

Abstract
Current cancer therapies exploit either differential metabolism or targeting to specific individual gene products that are overexpressed in aberrant cells. The work described herein proposes an alternative approach—to specifically target combinations of cell-surface receptors using heteromultivalent ligands (“receptor combination approach”). As a proof-of-concept that functionally unrelated receptors can be noncovalently cross-linked with high avidity and specificity, a series of heterobivalent ligands (htBVLs) were constructed from analogues of the melanocortin peptide ligand ([Nle4, DPhe7]-α-MSH) and the cholecystokinin peptide ligand (CCK-8). Binding of these ligands to cells expressing the human Melanocortin-4 receptor and the Cholecystokinin-2 receptor was analyzed. The MSH(7) and CCK(6) were tethered with linkers of varying rigidity and length, constructed from natural and/or synthetic building blocks. Modeling data suggest that a linker length of 20–50 Å is needed to simultaneously bind these two different G-protein coupled receptors (GPCRs). These ligands exhibited up to 24-fold enhancement in binding affinity to cells that expressed both (bivalent binding), compared to cells with only one (monovalent binding) of the cognate receptors. The htBVLs had up to 50-fold higher affinity than that of a monomeric CCK ligand, i.e., Ac-CCK(6)-NH2. Cell-surface targeting of these two cell types with labeled heteromultivalent ligand demonstrated high avidity and specificity, thereby validating the receptor combination approach. This ability to noncovalently cross-link heterologous receptors and target individual cells using a receptor combination approach opens up new possibilities for specific cell targeting in vivo for therapy or imaging.
INTRODUCTION
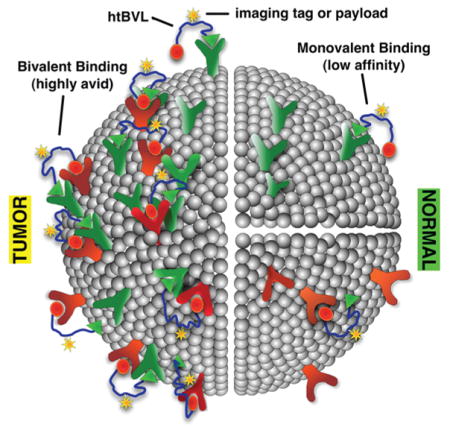
Multivalent ligands (MVLs) are characterized by the simultaneous binding of an entity that displays multiple molecular recognition elements to multiple receptors/epitopes. Such multiple interactions significantly enhance the affinity (avidity) of an agent whose constituent ligands may otherwise exhibit weak binding.1,2 For example, we demonstrated earlier that linear homobivalent and trivalent ligands of the MSH pharmacophore bound with higher affinities than the corresponding monovalent ligands to the human melanocortin-4 receptor (hMC4R).3,4 Simultaneous targeting of multiple cell-surface receptors with homomultivalent ligands (hmMVLs) has been the subject of intensive research in recent years.2,5–7 However, hmMVLs cannot differentiate among different binding modes, such as the chelation effect, the receptor clustering effect, or the statistical effect.8 Further, although homomultivalent binding can give rise to tissue-level specificity, there is no inherent specificity at the molecular binding event. Thus, its utility in tumor-specific targeting can be limited.7,9,10 The clustering effect is the only binding paradigm that takes advantage of protein expression combinations. A multivalent construct with at least two distinct binding pharmacophores spaced adequately apart is needed to demonstrate noncovalent cross-linking (i.e., clustering) of receptors.
We hypothesize that, by combining one or more copies of different binding moieties into a heteromultivalent ligand (htMVL), it should be possible to create compounds that will specifically and selectively bind to cells bearing the appropriate combination of complementary cell-surface receptors (Figure 1A).11,12 Further, we have manually curated the Agilent whole genome array and have identified 2408 genes encoding for cell-surface proteins (Morse et al., unpublished) that can potentially be targeted. The number of possible combinations of n genes organized into sets of x is [n!/(n − x)!] ÷ x!, indicating that there are ~2.9 × 106 potential two-gene combinations and ~2.3 × 109 potential three-gene combinations (and thus potential targets). Thus, it is increasingly likely that specific combinations of cell-surface proteins can be found that are expressed on a target (cancer) cell but not on any healthy cell (e.g., see ref 13 for gene expression profiling-based identification of receptor combination targets in pancreatic cancer). To exploit this potential, these combinations will have to be targeted with heteromultivalent ligands carrying imaging and/or therapeutic payloads (Figure 1A). Such constructs could also be used to investigate ligand–receptor or cell–cell interactions, investigate cellular signaling complexes such as “immune synapse”, mimic the multivalent binding of antibodies to antigens, and use them for combined in vivo imaging and therapeutic targeting.5
Figure 1.

(A) Receptor combination approach: predicted avidities of an htBVL binding to cells expressing one or two complementary receptors, and the number of possible combinations for potential targeting. High avidity and specificity can be expected for heterobivalent binding. (B) GPCR modeling. The two receptors can pack in any number of orientations, and the distance span between the two binding pockets could be up to 100 Å long depending on the dimer packing, domain swapping, or lipid rafts involvement (also see refs 3, 14, 18, 22, and 23).
In this report, we provide proof-of-principle studies, i.e., htMVL-directed noncovalent cross-linking of desired cell-surface proteins with high avidity and specificity. Further, we unambiguously prove the utility of this “receptor combination approach” in specific targeting with in vitro imaging of a fluorescently tagged htBVL. We present here the design, synthesis, and biological evaluation of a set of synthetic htBVLs composed of ligand motifs that target the human melanocortin-4 receptor (hMC4R) and the cholecystokinin-2 receptor (CCK-2R). For this purpose, MSH(7), a seven residue fragment of NDP-α-MSH ([Nle4, DPhe7]- α-MSH) and CCK(6), a six residue fragment of [Nle28, Nle31]CCK-8, respectively, were used (Scheme 1, inset). These ligands were conjoined with linkers of varying rigidity and length, which ranged from 18 to 148 atoms and spanned a distance of up to ca. 100 Å.3,4,14 The htBVLs were tested for their ability to compete against europium-labeled “lanthaligands”15 (Eu-DTPA-NDP-α-MSH and Eu-DTPA-CCK-8) for binding to cells that expressed one or both complementary receptors.
Scheme 1a. Synthetic Route for Heterobivalent Ligands (htBVLs).

aThe inset shows the sequence of MSH(7) and CCK(6) ligands and the structure of the PEGO linker (20 atoms). The reagents for each step are as follows: (i) Fmoc-amino acid-OH (3 equiv), HOCt (3 equiv), diisopropylcarbodiimide (3 equiv); (ii) 50% Ac2O in pyridine; (iii) 20% piperidine/DMF; (iv) (a) diglycolic anhydride (10 equiv) in DMF, (b) CDI (20 equiv) in anhyd. DMF, (c) 4,7,10-trioxa-1,13-tridecaneamine (20 equiv) in anhyd. DMF; (v) TFA/H2O/Thioanisole/Triisopropylsilane/Ethanedithiol 82.5:5:5:5:2.5; m is 3, 6, 9, 12, or 15; n is 1 or 2; o is 3, 6, 12, or 18.
EXPERIMENTAL PROCEDURES
General
All solvents and reagents were purchased from commercial sources and used without further purification. Peptides were synthesized on Tentagel Rink amide resin (initial loading: 0.2 mmol/g) using Nα-Fmoc protecting groups and a standard DIC/HOCt or HBTU/HOBt activation strategy, as published previously.4,16 The purity of final products was analyzed using Waters high-performance liquid chromatography (HPLC) apparatus and with a Vydac C18 reverse-phase column (diam × length, 4.6 mm × 150 mm; pore size: 3 μm). Purification of compounds was achieved using a Waters 600 HPLC apparatus equipped with a Vydac C18 reverse phase column (22 × 250 mm, 5 μm) under optimized gradients and flow rates, and monitored at 230 and 280 nm. Size exclusion chromatography was performed on a borosilicate glass column (2.6 × 250 mm, Sigma, St. Louis, MO) filled with medium-sized Sephadex G-25 or G-10. Solid-phase extraction (SPE) was employed where simple isolation of final compound was needed from excess salts and buffers, using C-18 Sep-Pak cartridges. Mass spectra of positive ions were recorded either with a single stage reflectron MALDI-TOF mass spectrometer (Bruker Rexlex III, Bruker Daltonics, Billerica, MA; α-cyanocinnamic acid as a matrix) in reflectron mode or with a low-resolution ESI mass spectrometer (Finnigan, Thermoquest LCQ ion trap instrument, Lake Forrest, CA) and/or using high-resolution Fourier transform mass spectrometer (FT-ICR MS, Bruker Apex Qh, Bremen, Germany) equipped with an ESI source. The peptide concentrations were determined by monitoring absorbance of peptides against 0.5 mM solution of tryptophan (D or L) in DMSO at 280 nm. (See Supporting Information for details.)
Molecular Modeling and Circular Dichroism of Linkers
The computation experiments were carried out using Macro-Model 9.1 implemented under Maestro 7.5 interface on a Linux Dell workstation, and with MacroModel implementations of AMBER*, MMFF, and OPLS all-atom force fields and GB/SA continuum model for water.17 GPCR intra- and inter-receptor distance estimations were carried out using PDB file 1GZM18 loaded in PyMOL program. CD measurements were carried out on a Jasco model J-710 spectropolarimeter using thermostatted quartz cuvettes (0.1 cm path length) (see ref 3 and Supporting Information).
Cloning, Transfection, and Cell Culture
HEK293 cells over-expressing either or both of the human melanocortin-4 receptor (hMC4R) and the cholecystokinin-2 (CCK-2) receptor were used to assess the binding. HEK293/hMC4R cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) with 10% FBS. Monovalent CCK binding was tested on HEK293 cells with stable expression of CCK-2 receptor. These cells were grown in DMEM supplemented with 10% FBS and were maintained under selection with 100 μg/mL Zeocin. Evaluation of bivalent ligand binding was completed on HEK293/hMC4R cells with transient expression of CCK-2R (referred to as HEK293/hMC4R/tCCK cells). For transient receptor expression, cells were plated at a density of 10 000 cells/well in Wallac B&W Isoplate TC (Wallac/PerkinElmer, 1450–583) 96-well plates. The day after plating, cells were transfected with CCK-2R using FuGENE 6 Transfection Reagent (Roche, 1814–443). A 3:1 ratio of FuGENE to DNA was used, as per reagent protocol. Reagents (30 μL media, 0.15 μL FuGENE 6, and 0.05 μg CCK-2R DNA per well) were added to a sterile microtube and incubated at room temp for 15 min. The media used during the incubation was antibiotic-free, serum-free DMEM. After the 15 min incubation, the reaction mixture (30 μL) was added to the cells in their normal media. It was determined that 72 h post-transfection was optimal for high surface expression of CCK-2R; thus, all binding assays were performed 72 h post-transfection. The presence of both receptors on a dual-expressing cell line was ascertained with antibody staining (nearly 100% coverage of cells with both receptors was observed). (See ref 19 on more details of various cell lines generated and protocols.)
Receptor Number Determination
The number of receptors present on the cell surface was determined with saturation binding analysis followed by correlation of fluorescent signal achieved at Bmax. Increasing amounts of Eu-labeled ligand were added to cells until saturation was achieved. For hMC4R, the Bmax was determined to be 95 200 ± 2500 AFU (Figure 2A). A standard curve relating the fluorescent signal to the concentration of Eu-labeled ligand present in the well was produced and used to determine the concentration of Eu-ligand present at saturation (Figure 2B). In case of hMC4R, a signal of 95 200 AFU correlates with 380 fmol/well. Assuming that during the saturation study that each receptor is bound by a single Eu-labeled ligand, this correlates with 2.3 × 1011 receptors/well. The average number of cells/well was obtained via counting with a hemocytometer and this number (61 800 cells/well, n = 10) was then used to determine the number of hMC4R receptors per cell. The same process was followed for determining the number of CCK-2R per cell (also see Supporting Information Figure S6 for more details).
Figure 2.

Receptor number determination and representative binding curves. (A) Saturation binding analysis of Eu-DTPA-NDP-α-MSH binding to HEK293/hMC4R/tCCK cells where (■), (○), and (▲) indicate total, specific, and nonspecific binding, respectively. From these data, Kd = 1.30 ± 0.14 nM and Bmax = 95 200 ± 2500 AFU. (B) Standard curve relating [Eu-DTPA-NDP-α-MSH] to fluorescent signal. For binding to the hMC4R, the Bmax was determined to be 95 200 ± 2500 AFU, which correlates to 380 fmol/well. (C) The ligands were evaluated for their monovalent and bivalent binding by comparing them against Eu-labeled lanthaligands. Single plot IC50 values were determined where data from all n measurements were pooled first and nonlinear regression analysis performed. Binding of ligand 12c compared with 0.1 nM Eu-DTPA-CCK8 in HEK293/CCK cells, with an IC50 of 130 nM. (D) Binding of ligand 12c compared with 0.1 nM Eu-DTPA-CCK8 in HEK293/hMC4R/CCK cells, with an IC50 of 38 nM.
Lanthanide-Based Binding Assays
Lanthanide-based competitive binding assays were conducted according to the method which has been previously described.19–21 (See Supporting Information for detailed protocols and Figure 2 and S7 for representative binding curves.)
Digital Imaging Microscopy
Cells were grown on #1 cover-slips harbored in individual wells of six-well culture dishes. Individual slides were mounted in a chamber maintained at 37 °C on the stage of an inverted Olympus IX70 microscope equipped with a 40× 1.35 NA objective. For excitation of Cy5 fluorescence, white light emitted from a 150 W Xe lamp was passed through a 10 nm band-pass filter centered at 640 nm. The emitted light was selected using a band-pass filter centered at 680 nm, and subsequently imaged onto a CCD camera (Photometrics CH-350; TEK-512 chip). Baseline control images were acquired at 5 min intervals prior to addition of the labeled htBVL to incubation medium. Following 3 min of ligand addition, the medium was replaced with ligand-free medium prior to further image acquisition. Control images were subtracted from images acquired from the same ligand bound cells. Image analysis was performed on a SGI Indy-2 workstation using customized software.
RESULTS AND DISCUSSION
Ligand Design
In order to test the proof-of-hypothesis that heteromultivalent ligands (htMVL) will specifically and selectively bind (noncovalently) to cells bearing the appropriate combination of complementary heterologous cell-surface receptors (Figure 1A), two model receptors—hMC4R and CCK-2R—were chosen. To estimate the linker length required to engage the two proof-of-principle receptors used in this study, G-protein coupled receptors (GPCRs) were modeled as dimers, based on the crystal structure of rhodopsin in a trigonal crystal form with two protein molecules per asymmetric unit.18 The width of a GPCR dimer was estimated to be 70 Å along the longer elliptical axis in a closely packed form. The centers of the binding pockets in each case were located approximately 10–15 Å from the nearest edge of the elliptical seven-membrane bundle (off-center). Thus, depending upon the orientation of binding pockets, the inter-receptor distance could span 20–50 Å (Figure 1B; also see Supporting Information Figure S3 for details). Any association of annular lipids with GPCRs may add 8 Å per layer of lipid molecules.22 Thus, in the case of loosely packed dimers, the inter-receptor distance could extend up to 100 Å or more (also see refs 3 and 14 for discussion on GPCR dimer modeling). These estimations assume that, upon dimerization, the two receptors do not participate in domain swapping.23 Upon the basis of these estimates, three families of htBVLs were designed composed of semirigid Pro-Gly linkers and/or flexible PEGO linkers (see Scheme 1 and Table 1). The design of these htBVLs required placement of the α-MSH peptide at the N-termini and the CCK peptide at the C-termini of the linkers because the CCK peptide requires the free amide form of the carboxyl terminus for optimal binding.24 Moderately weak ligands, namely, MSH(7) and CCK(6) (Scheme 1, inset), were used in place of more potent analogues, as it was anticipated that a more pronounced enhancement might be achieved with weaker ligands in a bivalent binding mode.25,26 The binding affinities of monovalent Ac-MSH(7)-NH2 [Ac-Ser-Nle-Glu-His-DPhe-Arg-Trp-NH2] and Ac-CCK(6)-NH2 [Ac-Gly-Nle-Trp-Nle-Asp-Phe-NH2] ligands, in terms of IC50, are 39 ± 4 nM and 26 ± 4 nM, respectively.
Table 1.
Binding Data of Heterobivalent Ligands Tested for Cross-Linking in HEK293/hMC4R/tCCK-2R (hMC4R > CCK-2R)a
| no. | linker | linker length (atoms) | estimated linker length (Å) | IC50 (nM) of CCK(6)b
|
fold increase | IC50 (nM) of MSH(7)b
|
fold increase | ||
|---|---|---|---|---|---|---|---|---|---|
| CCK-2Rc | dual expressione | hMC4Rd | dual expressione | ||||||
| 5a | -[PG]3- | 18 | ≤13 | 11 (8.3–15) | 1.4 (1.0–2.0) | 8 | 11 (8.9–12) | 1300 (880–1900) | 0 |
| 5b | -[PG]6- | 36 | ≤25 | 46 (40–52) | 2.3 (1.8–2.9) | 20 | 110 (85–140) | 1000 (690–1500) | 0.1 |
| 5c | -[PG]9- | 54 | ≤35 | 320 (240–420) | 170 (130–240) | 2 | 460 (250–850) | 530 (180–1500) | 0.9 |
| 5d | -[PG]12- | 72 | ≤45 | 50 (43–58) | 6.9 (5.6–8.6) | 7 | 28 (23–33) | 980 (250–3900) | 0 |
| 5e | -[PG]15 | 90 | ≤55 | 230 (190–290) | 55 (43–70) | 4 | 680 (310–1500) | 3500 (1300–9500) | 0.2 |
| 8a | -PEGO- | 20 | ≤18 | 11 (9.0–13) | 0.50 (0.31–0.82) | 22 | 28 (22–34) | 250 (210–300) | 0.1 |
| 8b | -PEGO-PEGO- | 40 | ≤36 | 50 (41–62) | 4.5 (3.4–5.8) | 11 | 60 (49–75) | 320 (260–400) | 0.2 |
| 12a | -PEGO-[PG]3-PEGO- | 58 | ≤46 | 150 (120–180) | 6.3 (5.0–7.9) | 24 | 390 (89–1700) | 1600 (560–4700) | 0.2 |
| 12b | -PEGO-[PG]6-PEGO- | 76 | ≤56 | 72 (56–94) | 20 (15–26) | 4 | 230 (160–340) | 21 (17–27) | 11 |
| 12c | -PEGO-[PG]12-PEGO- | 112 | ≤76 | 130 (110–150) | 38 (29–50) | 3 | 10 (6.7–16) | 32 (25–39) | 0.3 |
| 12d | -PEGO-[PG]18-PEGO- | 148 | ≤96 | 160 (130–200) | 93 (78–110) | 2 | 35 (26–47) | 28 (21–40) | 1.3 |
The IC50 values of Ac-MSH(7)-NH2 and Ac-CCK(6)-NH2 are 39 ± 4 nM and 26 ± 4 nM, respectively. “Fold Increase” is the ratio of IC50 values between single- and dual-expressing cell lines.
IC50 concentration in nM from at least 4 independent experiments reported. Values in parentheses represent the 95% confidence intervals of the IC50 values.
Binding data from competition with Eu-DTPA-CCK-8 (unsulfated) against CCK-2R expressing cells.
Binding data from competition with Eu-DTPA-NDP-α-MSH against hMC4R expressing cells.
Binding data from competition with either Eu-DTPA-CCK-8 (unsulfated) or Eu-DTPA-NDP-α-MSH against cells expressing both CCK-2R and hMC4R.
Linker Design and Structural Features
In our search for an ideal linker, we designed one based on polyproline helices that were modified appropriately for desirable characteristics. The polyproline helix type II (PPII) forms a highly rigid rod-like structure. Although the pitch/residue ratio of this helix may be appropriate for building a linker, ligand presentation may be conformation-dependent. The poly(Pro-Gly) linker presented here lacks residues with bulky aliphatic side chains and eliminates any functional groups to render it inert. Further, glycine at alternate positions provides flexibility in the linker backbone, compared to PPII. Remarkably, these two residues have opposing rigidity/flexibility characteristics giving the linker a semirigid structure. A representative set of computational analyses on the poly(Pro-Gly) linker and experimental confirmation is shown in Figure 3 (and Supporting Information Figure S4). Briefly, conformational analysis and molecular dynamic (MD) simulation studies on this linker predicted that the alternating Pro-Gly sequence would assume a relaxed helical conformation (Figure 3A) with a characteristic triangular shape (viewed down the radial axis) similar to PPII. However, the helix is much broader than PPII and, thus, takes a shorter pitch of 5–12 Å per turn of six residues with an average of 8 Å in the sampled MD set. As expected, the range of excursions is higher for smaller linker length. Circular dichroic (CD) analyses of 100 μM of Ac-[PG]6-NH2 in water exhibited a characteristic negative absorption band around 202 nm.27 This is slightly blue-shifted. Notably, the slight positive band at 225 nm usually seen in PPII is barely noticeable here (Figure 3C; also see Supporting Information Figure S5). This indicates that the poly(Pro-Gly) sequence does not have a predominantly rigid PPII helical conformation (about ~40% content; data not shown) but may acquire some random or other secondary structure components as well, thus, highlighting a semirigid backbone structure. This study was used as a guide to estimate the maximum linker lengths in each BVL (see Figure 3B for htBVL 12b and Table 1 for the complete set).
Figure 3.

Studies on linkers and heterobivalent ligand 12b. (A) Computationally generated structure of [PG]9 linker. (B) One of the conformations of htBVL 12b during MD simulation indicating the semirigid Pro-Gly backbone, the flexible PEGO ends, and β-turn features of MSH(7) and CCK(6) ligands with appropriate distances. (C) CD spectra of 100 μM of Ac-[PG]6-NH2 in water (pH 7) at different temperatures. The spectra reproduce a typical polyproline type II spectrum,27 albeit the positive band is not prominent.
Synthetic Strategy
The synthesis of htBVLs 5a–15 consisting of MSH(7) and CCK(6) ligands connected in a head-to-tail fashion by PEGO and/or Pro-Gly linkers are depicted in Scheme 1. The htBVLs were synthesized using a modular strategy based on parallel solid-phase synthesis.14,16 This stepwise strategy allows easy modification at any step with functional handles for further incorporation of imaging and therapeutic tags (e.g., dyes, lanthanide chelates, toxins, etc.). (See Supporting Information Experimental Procedure for details.)
Parallel Library Synthesis
Following hexapeptide CCK(6) assembly on resin, resin 2 was split into two portions. To one portion, a PEGO linker was coupled on the N-terminus following published procedures.4 The resin 6a containing a PEGO unit was again proportionally split for the synthesis of compounds 8a–b and 12a–d. At this stage, proline and glycine residues were added alternatively for resins 3a–e and 9a–d. After the final proline addition for these compounds, the Nα-Fmoc protecting group was removed. A second PEGO unit was coupled to the resins 6a and 9a–d for 6b and 10a–d, respectively. The free amine terminals of all resins were coupled with Fmoc-Trp(Boc)-OH and syntheses continued to complete the MSH(7) sequence, the Nα-terminus deprotected, then acetylated to give 4a–e, 7a–b, and 11a–d. Following cleavage with TFA cocktail, the crude peptides 5a–e, 8a–b, and 12a–d were isolated from the resin by filtration, the filtrate was reduced to low volume by evaporation using a stream of nitrogen, and the peptides were precipitated in ice-cold diethyl ether, centrifuged, washed several times with ether, dried, dissolved in water, and lyophilized to give off-white solid powders that were stored at −20 °C until purified. The final compounds were purified by size-exclusion chromatography and RP-HPLC, and characterized by ESI-MS and/or MALDI-TOF and/or FT-ICR. The yields of the crude peptides were 50–80% based on the resin weight gain, and overall, the purified yields for the syntheses were 5–30% based on the loading of the resin.
Labeled Ligands
The Cy5 label was introduced using lysine as a functional handle. After the first PEGO linker incorporation in resin 6a, an Nα-Fmoc-Nε-Mtt protected lysine was incorporated into the sequence (13) and the peptide synthesis continued to the end as above; the peptide was then cleaved from the resin and purified using preparative HPLC to give 14. For Cy5 labeling, the purified peptide was dissolved in DMSO, Cy5-NHS ester was added, and the reaction was monitored using analytical HPLC at 280 nm. The labeled peptide 15 was separated using size exclusion chromatography and lyophilized to yield a blue amorphous product. Finally, lantha-ligands (Eu-DTPA labeled NDP-α-MSH and CCK-8) were synthesized for competition binding assays as described previously.20,28
Receptor Binding Assay in Cells Co-Expressing hMC4R and CCK-2R
In order to assess the binding of htBVLs, a series of three cell lines was established. All cell lines originated from a human embryonic kidney, HEK293, parental cell line and were transfected to express either hMC4R or CCK-2R. Stable surface expression was selected and maintained by growing cells in appropriate selection media. Cell surface expression was validated with immunocytochemistry (data not shown). In order to assess htBVL binding at both receptors simultaneously, HEK293/hMC4R cells were transiently transfected with CCK-2R (referred to here as HEK293/hMC4R/tCCK-2R). High surface expression of CCK-2R was observed 72 h post-transfection, and at this time, the receptor ratio for hMC4R and CCK-2R was approximately 11:1. This time point was used to assess ligand binding to cells with dual expression using a time-resolved fluorescence (TRF) based competitive lanthaligand binding assay.19,20,29
The htBVLs were evaluated for monovalent binding in cell lines stably expressing only one of the complementary receptors (HEK293/hMC4R and HEK293/CCK-2R) and compared to bivalent binding in HEK293/hMC4R/tCCK-2R cells (see Figure 4A for assay scheme, Table 1 for cumulative data, and Figure 2C,D, Supporting Information Figure S7 for representative binding curves). The htBVLs 5b, 8a, and 12a with a semirigid Pro-Gly linker, a flexible PEGO linker, and a combination of both, respectively, showed over 20–24-fold enhancement in bivalent binding affinity as compared to monovalent binding (Figure 4B). Enhancement decreased when the length of linker was increased as evident from the series of compounds 5c–e, 8b, and 12b–d. As expected, when only one receptor was available for binding (i.e., monovalent binding), the IC50 values of CCK-(6) and MSH(7) binding motifs in the htBVLs were higher than the parent ligands, Ac-CCK(6)-NH2 and Ac-MSH(7)-NH2. This is presumably due to the entropic cost of the linker region. However, once bivalent constructs were able to bind through both ends simultaneously, the IC50’s of CCK ligand decreased, and thus, the binding affinity increased nearly 50-fold, when compared to Ac-CCK(6)-NH2 peptide (cf. bivalent IC50 of 0.5 nM for htBVL 8a vs 26 nM for Ac-CCK(6)-NH2). We hypothesize that this was due to a slower off-rate of the htBVL construct in the bivalent binding mode and the enthalpic gain provided by the apparent cooperativity effect from simultaneous binding of the MSH ligand.
Figure 4.

Binding analysis of htBVLs. (A) The ligands are tested for bivalent binding on cells expressing both complementary receptors (left), and for monovalent binding by either saturating (blocking) one of the receptors on dual-expressing cells (middle), or on cells expressing single complementary receptor (right). A cross-linking event would result in higher affinity for each ligand. (B) Plot of monovalent and bivalent IC50 of CCK(6) ligand for the htBVLs 5b, 8a, and 12a displaying up to 24-fold higher bivalent affinity. (C) Plot of monovalent and bivalent IC50 values of MSH(7) ligand for the same htBVLs revealing decrease in bivalent affinity. (D) The lack of enhancement at the more abundant receptor could be explained on the basis that (left) at low concentrations, all the high affinity cross-linked sites are occupied corresponding to the less abundant receptor; (right) any subsequent binding event for the more abundant receptor is, therefore, monovalent resulting in no significant binding enhancement.
Of particular note is the consistent lack of affinity enhancement at the hMC4R (Table 1 and Figure 4C). This phenomenon of enhancement at one receptor but not at the other appears to be related to the receptor expression ratio where only the less abundant receptor shows enhanced binding. At low nM concentration of htBVLs, the lower-abundant CCK-2 receptors get saturated with bound htBVLs and yet occupy only a small fraction of the hMC4R pool (Figure 4D, left). At higher concentrations, only hMC4R remains available for binding (as monovalent); thus, htBVLs exhibit a lack of binding enhancement (Figure 4D, right). Further, the phenomenon here may reflect the ligands’ binding to “low affinity states” of the more abundantly expressed receptor, once all the high affinity states have been occupied in these overexpressed systems.30 The results display a similar trend as was seen earlier with the δ-opioid receptor (δ-OR)/hMC4R co-expression system (6:1 receptor ratio), where a 50-fold maximum enhancement was noticed again only at the lower-abundant receptor (hMC4R in this case).14 Therefore, irrespective of the receptor pairs investigated in our studies, the reversal of the MSH(7) binding pattern in these two systems clearly shows that the effect was not linked to a particular receptor, nor was it ligand-dependent. Similarly, in our latest studies with the hMC4R/CCK-2R system with opposite receptor ratio (CCK-2R/hMC4R = 2:1), no binding enhancement was observed on CCK-2R, but up to 80-fold enhancement was seen for the hMC4R, the lower-abundant receptor in this case (data not shown).
The aim of systematically studying linker lengths in this and our previous study on hMC4R/δOR14 was twofold: (a) to estimate a general length requirement for cross-linking any two receptors with the type of linkers used here and (b) to test the broad applicability of rigid, flexible, or semirigid linkers when different receptor pairs are considered. Notably, the estimated inter-receptor distance of 25 ± 10 Å from GPCR modeling studies generally fits well with predicted linker lengths in the three best compounds here and is in agreement with reports in the literature31 (although slightly longer linker requirement was noticed with hMC4R/δOR system). Though PEGO-[PG] n-PEGO linker provided no additional gain in affinity enhancement than the [PG] or PEGO linkers alone, it was observed to cross-link the receptors at shorter [PG] lengths in both hMC4R/CCK-2R and hMC4R/δOR systems. Thus, from our studies conducted so far, PEGO-[PG]n-PEGO, where n = 3 or 6, seems to be adequate for initial investigation of optimized linkers for any cell-surface receptor pair. Further, the use of shorter [PG] sequences and flexible PEGO chains aid in synthetic ease and solubility of compounds.
It must be emphasized here that whereas enhancement through cross-linking two subsites on a protein (subsite effect) or through chelate effect (e.g., vancomycin model) can be three or more orders of magnitude,32,33 synthetic multivalent effectors binding multiple cell-surface proteins have exhibited much smaller improvements. The enhancement through (homo)bivalency has been reported to be generally 1–2 orders of magnitude.3,4,31,34 This is presumably due to the different on-/off-rates of the individual binding events relative to the limiting diffusion rates of receptors across the membrane. Also, it is notable that the lack of affinity enhancement on the more abundant receptor does not invalidate our model to achieve specificity using receptor combinations. The goal of increased affinity was shown by the CCK(6) binding data for hMC4R > CCK-2R expression system, and by MSH(7) binding data for δ-OR > hMC4R.14 Nonetheless, the results are mechanistically revealing, i.e., since the avidity changes were inferred from the measurement of affinity changes at each ligand motif by competition assays, the observed effect is an attribute of the assay scheme used. Labeled ligands, on the other hand, should display enhanced bivalent avidity on both receptor sites, as demonstrated below.
Microscopic Observation of Cell-Surface Labeling using the Cy5 Conjugated htBVL 15
In order to confirm the bivalent binding of these multivalent constructs and test the targeting specificity, a fluorescently labeled htBVL 15 was synthesized and evaluated for differential labeling of cells expressing one or two receptor types. The htBVL 12a, the most promising compound in the series, was modified in the linker region to incorporate a Cy5 dye (far-red emission profile) yielding htBVL 15. This ligand was chosen as a template since the attachment of label in between the PEGO and [PG] linker units provides necessary spacing of the label from the ligands, thus minimizing any influence on ligand binding. Figure 5A,B shows cell-surface labeling of cells expressing both hMC4R and CCK-2R at a ligand concentration of 0.2 nM. Figure 5C shows labeling of cells expressing CCK-2R only (at 0.2 nM). Figure 5D shows labeling of cells expressing hMC4R only (at 0.8 nM). The labeled ligand bound to the dual-expressing cells with high avidity, as demonstrated by the high fluorescence intensity even at subnanomolar concentrations (Figure 5A). Moreover, the ligand displayed weak binding to the cell lines expressing only single receptor at these concentrations (compare Figure 5C,D with B with same contrast scale). Since only cells that expressed both receptors bound the multivalent ligand with high affinity (Table 1), the enhanced labeling must be characteristic of dual receptor expression and bivalent cross-linking. These imaging data provide a direct visualization of the binding results and a clear demonstration that the labeled htBVL bound rapidly with relatively high avidity and with enhanced specificity to target cells that expressed the complementary receptor combination.
Figure 5.

Cell-surface labeling with the Cy5 conjugated htBVL 15. (A,B) Binding of Cy5-labeled htBVL 15 (0.2 nM) to cells expressing both hMC4R and CCK-2R. (C) Binding of 15 (0.2 nM) to cells expressing CCK-2R only. (D) Binding of 15 (0.8 nM) to cells expressing hMC4R only. Image (B) is the same as (A) with contrast and background set to levels shown in (C) and (D) to highlight and compare absolute levels of binding between dual receptor-expressing cells and monoreceptor expressing cells. The intensity differential underscores the utility of targeting receptor combinations to enhance specificity of binding of the imaging/therapeutic agent to tumor cells. Scale bar = 25 μM.
CONCLUSION
In summary, this study demonstrated that htBVLs targeted to two heterologous receptors—MSH and CCK receptors—were able to simultaneously bind them resulting in avidity enhancement. The optimized heterobivalent constructs exhibited higher “apparent” affinities (20–24-fold increase and low nanomolar range affinities, 0.5–10 nM) on cells expressing both receptors, when compared to their binding on cells with only one of the receptor. It is our contention that htBVL will specifically target cells expressing the two-receptor combination when applied at low (nM) concentrations. This is clearly borne out from studies with the Cy5 labeled htBVL 15, which showed high avidity and specificity to dual-expressing cells at subnanomolar concentrations (0.2–0.8 nM). The observed affinity gain is most likely the result of clustering of receptors (cluster effect).35 Note that the concept presented here is fundamentally different in nature and purpose from the bispecific (or bifunctional) ligands reported in literature,31 which are often overlapping pharmacophores or two pharmacophore units connected via very short linkers. Using a “receptor combination approach”, payloads attached to multivalent ligands could be targeted to cell surface receptor combinations that are expressed in cancer but are absent in normal cells.13 These payloads could be cell-specific toxins (“magic bullets”) or could have more regional effects (“smart bombs”). We believe this approach can provide novel targets and agents for various malignancies that are currently not feasible with single-receptor approaches. This may also address some of the serious concerns with conventional approaches, e.g., it is rare that the target protein is expressed only in the aberrant cells, few good single protein targets have been identified, and the fact that the expression of a single target is heterogeneous in many cancers.12 Although much remains to be studied regarding the behavior of these ligands and receptor combinations in terms of bioavailability, immunogenicity (albeit the ligands are relatively smaller in size as compared to antibodies), internalization properties, receptor densities, lateral diffusion of receptors, etc., the present data indicate that these combinations should provide a remarkably higher degree of cell-surface differentiation, and must be further explored. To this end, our current studies are directed toward the in vivo potential of this approach with the receptor systems described here.
Supplementary Material
Acknowledgments
We thank Ms. Lucinda Begay, Mrs. Renata Patek, and Ms. Tiffany Bialis for HPLC and technical assistance. This work was supported by grants R33 CA 95944 and RO1 CA 97360 from the National Cancer Institute.
ABBREVIATIONS
- AFU
Arbitrary Fluorescence Units
- AMBER
Assisted Model Building Energy Refinement
- CCK-2R
Cholecystokinin Receptor subtype 2
- CCK-6
Nle-Gly-Trp-Nle-Asp-Phe-NH2
- CCK-8
Asp-Tyr-Nle-Gly-Trp-Nle-Asp-Phe-NH2
- CD
Circular Dichroism
- CDI
N,N′-carbonyldiimidazolide
- Cy5
Cyanine 5 dye
- δ-OR
delta-opioid receptor
- DCM
dichloromethane
- DIC
N,N′-diisopropylcarbodiimide
- DIEA
diisopropylethylamine
- DMF
N,N′-dimethylformamide
- DMSO
dimethylsulfoxide
- DTPA
diethylenetriamine-N1,N2,N3,N4-pentaacetic acid
- ESI-MS
Electrospray ionization - mass spectrometry
- FT-ICR
Fourier Transform -Ion Cyclotron Resonance
- HBTU
2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HEK
human embryonic kidney
- hMC4R
human melanocortin-4-receptor
- HOBt
N-hydroxybenzotriazole
- HOCt
6-chloro-1H-hydroxybenzotriazole
- htBVL
heterobivalent ligands
- htMVL
heteromultivalent ligand
- MALDI
Matrix Assisted Laser Desorption Ionization - Time of Flight
- MD
Molecular Dynamics
- MMFF
Merck Molecular Force Field
- MSH
melanocyte-stimulating hormone
- MSH-7
Ser-Nle-Glu-His-DPhe-Arg-Trp
- Mtt
N-methyltrityl
- NDP-α-MSH
Ac-Ser-Tyr-Ser-Nle-Glu-His-DPhe-Arg-Trp-Gly-Lys-Pro-Val-NH2
- NHS
N-hydroxysuccinimide
- OPLS
Optimized Potentials for Liquid Simulations
- PEGO
19-amino-5-oxo-3,10,13, 16-tetraoxa-6-azanonadecan-1-oic acid
- RP-HPLC
reverse-phase high performance liquid chromatography
- SPPS
solid-phase peptide synthesis
- SD
stochastic dynamics
- THF
tetrahydrofuran
- TFA
trifluoroacetic acid
- TRF
time-resolved fluorescence
Footnotes
Supporting Information. Additional experimental data and details can be found in Table S1 and Figures S1–S7. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hornick CL, Karush F. Antibody affinity. III. The role of multivalance. Immunochemistry. 1972;9:325–340. doi: 10.1016/0019-2791(72)90096-1. [DOI] [PubMed] [Google Scholar]
- 2.Mammen M, Choi S-K, Whitesides GM. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew Chem, Int Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 3.Handl HL, Sankaranarayanan R, Josan JS, Vagner J, Mash EA, Gillies RJ, Hruby VJ. Synthesis and evaluation of bivalent NDP-alpha-MSH(7) peptide ligands for binding to the human melanocortin receptor 4 (hMC4R) Bioconjugate Chem. 2007;18:1101–9. doi: 10.1021/bc0603642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vagner J, Handl HL, Gillies RJ, Hruby VJ. Novel targeting strategy based on multimeric ligands for drug delivery and molecular imaging: homooligomers of alpha-MSH. Bioorg Med Chem Lett. 2004;14:211–5. doi: 10.1016/j.bmcl.2003.09.079. [DOI] [PubMed] [Google Scholar]
- 5.Kiessling LL, Gestwicki JE, Strong LE. Synthetic multivalent ligands as probes of signal transduction. Angew Chem, Int Ed. 2006;45:2348–2368. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma SD, Granberry ME, Jiang J, Leong SP, Hadley ME, Hruby VJ. Multivalent melanotropic peptide and fluorescent macromolecular conjugates: new reagents for characterization of melanotropin receptors. Bioconjugate Chem. 1994;5:591–601. doi: 10.1021/bc00030a015. [DOI] [PubMed] [Google Scholar]
- 7.Sharma SD, Jiang J, Hadley ME, Bentley DL, Hruby VJ. Melanotropic peptide-conjugated beads for microscopic visualization and characterization of melanoma melanotropin receptors. Proc Natl Acad Sci U S A. 1996;93:13715–20. doi: 10.1073/pnas.93.24.13715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiessling LL, Gestwicki JE, Strong LE. Synthetic multivalent ligands in the exploration of cell-surface interactions. Curr Opin Chem Biol. 2000;4:696–703. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 9.Ghaghada KB, Saul J, Natarajan JV, Bellamkonda RV, Annapragada AV. Folate targeting of drug carriers: a mathematical model. J Controlled Release. 2005;104:113–28. doi: 10.1016/j.jconrel.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 10.Saul JM, Annapragada A, Natarajan JV, Bellamkonda RV. Controlled targeting of liposomal doxorubicin via the folate receptor in vitro. J Controlled Release. 2003;92:49–67. doi: 10.1016/s0168-3659(03)00295-5. [DOI] [PubMed] [Google Scholar]
- 11.Caplan MR, Rosca EV. Targeting drugs to combinations of receptors: a modeling analysis of potential specificity. Ann Biomed Eng. 2005;33:1113–24. doi: 10.1007/s10439-005-5779-1. [DOI] [PubMed] [Google Scholar]
- 12.Gillies RJ, Hruby VJ. Expression-driven reverse engineering of targeted imaging and therapeutic agents. Expert Opin Ther Targets. 2003;7:137–9. doi: 10.1517/14728222.7.2.137. [DOI] [PubMed] [Google Scholar]
- 13.Balagurunathan Y, Morse DL, Hostetter G, Shanmugam V, Stafford P, Shack S, Pearson J, Trissal M, Demeure MJ, Von Hoff DD, Hruby VJ, Gillies RJ, Han H. Gene expression profiling-based identification of cell-surface targets for developing multimeric ligands in pancreatic cancer. Mol Cancer Ther. 2008;7:3071–80. doi: 10.1158/1535-7163.MCT-08-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vagner J, Xu L, Handl HL, Josan JS, Morse DL, Mash EA, Gillies RJ, Hruby VJ. Heterobivalent ligands crosslink multiple cell-surface receptors: the human melanocortin-4 and delta-opioid receptors. Angew Chem, Int Ed. 2008;47:1685–8. doi: 10.1002/anie.200702770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.We have coined the term “lanthaligand” to denote a lanthanide chelate labeled ligand. See Josan JS. PhD Thesis. The University of Arizona; Tucson: 2008. Heteromultivalent ligands directed targeting of cell-surface receptors - Implications in cancer diagnostics & therapeutics.
- 16.Josan JS, Vagner J, Handl HL, Sankaranarayanan R, Gillies RJ, Hruby VJ. Solid-phase synthesis of hetero-bivalent ligands targeted to melanocortin and cholecystokinin receptors. Int J Pept Res Ther. 2008;14:293–300. doi: 10.1007/s10989-008-9150-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohamadi F, Richards NGJ, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. Macromodel - an integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J Comput Chem. 1990;11:440–467. [Google Scholar]
- 18.Li J, Edwards P, Burghammer M, Villa C, Schertler G. Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol. 2004;343:1409–1438. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- 19.Xu L, Vagner J, Josan JS, Lynch RM, Morse DL, Baggett B, Han H, Mash EA, Hruby VJ, Gillies RJ. Enhanced targeting with heterobivalent ligands. Mol Cancer Ther. 2009;8:2356–2365. doi: 10.1158/1535-7163.MCT-08-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Handl HL, Vagner J, Yamamura HI, Hruby VJ, Gillies RJ. Lanthanide-based time-resolved fluorescence of in cyto ligand–receptor interactions. Anal Biochem. 2004;330:242–250. doi: 10.1016/j.ab.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 21.Handl HL, Gillies RJ. Lanthanide-based luminescent assays for ligand-receptor interactions. Life Sci. 2005;77:361–371. doi: 10.1016/j.lfs.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 22.Carnie S, Israelachvili JN, Pailthorpe BA. Lipid packing and transbilayer asymmetries of mixed lipid vesicles. Biochim Biophys Acta. 1979;554:340–57. doi: 10.1016/0005-2736(79)90375-4. [DOI] [PubMed] [Google Scholar]
- 23.Hadac EM, Ji Z, Pinon DI, Henne RM, Lybrand TP, Miller LJ. A peptide agonist acts by occupation of a monomeric G protein-coupled receptor: dual sites of covalent attachment to domains near TM1 and TM7 of the same molecule make biologically significant domain-swapped dimerization unlikely. J Med Chem. 1999;42:2105–11. doi: 10.1021/jm980732q. [DOI] [PubMed] [Google Scholar]
- 24.Fourmy D, Escrieut C, Archer E, Galès C, Gigoux V, Maigret B, Moroder L, Silvente-Poirot S, Martinez J, Fehrentz JA, Pradayrol L. Structure of cholecystokinin receptor binding sites and mechanism of activation/inactivation by agonists/antagonists. Pharmacol Toxicol. 2002;91:313–20. doi: 10.1034/j.1600-0773.2002.910608.x. [DOI] [PubMed] [Google Scholar]
- 25.Handl HL, Vagner J, Han H, Mash EA, Hruby VJ, Gillies RJ. Hitting multiple targets with multimeric ligands. Expert Opin Ther Targets. 2004;8:565–86. doi: 10.1517/14728222.8.6.565. [DOI] [PubMed] [Google Scholar]
- 26.Vagner J, Handl HL, Monguchi Y, Jana U, Begay LJ, Mash EA, Hruby VJ, Gillies RJ. Rigid linkers for bioactive peptides. Bioconjugate Chem. 2006;17:1545–50. doi: 10.1021/bc060154p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ladokhin AS, Selsted ME, White SH. CD spectra of indolicidin antimicrobial peptides suggest turns, not polypro-line helix. Biochemistry. 1999;38:12313–12319. doi: 10.1021/bi9907936. [DOI] [PubMed] [Google Scholar]
- 28.Reubi JC, Waser B, Schaer JC, Laederach U, Erion J, Srinivasan A, Schmidt MA, Bugaj JE. Unsulfated DTPA-and DOTA-CCK analogs as specific high-affinity ligands for CCK-B receptor-expressing human and rat tissues in vitro and in vivo. Eur J Nucl Med. 1998;25:481–490. doi: 10.1007/s002590050247. [DOI] [PubMed] [Google Scholar]
- 29.Handl HL, Vagner J, Yamamura HI, Hruby VJ, Gillies RJ. Development of a lanthanide-based assay for detection of receptor–ligand interactions at the δ-opioid receptor. Anal Biochem. 2005;343:299–307. doi: 10.1016/j.ab.2005.05.040. [DOI] [PubMed] [Google Scholar]
- 30.Huang J, Dong L, Lebreton G. Mass-dependent signaling between G protein coupled receptors. Cell Signal. 2006;18:564–576. doi: 10.1016/j.cellsig.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 31.Portoghese PS. From models to molecules: opioid receptor dimers, bivalent ligands, and selective opioid receptor probes. J Med Chem. 2001;44:2259–69. doi: 10.1021/jm010158+. [DOI] [PubMed] [Google Scholar]
- 32.Rao J, Lahiri J, Isaacs L, Weis RM, Whitesides GM. A trivalent system from vancomycin. DAla-DAla with higher affinity than avidin.biotin. Science. 1998;280:708–11. doi: 10.1126/science.280.5364.708. [DOI] [PubMed] [Google Scholar]
- 33.Shrivastava A, Von Wronski M, Sato A, Dransfield D, Sexton D, Bogdan N, Pillai R, Nanjappan P, Song B, Marinelli E, Deoliveira D, Luneau C, Devlin M, Muruganandam A, Abujoub A, Connelly G, Wu Q, Conley G, Chang Q, Tweedle M, Ladner R, Swenson R, Nunn A. A distinct strategy to generate high-affinity peptide binders to receptor tyrosine kinases. Protein Eng Des Sel. 2005;18:417–424. doi: 10.1093/protein/gzi049. [DOI] [PubMed] [Google Scholar]
- 34.Carrithers MD, Lerner MR. Synthesis and characterization of bivalent peptide ligands targeted to G-protein-coupled receptors. Chem Biol. 1996;3:537–42. doi: 10.1016/s1074-5521(96)90144-1. [DOI] [PubMed] [Google Scholar]
- 35.Zheng Y, Akgun E, Harikumar KG, Hopson J, Powers MD, Lunzer MM, Miller LJ, Portoghese PS. Induced Association of μ Opioid (MOP) and Type 2 Cholecystokinin (CCK 2) Receptors by Novel Bivalent Ligands. J Med Chem. 2009;52:247–258. doi: 10.1021/jm800174p. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.