significant (data not shown).
4 DISCUSSION
In this article, we developed a data integration method for two-stage case control studies where a portion of cases are sequenced to discover variants, and the identified variants are genotyped in the remaining sample. The SEQCHIP method performs a correction on the variant genotypes observed in sequenced cases, such that the corrected sequence genotypes follow approximately the same distribution as that of the genotyped samples. The integrated dataset can be analyzed by all existing rare variant association tests that can handle genotypes with uncertainties (e.g. imputed genotypes). SEQCHIP can also be used with regression-based methods for detecting primary or secondary traits associations (Lin and Zeng, 2009; Liu and Leal, 2011), where confounders such as population substructures can be controlled. Through extensive simulations, we demonstrate that when SEQCHIP is used to integrate sequence and genotype data, all rare variant association tests have controlled type-I errors. The power can be substantially improved compared with using other data integration strategies, i.e. ROPS and GSO.
The method is mainly developed for combining sequence and genotype data when only cases were sequenced for variant discoveries. A popular alternative two-stage study design is to sequence a combination of selected cases and controls for variant discovery, and genotype the identified variants in the rest of the samples. Under this study design, sequenced and genotyped samples can be separately analyzed and combined using standard meta-analyses methods, which will have controlled type-I error rates. When both cases and controls are sequenced, protective variants for the disease phenotype may be uncovered with higher probability (Rivas et al., 2011). For a two-stage study that combines sequence and genotype data, given a small fixed number of samples that are sequenced, sequencing only cases can be more powerful for detecting causal variant associations than sequencing a balanced number of cases and controls.
In practice, it is of interest to know the optimal fraction of cases to sequence to maximize power. Although sequencing a larger number of samples allows discovering a higher number of variant sites, it does not necessarily lead to improved power. This is because the frequencies of very rare variants identified in sequenced cases can be slightly underestimated by ROPS and SEQCHIP methods, which reduces power. The optimal number depends on the underlying disease model, the size of the cohort, and the proportion of the cases that are sequenced, which will need to be examined on a case by case basis.
Although the cost of sequencing is quickly dropping, genotyping still has a clear cost advantage. The two-stage study design of sequencing cases and genotyping the remaining sample allows extracting genetic information from a much larger number of samples, which can be more powerful than one stage study design given a fixed cost constraint. SEQCHIP is a very useful method for integrating data in a two-stage study design and will greatly accelerate the process of identifying variants involved in complex trait etiologies.
Supplementary Material
5 ACKNOWLEDGEMENTS
The authors thank Dr Shamil Sunyaev for sharing the simulated genetic datasets; and also thank Dr Walter Bodmer and Ms. Nicola Fearnhead for sharing the colorectal adenomas dataset.
Funding: National Institutes of Health [HL102926, MD005964, HG006493 to S.M.L. and MH084676 to Shamil Sunyaev]; Cancer Research, UK.
Conflict of Interest: none declared.
REFERENCES
- Adams A.M., Hudson R.R. Maximum-likelihood estimation of demographic parameters using the frequency spectrum of unlinked single-nucleotide polymorphisms. Genetics. 2004;168:1699–1712. doi: 10.1534/genetics.104.030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahituv N., et al. Medical sequencing at the extremes of human body mass. Am. J. Hum. Genet. 2007;80:779–791. doi: 10.1086/513471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S., Pan W. Comparison of statistical tests for disease association with rare variants. Genet. Epidemiol. 2011;35:606–619. doi: 10.1002/gepi.20609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia G., et al. A covering method for detecting genetic associations between rare variants and common phenotypes. PLoS Comput. Biol. 2010;6:e1000954. doi: 10.1371/journal.pcbi.1000954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer W., Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J.C., et al. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305:869–872. doi: 10.1126/science.1099870. [DOI] [PubMed] [Google Scholar]
- Cohen J.C., et al. Multiple rare variants in NPC1L1 associated with reduced sterol absorption and plasma low-density lipoprotein levels. Proc. Natl Acad. Sci. USA. 2006;103:1810–1815. doi: 10.1073/pnas.0508483103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards T.L., et al. Enriching targeted sequencing experiments for rare disease alleles. Bioinformatics. 2010;27:2112–2118. doi: 10.1093/bioinformatics/btr324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnhead N.S., et al. Multiple rare variants in different genes account for multifactorial inherited susceptibility to colorectal adenomas. Proc. Natl Acad. Sci. USA. 2004;101:15992–15997. doi: 10.1073/pnas.0407187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frayling I.M., et al. The APC variants I1307K and E1317Q are associated with colorectal tumors, but not always with a family history. Proc. Natl Acad. Sci. USA. 1998;95:10722–10727. doi: 10.1073/pnas.95.18.10722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y., Stephens M. Practical issues in imputation-based association mapping. PLoS Genet. 2008;4:e1000279. doi: 10.1371/journal.pgen.1000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han F., Pan W. A data-adaptive sum test for disease association with multiple common or rare variants. Hum. Hered. 2010;70:42–54. doi: 10.1159/000288704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionita-Laza I., et al. A new testing strategy to identify rare variants with either risk or protective effect on disease. PLoS Genet. 2011;7:e1001289. doi: 10.1371/journal.pgen.1001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji W., et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 2008;40:592–599. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.C., et al. The E-cadherin gene (CDH1) variants T340A and L599V in gastric and colorectal cancer patients in Korea. Gut. 2000;47:262–267. doi: 10.1136/gut.47.2.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryukov G.V., et al. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am. J. Hum. Genet. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryukov G.V., et al. Power of deep, all-exon resequencing for discovery of human trait genes. Proc. Natl Acad. Sci. USA. 2009;106:3871–3876. doi: 10.1073/pnas.0812824106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladouceur M., et al. The empirical power of rare variant association methods: results from sanger sequencing in 1,998 individuals. PLoS Genet. 2012;8:e1002496. doi: 10.1371/journal.pgen.1002496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamlum H., et al. Germline APC variants in patients with multiple colorectal adenomas, with evidence for the particular importance of E1317Q. Hum. Mol. Genet. 2000;9:2215–2221. doi: 10.1093/oxfordjournals.hmg.a018912. [DOI] [PubMed] [Google Scholar]
- Li B., Leal S.M. Discovery of rare variants via sequencing: implications for the design of complex trait association studies. PLoS Genet. 2009;5:e1000481. doi: 10.1371/journal.pgen.1000481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., et al. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D.Y., Zeng D. Proper analysis of secondary phenotype data in case-control association studies. Genet. Epidemiol. 2009;33:256–265. doi: 10.1002/gepi.20377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton L., et al. Germline mutations in the TGF-beta and Wnt signalling pathways are a rare cause of the "multiple" adenoma phenotype. J. Med. Genet. 2003;40:e35. doi: 10.1136/jmg.40.4.e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.J., Leal S.M. A novel adaptive method for the analysis of next-generation sequencing data to detect complex trait associations with rare variants due to gene main effects and interactions. PLoS Genet. 2010a;6:e1001156. doi: 10.1371/journal.pgen.1001156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.J., Leal S.M. Replication strategies for rare variant complex trait association studies via next-generation sequencing. Am. J. Hum. Genet. 2010b;87:790–801. doi: 10.1016/j.ajhg.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.J., Leal S.M. A flexible likelihood framework for detecting associations with secondary phenotypes in genetic studies using selected samples: application to sequence data. Eur. J. Hum. Genet. 2011;20:449–456. doi: 10.1038/ejhg.2011.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longmate J.A., et al. Three ways of combining genotyping and resequencing in case-control association studies. PLoS ONE. 2010;5:e14318. doi: 10.1371/journal.pone.0014318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen B.E., Browning S.R. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5:e1000384. doi: 10.1371/journal.pgen.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris A.P., Zeggini E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet. Epidemiol. 2010;34:188–193. doi: 10.1002/gepi.20450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munafo M.R., Flint J. Meta-analysis of genetic association studies. Trends Genet. 2004;20:439–444. doi: 10.1016/j.tig.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Neale B.M., et al. Testing for an unusual distribution of rare variants. PLoS Genet. 2011;7:e1001322. doi: 10.1371/journal.pgen.1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price A.L., et al. Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet. 2010;86:832–838. doi: 10.1016/j.ajhg.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S., et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat. Genet. 2011;43:1232–1236. doi: 10.1038/ng.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas M.A., et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 2011;43:1066–1073. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo S., et al. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat. Genet. 2007;39:513–516. doi: 10.1038/ng1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo S., et al. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J. Clin. Invest. 2009;119:70–79. doi: 10.1172/JCI37118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna S., et al. Fine mapping of five loci associated with low-density lipoprotein cholesterol detects variants that double the explained heritability. PLoS Genet. 2011;7:e1002198. doi: 10.1371/journal.pgen.1002198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M.C., et al. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawistowski M., et al. Extending rare-variant testing strategies: analysis of noncoding sequence and imputed genotypes. Am. J. Hum. Genet. 2010;87:604–617. doi: 10.1016/j.ajhg.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J., et al. A comparison of approaches to account for uncertainty in analysis of imputed genotypes. Genet. Epidemiol. 2011;35:102–110. doi: 10.1002/gepi.20552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.